
Abstract
Background
Hypertrophic olivary degeneration (HOD) is a unique type of neuronal degeneration presenting as hypertrophy, in contrast to atrophy as seen in most cases. It presents with classical characteristic clinical features due to involvement of dentate-rubral-olivary pathway, also described as triangle of Guillain and Mollaret formed in midbrain, pons and cerebellum. It can be idiopathic or secondary to infarction, bleeding, tumours, trauma or demyelination. However, the mechanism is still unclear. Herein, we present a case of HOD that had developed after post-traumatic pontine and midbrain haemorrhagic contusion.
Case presentation
A young male patient presented with progressively increasing tremors of both hands, inability to walk and multiple cranial nerve palsy. Magnetic resonance imaging demonstrated bilateral inferior olivary nucleus enlargement and signal changes seen as T2 and T2-FLAIR hyperintensities and non-enhancing T1 iso-intensities. Based on these features, diagnosis of HOD was made. Patient was kept on conservative management and his condition improved.
Conclusions
Hypertrophic olivary degeneration is a unique neuronal degeneration with typical clinical manifestations and distinct imaging features. Proper and early recognition and multidisciplinary treatment approach can result in the best outcomes for the patient.
Similar content being viewed by others

Background
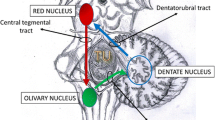
Dentatorubro-olivary pathway (DROP) is a circuit formed by synapses in the brainstem and cerebellum involving inferior olivary nucleus (ION) and red nucleus (RN) on the same side and dentate nucleus (DN) from the opposite side forming a triangle also known as Guillain–Mollaret triangle (GMT)[1] (Fig. 1). It basically modulates the motor activity mediated by spinal cord. It was first described by Oppenheim in 1887 in a post-mortem study [2]. In 1931, Guillain and Mollaret studied this pathway extensively [3]. The arms of the GMT are formed by central tegmental tract (CTT) connecting the RN with ION, inferior cerebellar peduncle (ICP) connecting the DN and ION and superior cerebellar peduncle (SCP) connecting the DN and RN. Patients with hypertrophic olivary degeneration (HOD) present with classical signs and symptoms of nystagmus (ocular myoclonus), cerebellar ataxia, palatal myoclonus and Holmes tremor (affecting upper limbs) [4,5,6]. Signal alterations in the ION can be seen on MRI which is the preferential imaging method [7]. Sometimes metastasis or tumour recurrence may mimic these signal changes rendering the diagnosis difficult. In patients with injury to brainstem and cerebellum, if these clinical features present during the course of treatment or improvement, the possibility of HOD should be considered. The management of HOD is mainly pharmaceutical. Rehabilitation can be beneficial for patients with limb dysfunction or in patient of post-traumatic brain injury [8]. Herein, we present a case of HOD which had developed after head injury.

Guillain–Mollaret triangle: (RN—red nucleus, DN—dentate nucleus, ION—inferior olivary nucleus)
Case presentation
A twenty-four-year-old male patient presented in Neurology department with complaints of progressively increasing tremors of both hands for past three months exaggerated by posture and activity, along with bilateral vertical nystagmus, inability to walk and multiple cranial nerve palsy. The patient also complained of increased involuntary movement of the tongue and mouth. On clinical examination, both hands exhibited persistent rhythmic and involuntary tremors classically known as Holmes tremors (Video 1), the left eyeball slightly clustered with medial deviation, left-sided deviation of tongue on protrusion (Video 2), instability on left hand finger-nose test, instability on walking and dysdiadochokinesia.
