
Abstract
Background
Lipocalin-2 (LCN2), a neutrophil gelatinase-associated protein, plays an important role in iron homeostasis, infection, and inflammation. Polymorphism in the LCN2 gene is linked to various diseases such as cardiovascular disease, renal damage, and colorectal and pancreatic cancer. Identifying deleterious functional non-synonymous SNPs in the LCN2 gene is crucial in understanding how these genetic variations affect its structure and function.
Methods
Several in silico tools such as SIFT, Polyphen-2, PROVEAN, PREDICT SNP, MAPP, and SNAP2 followed by I-MUTANT 2.0, MUpro, ConSurf, and NetsurfP-2.0, secondary structure of the protein by SOPMA and PSIPRED, while its interaction with other genes and proteins was analyzed using GeneMANIA and STRING, respectively, and AlphaFold for protein's 3D structure prediction.
Results
The study identified 6 potentially harmful nsSNPs (rs11556770, rs139418967, rs142623708, rs200107414, rs201365744, rs368926734) and their structure and function were analyzed using prediction tools. I-MUTANT 2.0 predicted an increase in stability with the nsSNPs rs139418967, while the other shows decrease in protein stability with the 6 nsSNPs (rs11556770, rs139418967, rs142623708, rs200107414, rs201365744, rs368926734) which was validated using MUpro. ConSurf identified the 6 high-risk nsSNPs to be in the conserved regions of the protein. The result showed that rs11556770, rs139418967, rs142623708, rs200107414, rs201365744, and rs368926734 were found to be highly conserved and the variant amino acids. According to NetsurfP-2.0 server, the result showed that rs11556770 (Q39H), rs139418967 (L6P), rs368926734 (Y135H) were predicted to be exposed and rs142623708 (M71I), rs200107414 (Y52C), rs368926734 (Y135) were buried. The PSIPRED server analysis indicated that the predominant secondary structure was a strand, with lesser occurrences of coil and helix.
Conclusion
Overall, the study identified detrimental nsSNPs of LCN2 using computational analysis which could be used for large population-based investigations and diagnosis.
Similar content being viewed by others
Background
Genetic polymorphisms like single nucleotide polymorphisms (SNPs) are inherited variations in the DNA sequence that contribute to phenotypic diversity and can influence disease susceptibility by affecting gene expression and function [1, 2]. Recent advancements in gene expression analyses, high-throughput single nucleotide polymorphism genotyping, and association studies have identified genetic loci or genes that influence immune abnormalities in autoimmune disease [2]. Non-synonymous single nucleotide polymorphisms (nsSNPs) within protein-coding regions induce protein modification through amino acid substitution. Detrimental nsSNPs cause unstable protein structures, alter gene regulation, modify ligand-binding sites, and change protein hydrophobicity. The other adverse impacts of nsSNPs manifest in geometry, charge, dynamics, stability, protein–protein interactions, altering translation and threatening cellular integrity [3]. These variations have the capacity to modulate protein function and serve as crucial indicators for elucidating the mechanisms underlying various diseases [4]. In silico analysis predicts the harmful effects of these mutations and its effect on the structure and function of genes more quickly and cost-effectively than experimental methods [5].
Lipocalin-2 (LCN2) is a novel 198 amino acid adipocytokine also known as neutrophil gelatinase-associated lipocalin (NGAL) which was first isolated in neutrophil granules of humans [6], and these proteins circulate and transport hydrophobic compounds (steroid, free fatty acids, prostaglandins, and hormones) to target organs after binding to megalin/glycoprotein and GP330 SLC22A17 or 24p3R LCN2 receptors. LCN2 has been used as a biomarker to assess acute and chronic damage to the renal system [7], and it has been shown to prevent carcinogenesis in colorectal and pancreatic cancer, whereas it induced tumorigenesis in breast and prostate cancer [8]. LCN2 has been discovered as a key regulator of oxidative stress and inflammation in the pathogenesis of cardiovascular disease [9] and used as markers of tissue damage, particularly in the kidneys, and is also associated with cardiovascular disease symptoms such as hypertensive cardiac enlargement and heart failure [10]. Recent studies have shown that LCN2 levels are elevated in obese and type 2 diabetic patients [7] suggesting its potential as a biomarker for early detection of pulmonary hypertension in children with congenital heart disease [11]. Lipocalin-2-induced cardiomyocyte apoptosis affects intracellular iron levels, contributing to obesity-related heart failure. It causes cardiomyocyte death by increasing intracellular iron, which detrimentally impacts cardiac function [12]. The association of single nucleotide polymorphisms (SNPs) in the LCN2 gene may influence blood pressure without causing hypertension, yet still increase the risk of cardiovascular disease due to the continuous relationship between blood pressure and cardiovascular risk. Specifically, the SNP rs3814526 is associated with elevated blood pressure, indicating that lipocalin-2 may impact hypertension through inflammatory pathways [13]. In this study, we focused on investigating the missense nsSNPs of the LCN2 gene using bioinformatics tools to assess its potential detrimental effects and understand the structural and functional significance of the LCN2 protein.