Patient had past history of road traffic accident one year back, for which he was admitted in the Emergency medicine department of our institute. From the Hospital Information system of our institute, we retrieved clinical details of his previous hospitalization. At the time of admission, he was unconscious with Glasgow Coma score of E2V2M5 and right-sided hemiparesis. On clinical examination, there was left third and sixth nerve palsy. The rest of the cranial nerves could not be examined. A clinical diagnosis of diffuse axonal injury was made. Non-contrast computed tomography performed at that time revealed haemorrhagic contusion with adjacent oedema on the left side of the midbrain and pons (Fig. 2). Magnetic resonance imaging (MRI) was performed for further evaluation which revealed same findings, i.e. haemorrhagic contusion involving left side of mid brain and pons (Fig. 2). No other positive finding was present in the rest of the brainstem, cerebrum or cerebellum. Patient was treated conservatively and discharged after one week in stable condition on Ryle’s tube feeding under supportive care. He regained consciousness in one month time. As per history given by relatives, his hemiparesis also improved and he started walking with support. His condition gradually improved until he started developing complaining involuntary hand movements for which he had recently presented.

Non-contrast CT performed few hours after road traffic accident showed hyperdense haemorrhagic contusion in the midbrain and pons on left side (arrow)
MRI was advised for evaluation of the cause of tremors. It revealed haemosiderin staining (as a result of old contusion) in midbrain and pons with surrounding gliosis (Fig. 3). Both inferior olivary nuclei (ION) were hypertrophied along with T2 and T2-FLAIR hyperintensities and non-enhancing T1 iso-intensity. No evidence of diffusion restriction or blooming on Gradient echo sequence was noted (Figs. 4, 5). The diagnosis was challenging; however, after careful consideration of clinical manifestations and imaging findings, a diagnosis of HOD was made.

MRI performed next day, demonstrated T2 and T1 hypointense haemorrhagic contusion in midbrain and pons on left side with surrounding oedema (arrow)

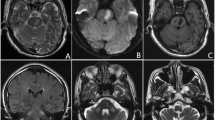
MRI performed 1 year later, at the level of the midbrain shows changes of chronic bleed [marked with arrow] on A T2WI, B T1WI, C diffusion-weighted image, D GRE sequence and E post-contrast fat supressed T1WI. MRI at the level of the medulla shows F hyperintensity and enlargement of ION on T2WI, G iso-intense on T1WI, H no hyperintensity on DWI, I no blooming on GRE and J no post-contrast enhancement

Hypertrophic olivary degeneration MRI: T2 hyperintensity with enlargement of the inferior olivary nucleus in axial and coronal planes (A, B)
On admission, clonazepam and levetiracetam were added to suppress tremors. Patient was kept on medications and rehabilitation for one month, which was ensured by relatives, his tremors were reduced and only slight nystagmus was noted. There were no adverse and unanticipated events throughout this period. He was advised to continue with medication and supporting care and remain in regular follow-up.
Discussion
HOD is due to neuronal degeneration in the form of hypertrophy of neurons rather than atrophy which is commonly seen. The pathophysiology of HOD can be understood by its development in six stages [9, 10]. In the first stage, no abnormality is detected in the ION. Second stage shows ION degeneration seen as mild neuronal hypertrophy without glial proliferation on microscopy during 3 weeks, In the third stage, ION hypertrophy is seen due to both proliferation of both neurons and astrocyte during 6 months which can be attributed to the loss of GABA (Gamma-aminobutyric acid)-mediated inhibition of RN over ION. Fourth stage shows ION enlargement and fifth stage shows persistent pseudohypertrophy (with dissolution of neurons) which remains up to 3–4 years. In the final stage after few years, neuronal loss leads to atrophy [11, 12]. HOD can be unilateral or bilateral. Usually in unilateral HOD, lesion and affected ION show causal association but no attributable structural lesions found in most of the bilateral HOD [13, 14]. Causes of unilateral and bilateral HOD may or may not be similar. Commonly DROP lesion can lead to unilateral HOD. Unilateral HOD can be attributed either due to ipsilateral primary lesion affecting RN/ CTT or due to contralateral primary lesion affecting DN or SCP. Bilateral HOD can be either idiopathic or due to lesions involving bilateral DROP pathway [7]. In a study, in patients with HOD undergoing MRI, approximately 44% patients had no identifiable lesions in the DROP and out of them; approx. 45% even had no lesions outside the DROP pathway [13]. This means that bilateral HOD is usually due to neurodegeneration, which can be consolidated by a study of paediatric cases with ION involvement showing neurodegeneration [15]. In our case, haemorrhagic contusion in the left midbrain and pons caused bilateral HOD. Few gene mutations like polymerase gamma gene (POLG), SIRF1, TTC19 and AIFM1 encoding mitochondria have shown associations with bilateral HOD [16,17,18,19,20,21].