Methods
SNP data mining
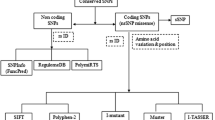
The LCN2 variants with (Accession: NP_005555.2) were retrieved from National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/projects/SNP)and primary sequence of protein were retrieved from UniProt database (https://www.uniprot.org/) (UniProtKB—P80188 [(NGAL_HUMAN)]. Additionally, SNPs of the LCN2 gene were retrieved from the ENSEMBL database to assess the impact of amino acid changes on protein function [14] (Fig. 1).

In silico analysis of LCN2
Prediction of deleterious of SNPs
Several online bioinformatics tools were used to identify damaging missense nsSNPs of the LCN2 gene. First, nsSNPs of the LCN2 gene were subjected to Sorting Intolerant from Tolerant (SIFT) and Polymorphism Phenotyping v2 (Polyphen-2) tools. SIFT, a web-based tool (https://sift.bii.a-star.edu.sg/), was employed to distinguish between harmful and tolerated SNPs by assessing their sequence homology. The predictive scoring system spanned a spectrum of values, wherein a score of ≤ 0.05 signified adverse impacts, while a score of ≥ 0.05 indicated tolerance [14]. Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) was used to predict the effects of amino acid substitutions on protein structure and function, categorizing mutations as “Possibly Damaging” (probability score > 0.15), “Probably Damaging” (probability score > 0.85), or “Benign” based on analysis of the protein sequence and variant position [15]. The nsSNPs identified by SIFT and Polyphen-2 were then subjected to Protein Variation Effect Analyzer (PROVEAN; http://provean.jcvi.org/), (PREDICTSNP; https://loschmidt.chemi.muni.cz/predictsnp1/), Multivariate Analysis of Protein Polymorphism (MAPP;http://www.ngrl.org.uk/Manchester/page/mapp-multivariate-analysis-proteinpolymorphism.html), Screening for non-acceptable polymorphism 2 (SNAP2; https://rostlab.org/services/snap2web/). PROVEAN predicts the detrimental effects of protein variations, including in-frame insertions, deletions, and several amino acid changes as well as individual amino acid changes. A score of − 2.5 or greater is deemed deleterious, whereas all other levels are neutral [16]. PREDICTSNP integrates data from multiple tools to predict the effect of a single amino acid changes, efficiency, and accuracy through a consensus prediction. MAPP evaluates the physiochemical alterations in each protein sequence alignment to predict the impact of amino acid substitutions on protein function [17]. SNAP2 utilizes a neural network to categorize genetic variations. The prediction method evaluates alterations induced by nsSNPs on the secondary structure and contrasts the solvent accessibility of native and mutated proteins to categorize them as either effect (+100, strongly predicted) or neutral (− 100, strongly predicted) [18]. The FASTA sequence of the LCN2 protein was used for input.
Analyzing the impact on protein stability
I-MUTANT 2.0
I-MUTANT2.0 (http://gpcr.biocomp.unibo.it/cgi/predictors/I-Mutant2.0/I-Mutant2.0.cgi) predicts changes in the stability of a mutant protein structures, estimating alterations in protein sequence that affect the stability of folded protein. I-MUTANT 2.0 utilizes support vector machines (SVMs) to forecast alterations in protein stability and corresponding ΔΔG values [19]. Delta Delta G (ΔΔG) represents the difference in Gibbs free energy, indicating the change in free energy of folding derived from the variations in the free energies between the native and mutant structures [20].
MUpro
MUpro (https://mupro.proteomics.ics.uci.edu/) predicts changes in protein stability caused by non-synonymous SNPs. It predicts an energy change value, yielding a confidence score ranging from − 1 to 1. This score is used to calculate the prediction's confidence. Scores less than zero indicate that the substitution decreases protein stability, whereas scores > 0 indicate increased protein stability [21].
Conservation of amino acids using ConSurf
ConSurf (https://consurfdb.tau.ac.il/) is a widely used tool for identifying functional regions in macromolecules by analyzing the evolutionary patterns of amino/nucleic acid variations in related sequences [22]. This method utilizes an empirical Bayesian approach to assign conservation scores to each residue, with a confidence interval, categorizing them as variable (scoring 1–4), intermediate (scoring 5–6), or conserved (scoring 7–9) [4].
Relevant solvent prediction using NetsurfP-2.0
NetsurfP-2.0 (https://services.healthtech.dtu.dk/services/NetSurfP-2.0/) tool accurately predicts solvent accessibility, secondary structure, structural disorder, and backbone dihedral angles for every residue in a given sequence. It provides precise and fast analysis of local structural elements [23]. The FASTA sequence of the LCN2 was given as input format.
Predicting structural effects of nsSNPs and mutant analysis
The PSIPRED workbench (http://bioinf.cs.ucl.ac.uk/psipred/) provides a range of protein annotation tools. It functions as a protein structure prediction server employing artificial neural networks and PSI-BLAST alignments to predict secondary structure [24]. The FASTA sequence of the LCN2 protein was provided as an input format.
Predicting the secondary structure of LCN2
SOPMA, (https://npsa-prabi.ibcp.fr/NPSAHLP/npsahlp_secpredsopma.html) an enhanced iteration of the self-optimized prediction method, effectively forecasts the secondary structure (including α-helix, β-turn, and coil) for 69.5% of amino acids within a dataset of 126 non-homologous (less than 25% homology) protein chains. Both SOPMA and a neural network method correctly predict 82.2% of individual residues and 74% of predicted amino acids [25].
Protein–Protein interaction
Protein–protein interactions (PPI) play a vital role in determining the functional connections of all proteins in the cell. PPI network information for LCN2 protein was obtained from the Search Tool for the Retrieval of Interacting Genes database (STRING V11.0; https://string-db.org/). The STRING constructs a PPI network by establishing direct or indirect links between known proteins and other proteins [26].
Gene–Gene interaction
Following the identification of several disease-associated polymorphisms by whole-genome association analysis, there is an increasing interest in the detection of the effects of polymorphism due to interaction with other genetic factors [27]. The GeneMANIA uses different parameters including genetic and protein interaction, co-expression, co-localization, pathways, and protein domain similarities to predict the interaction of input gene with many other genes [28]. GeneMANIA predicted the gene–gene interaction network for the LCN2 gene.
3D structure prediction using AlphaFold
The 3D structure of LCN2 protein was predicted using AlphaFold (https://alphafold.ebi.ac.uk/) computationally with accuracy and speed. In addition to highly accurate domain structures, AlphaFold constructs highly accurate side chains [29]. The UniProt ID for the LCN2 protein served as the input for the AlphaFold model.
Results
Retrieval of SNP dataset from dbSNP database
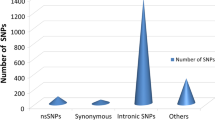
A total number of 2689 SNPs for the LCN2 gene were retrieved from the NCBI (https://www.ncbi.nlm.nih.gov/projects/SNP) dbSNP databases. Among these SNPs 180 were missense non-synonymous SNPs (nsSNPs), 1341 were introns SNPs, and 88 were synonymous SNPs, while the others belongs to different categories. The missense nsSNPs were selected for our study since deleterious nsSNPs could have structural and functional impact on the protein.
Prediction and functional analysis of nsSNPs in LCN2
Missense nsSNPs 180 were chosen for our study because they may have both structural and functional effects on proteins. Several in silico tools such as SIFT, Polyphen-2, PROVEAN, PREDICTSNP, MAPP, and SNAP2 were used to predict the deleterious effect on SNPs. Initially, 180 missense SNPs were loaded to SIFT server, which predicted 132 nsSNPs as deleterious or tolerated. Among them, 35 nsSNPs were predicted as deleterious with the score ≤ 0.05 and remaining 97 nsSNPs were tolerated. Then, nsSNPs were examined for Polyphen-2 server analysis which shows the nsSNPs as “Probably Damaging” with a score of 0.9–1, “Possibly Damaging” with a score of 0.7–0.9. The results from both SIFT and Polyphen-2 were combined to enhance the prediction accuracy. Further other bioinformatics tools PROVEAN, PREDICTSNP, MAPP, and SNAP2 were utilized. Based on the PROVEAN results, all 7 nsSNPs were predicted as deleterious. Through the PREDICTSNP results 6 nsSNPs were predicted as deleterious and 1nsSNPs were neutral. Moreover, Snap results 5 nsSNPs were predicted as disease causing and 2 nsSNPs were neutral. After prediction the using above-mentioned tools, 6 nsSNPs alone were found to be deleterious and are listed in Table 1.These potentially deleterious SNPs were considered to further analysis.