The classical clinical features of HOD are ataxic gait, nystagmus, myoclonus of palate, soft palate tremor and limb clonus. Out of these, palatal myoclonus may not be seen in all HOD patients. Terao et al. reported absence of palatal myoclonus in two patients [22]. Our patient presented with mouth and tongue myoclonus. Limb clonus is predominant in upper extremity, usually unilateral, with very rare truncal or lower extremity spread [23,24,25]. Some posture or intention may instigate them which do not appear in sleep [26]. The tremors are thought to be due amplification of low-frequency oscillations by cerebellum due to GMT injury [27, 28], while ION degeneration leads to abnormal function of cerebellum leading to ataxia. [29, 30].
Best imaging modality to diagnose HOD is MRI which usually shows increase bulk of olivary nucleus with hyperintense signal on T2-weighted imaging and non-enhancing iso-intense to hypointense signal on T1-weighted imaging. T2-weighted imaging can pick up signal changes from one month which can be seen well during 8–18 months before normalising completely around 3–4 years [31, 32]. Diffusion tensor imaging (DTI) has been found useful in determining the severity of disease by analysing fibres of tracts of white matters [33]. Cheng et al. described DTI findings in two patients of pontine haemorrhage which showed the interruption and sparse of the left cortico-pontine tracts [34]. HOD is differentiated from T2 hyperintensity of the ION on MRI. Imaging is not satisfactory for early detection of disease.
HOD was first described by the Oppenheim in 1887 based on his post-mortem study [2]. But it took years to comprehend the pathophysiology of this entity until Guillain and Mollaret described HOD in a patient with oculopalatal myoclonus and a lesion on the ION. They also detailed the anatomy of this condition and named it the Guillain–Mollaret triangle [3]. MRI findings in a case of HOD were first reported by Sperling in 1985. Since then, many case series and case reports have been published in the literature [35]. A series of 11 cases was published by Yun JH et al. They described development of HOD after surgical resection or gamma knife radiosurgery for brainstem cavernous malformations [36]. Hornyak M reported a series of four patients who developed delayed HOD after surgical resection of pontine lesions [37]. Carr et al. performed a retrospective study on 102 patients with radiology findings consistent with HOD. They found that 76% patients had bilateral ION lesions and in 44% of the cases, a lesion could not be identified to explain HOD. They also found that bilaterality was more common in the non-lesional group [14]. Tommaso et al. retrospectively evaluated 58 patients who underwent surgery for posterior cranial fossa tumours. They found T2 signal alterations in ION in 19 cases (15 bilateral and four unilateral) and olivary hypertrophy in only 7 cases. Dentate nucleus damage was present in all cases [38]. Yuki et al. reported MR findings of HOD after surgical resection of brain tumour in seven cases. All cases demonstrated T2 hyperintensity with olivary hypertrophy in 5 cases. Two patients showed both enlargement and T2-hyperintensity of the ION on initial follow-up MRI, whereas in three patients ION enlargement appeared on serial follow-up MR images [10]. The only limitation of our study is that it describes a single case.
Usually, conservative management of HOD results in spontaneous resolution of symptoms. Some drugs likely levodopa, carbamazepine, propranolol, tiapride and clonazepam are used for treatment. Levetiracetam is antiepileptic drug which has shown to reduce tremor, dyskinesia and myoclonus [39]. Most drugs may not completely control tremor. In severe or refractory cases, deep brain stimulation has shown promising results [39]. In our case, after oral administration of clonazepam, levodopa and gabapentin, tremor was significantly reduced. Disturbance in motor control can be attributed to the balance and sensory disorders and ataxic gait in our patient which made difficulties in rehabilitation. Few cases of soft palate clonus have been managed successfully with local botulinum toxin injection [40,41,42].
Conclusions
Multidisciplinary approach is essential for successful diagnosis and management of this rare subset of neuronal degeneration. Hypertrophic olivary degeneration usually presents with classical clinical features which can be correlated to the imaging findings of increased volume and signal changes of inferior olivary nucleus. The disease can be idiopathic or may manifest months after the primary causal lesion (like haemorrhage in our case).
Availability of data and materials
The datasets used during the current study can be made available from the corresponding author on reasonable request.
Abbreviations
- CTT:
-
Central tegmental tract
- DN:
-
Dentate nucleus
- DROP:
-
Dentate-rubral-olivary pathway
- DTI:
-
Diffusion tensor imaging
- FLAIR:
-
Fluid-attenuated inversion recovery
- GMT:
-
Guillain–Mollaret triangle
- HOD:
-
Hypertrophic olivary degeneration
- ION:
-
Inferior olivary nucleus
- MRI:
-
Magnetic resonance imaging
- RN:
-
Red nucleus
- SCP:
-
Superior cerebellar peduncle
- T1WI:
-
T1-weighted imaging
- T2WI:
-
T2-weighted imaging
References
Murdoch S, Shah P, Jampana R (2016) The Guillain–Mollaret triangle in action. Pract Neurol 16:243–246. https://doi.org/10.1136/practneurol-2015-001142
Oppenheim H (1887) Olivendegeneration bei Atheromatese der basalen Hirnarterien [Article in German]. Berl Klin Wschr 638–639
Guillain G, Mollaret P (1931) Deus de myoclonies synchrones et rhythmees velopharyngolaryngo-oculo-diaphragmatiques [Article in French]. Rev Neurol 545:546
Sabat S, Mannering N, Agarwal A (2016) Hypertrophic olivary degeneration: case series and review of literature. J Neurol Sci 370:180–186. https://doi.org/10.1016/j.jns.2016.09.055
Konno T, Broderick DF, Tacik P, Caviness JN, Wszolek ZK (2016) Hypertrophic olivary degeneration: a clinico-radiologic study. Parkinsonism Relat Disord 28:36–40. https://doi.org/10.1016/j.parkreldis.2016.04.008
Blanco Ulla M, López Carballeira A, Pumar Cebreiro JM (2015) Magnetic resonance imaging of hypertrophic olivary degeneration [Article in English, Spanish]. Radiologia 57:505–511. https://doi.org/10.1016/j.rx.2014.12.008
Cachia D, Izzy S, Smith T, Ionete C (2013) A rare presentation of hypertrophic olivary degeneration secondary to primary central nervous system lymphoma. JAMA Neurol 70:1192–1193. https://doi.org/10.1001/2013.jamaneurol.218
Zhou J, Liu Y, Ali Shah SZ (2021) Hypertrophic olivary degeneration following cerebral trauma managed with rehabilitation—a case report. J Pak Med Assoc 71:1252–1254
Pandey P, Westbroek EM, Gooderham PA, Steinberg GK (2013) Cavernous malformation of brainstem, thalamus, and basal ganglia: a series of 176 patients. Neurosurgery 72: ion
Shinohara Y, Kinoshita T, Kinoshita F, Kaminou T, Watanabe T et al (2013) Hypertrophic olivary degeneration after surgical resection of brain tumors. Acta radiologica (Stockholm, Sweden: 1987) 54:462–466
Revel MP, Mann M, Brugieres P, Poirier J, Gaston A (1991) MR appearance of hypertrophic olivary degeneratIO after contralateral cerebellar hemorrhage. AJNR Am J Neuroradiol 12:71–72
Araujo NA, Raeder MT, da Silva Junior NA, Oshima MM, Parizotto LO, Reis F (2015) Hypertrophic olivary degeneration secondary to central tegmental tract injury. Radiol Bras 48:199–200. https://doi.org/10.1590/0100-3984.2014.0075
Gu CN, Carr CM, Kaufmann TJ, Kotsenas AL, Hunt CH, Wood CP (2015) MRI findings in nonlesional hypertrophic olivary degeneration. J Neuroimaging 25:813–817. https://doi.org/10.1111/jon.12267
Carr CM, Hunt CH, Kaufmann TJ, Kotsenas AL, Krecke KN, Wood CP (2015) Frequency of bilateral hypertrophic olivary degeneration in a large retrospective cohort. J Neuroimaging 25:289–295. https://doi.org/10.1111/jon.12118
Mirabelli-Badenier M, Morana G, Bruno C, Di Rocco M, Striano P, De Grandis E, Veneselli E, Rossi A, Biancheri R (2015) Inferior olivary nucleus involve ment in pediatric neurodegenerative disorders: does it play a role in neuro imaging pattern-recognition approach? Neuropediatrics 46:104–109
Van Goethem G, Dermaut B, Löfgren A, Martin JJ, Van Broeckhoven C (2001) Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 28:211–212. https://doi.org/10.1038/90034
Zoulis C, Engelsen BA, Telstad W, Aasly J, Zeviani M, Winterthun S, Ferrari G, Aarseth JH, Bindoff LA (2006) The spectrum of clinical disease caused by the A467T and W748S POLG mutations: a study of 26 cases. Brain 129:1685–1692. https://doi.org/10.1093/brain/awl097
Sonam K, Khan NA, Bindu PS, Taly AB, Gayathri N, Bharath MM, Govindaraju C, Arvinda HR, Nagappa M, Sinha S, Thangaraj K (2014) Clinical and magnetic resonance imaging findings in patients with Leigh syndrome and SURF1 mutations. Brain Dev 36:807–812. https://doi.org/10.1016/j.braindev.2013.10.012
Bindu PS, Taly AB, Sonam K, Govindaraju C, Arvinda HR, Gayathri N, Bharath MM, Ranjith D, Nagappa M, Sinha S, Khan NA, Thangaraj K (2014) Bilateral hypertrophic olivary nucleus degeneration on magnetic resonance imaging in children with Leigh and Leigh-like syndrome. Br J Radiol 87:20130478. https://doi.org/10.1259/bjr.20130478
Ghezzi D, Arzuffi P, Zordan M, Da Re C, Lamperti C, Benna C, D’Adamo P, Diodato D, Costa R, Mariotti C, Uziel G, Smiderle C, Zeviani M (2011) Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet 43:259–263. https://doi.org/10.1038/ng.761
Ardissone A, Piscosquito G, Legati A, Langella T, Lamantea E, Garavaglia B, Salsano E, Farina L, Moroni I, Pareyson D, Ghezzi D (2015) A slowly progressive mitochondrial encephalomyopathy widens the spectrum of AIFM1 disorders. Neurology 84:2193–2195. https://doi.org/10.1212/WNL.0000000000001613
Terao S, Sobue G, Takahashi M, Osano Y, Shimada N, et al (1994) [Chronological changes in MR imaging of inferior olivary pseudohypertrophy-report of two cases]. No to shinkei = Brain and nerve 46:1184–1189
Kim JS, Park JW, Kim YI, Han SJ, Kim HT et al (2006) Tremors associated with an inferior olivary lesion that developed after a pontine hemorrhage. Movement Disord 21(1539–1540):13
Menéndez DF, Cury RG, Barbosa ER, Teixeira MJ, Fonoff ET et al (2014) Hypertrophic olivary degeneration and holmes’ tremor secondary to bleeding of cavernous malformation in the midbrain. Tremor and other Hyperkinetic Movements (New York, NY) 4:264
Choi SM (2016) Movement disorders following cerebrovascular lesions in cerebellar circuits. J Movement Disord 9:80–88
Kim MK, Cho BM, Park SH, Yoon DY (2014) Holmes’ tremor associated with bilateral hypertrophic olivary degeneration following brain stem hemorrhage: a case report. J Cerebrovasc Endovasc Neurosurg 16:299–302
Zhang X, Santaniello S (2019) Role of cerebellar GABAergic dysfunctions in the origins of essential tremor. Proc Natl Acad Sci USA 116:13592–13601
Wright GC, Brown R, Grayton H, Livingston JH, Park SM et al (2020) Clinical and radiological characterization of novel FIG4-related combined system disease with neuropathy. Clin Genet 98:147–154
Nishie M, Yoshida Y, Hirata Y, Matsunaga M (2002) Generation of symptomatic palatal tremor is not correlated with inferior olivary hypertrophy. Brain 125:1348–1357
Shaikh AG, Hong S, Liao K, Tian J, Solomon D et al (2010) Oculopalatal tremor explained by a model of inferior olivary hypertrophy and cerebellar plasticity. Brain J Neurol 133:923–940
Goyal M, Versnick E, Tuite P, Cyr JS, Kucharczyk W et al (2000) Hypertrophic olivary degeneration: meta analysis of the temporal evolution of MR findings. AJNR Am J Neuroradiol 21(1073–1077):21
Blanco Ulla M, López Carballeira A, Pumar Cebreiro JM (2015) Magnetic resonance imaging of hypertrophic olivary degeneration. Radiologia 57:505–511
Schaller-Paule MA, Steidl E, Shrestha M, Deichmann R, Steinmetz H et al (2021) Multicenter prospective analysis of hypertrophic olivary degeneration following infratentorial stroke (HOD-IS): evaluation of disease epidemiology, clinical presentation, and MR-imaging aspects. Front Neurol 12:675123
Cheng S, Li X, Zhang W, Laakso E-L, Sun JW et al (2022) Delayed presentation of hypertrophic olivary degeneration following pontine hemorrhage: two case reports highlighting clinical presentation, diagnostics and multidisciplinary treatment. J Clin Images Med Case Rep 3(11):2133
Sperling MR, Herrmann C (1985) Jr Syndrome of palatal myoclonus and progressive ataxia: two cases with magnetic resonance imaging. Neurology 35:1212–1214
Yun JH, Ahn JS, Park JC, Kwon DH, Kwun BD, Kim CJ (2013) Hypertrophic olivary degeneration following surgical resection or gamma knife radiosurgery of brainstem cavernous malformations: an 11-case series and a review of literature. Acta Neurochir (Wien) 155(3):469–476. https://doi.org/10.1007/s00701-012-1567-y
Hornyak M, Osborn AG, Couldwell WT (2008) Hypertrophic olivary degeneration after surgical removal of cavernous malformations of the brain stem: report of four cases and review of the literature. Acta Neurochir (Wien) 150(2):149–156. https://doi.org/10.1007/s00701-007-1470-0
Tartaglione T, Izzo G, Alexandre A, Botto A, Di Lella GM, Gaudino S, Caldarelli M, Colosimo C (2015) MRI findings of olivary degeneration after surgery for posterior fossa tumours in children: incidence, time course and correlation with tumour grading. Radiol Med 120(5):474–482. https://doi.org/10.1007/s11547-014-0477-x
Sabat S, Mannering N, Agarwal A (2016) Hypertrophic olivary degeneration: Case series and review of literature. J Neurol Sci 370:180–186
Bouz P, Woods ROJ, Woods KRM (2013) The pathophysiological basis for hypertrophic olivary degeneration (HOD) following brainstem insult. JSM Neurosurg Spine 1:1004
Zhu M, Liu C, Ren S, Lin Z, Miao L, Sun W (2015) Fusion of a supernumerary tooth to right mandibular second molar: a case report and literature review. Int J Clin Exp Med 8:11890–21185
Ohazama A, Johnson EB, Ota MS, Choi HY, Porntaveetus T, Oommen S, Itoh N, Eto K, Gritli-Linde A, Herz J, Sharpe PT (2008) Lrp4 modulates extracellular integration of cell signaling pathways in development. PLoS ONE 3:e4092. https://doi.org/10.1371/journal.pone.0004092
Acknowledgements
Not applicable.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
NS was involved in the conceptualization and manuscript writing. JM contributed to the data collection and image formation. DKS performed proof reading of the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Written consent was taken from patient’s relative as patient was not able to write due to hand tremors.
Consent for publication
Written consent was taken from patient’s relative as patient was not able to write due to hand tremors.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, N., Thakur, K.K., Singh, D.K. et al. Hypertrophic olivary degeneration following head injury: a case report. Egypt J Radiol Nucl Med 55, 122 (2024). https://doi.org/10.1186/s43055-024-01292-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43055-024-01292-1