Prediction of the effect of nsSNPs on protein stability
MUpro and I-MUTANT 2.0 were used to analyze whether the selected missense nsSNPs predict the change of protein stability in LCN2 protein. According to I-MUTANT 2.0 server, nsSNPs rs11556770, rs142623708, rs200107414, rs201365744, rs368926734 were unstable and decreased the protein stability. In MUpro server, all nsSNPs rs147787222, rs11556770, rs139418967, rs142623708, rs200107414, rs201365744, rs368926734 decreased the stability of protein listed in Table 2
Analysis of deleterious nsSNPs conservation
According to phylogenetic conservation study, amino acids in conserved regions were significantly harmful than those in non-conserved regions. The ConSurf server was used to analyze the conservation profiles of amino acids in LCN2. The result showed that Q39H, L6P, M71I, Y52C, Y76H, and Y135 were found to be highly conserved and the variant amino acids were denoted in black boxes represented in Fig. 2. The result of ConSurf is shown in Table 2

Conservation analysis of LCN2 by ConSurf server. This figure represents the amino acids in conserved regions were significantly harmful than those in non-conserved regions. It found to be highly conserved, and the variant amino acids were denoted in black boxes represented
Prediction of relative solvent accessibility
NetsurfP-2.0 was employed to assess the solvent accessibility, stability, and predict secondary structure variations with high conservation scores identified in the ConSurf output. According to NetsurfP-2.0 server, the result showed that Q39H, L6P, Y135H were predicted to be exposed and M71I, Y52C, Y135 were buried. The results are displayed in Table 3
Predicting structural analysis of nsSNPs by PSIPRED software
PSIPRED projected the alpha-helix, beta-sheet, and coils that were distributed in the LCN2 secondary structure. The PSIPRED server analysis indicated that the predominant secondary structure was a strand, with lesser occurrences of coil and helix, as illustrated in Fig. 3. The PSIPRED predicted the transmembrane MEMSAT topology and the amino acid types. All of the transmembrane topology was cytoplasmic, the amino acid types were aromatic plus cysteine, and hydrophobic and polar are listed in Table 4.

Prediction of structural analysis by PSIPRED. PSIPRED examined the alpha-helix, beta-sheet, and coils that were distributed in the LCN2 secondary structure. This figure represents that PSIPRED revealed that the strand was the common secondary structure and less distribution of coil and helix
Secondary structural analysis of LCN2 by SOPMA
SOPMA analysis indicated that LCN2's secondary structure comprises distributions of alpha-helix, beta-sheet, and random coil. SOPMA secondary structure prediction for LCN2 is displayed in Fig. 4, where 21.21% of sites were alpha helixes, 51.52% were random coils, 3.54% were beta twists, and 23.74% were extended strands.

Prediction of secondary structure using SOPMA. This figures represent the LCN2's secondary structure as 21.21% of sites where alpha-helix, 3.54% beta-sheet, and 51.52% were random coil distributions
Protein interaction by STRING server
The STRING server result showed that LCN2 protein interacts with ten proteins including matrix mettaloproteinase-9 (MMP9), solute carrier family 22 member 17(SLC22A17), lacto transferrin (LTF), hepcidin-20 (HAMP), cytotoxic T-lymphocyte protein 4 (CTLA4), low-density lipoprotein receptor-related protein 2 (LRP2), gamma-secretase C-terminal fragment 50 (APP), fibronectin (FN1), cystatin-C (CST3), hepatitis A virus cellular receptor 1 (HAVCR1). Based on the analysis, CTAL4, LTF, SLC22A17, HAVCR1, MMP9, APP, HAMP proteins had direct interaction with which is shown in Fig. 5.

Protein–Protein interaction network of LCN2 gene. The network of protein–protein interactions is critical for understanding biological processes. Using STRING functional genomics data and structural assessment, functional and evolutionary aspects of the LCN2 protein were examined. Based on genomics data and fundamental assessment, functional CTLA4, LTF, SLC22A17, HAVCR1, MMP9, APP, HAMP these 7 proteins has strong and direct interaction with LCN2 protein
Gene–gene interaction by GeneMANIA
The GeneMANIA tool was used to analyze the gene interactions with the LCN2 protein. This server predicts that 9 genes matrix mettaloproteinase-9(MMP9), matrixmetallopeptidase2(MMP2), S100 calcium binding protein P (S100P), Lysozyme(LYZ), S100 calcium binding protein A8 (S100A8), GID complex subunit 8 homolog (GID8), LDL receptor-related protein 2(LRP2), Integrin subunit alpha 9 (ITGA9), L-2-hydroxyglutarate dehydrogenase(L2HGDH) has physical and genetic interactions. 7 genes WAP four-disulfide core domain 2(WFDC2), lacto transferrin (LTF), lysozyme (LYZ), secretory leukocyte peptidase inhibitor (SLP1), transcobalamin1 (TCN1), serpin family B member 5 (SERPINB5), peptidase inhibitor 3(P13) colocalized. 1 gene progestagen-associated endometrial protein (PAEP) shared protein domain and 6 genes MMP9, MMP2, LRP2, GID8, L2HGDH, ITGA9 were directly bound to LCN2 gene as shown in Fig. 6

Gene–gene interaction of LCN2 gene. GeneMANIA facilitates the identification of functional interactions between 6 genes: MMP9, MMP2, LRP2, GID8, L2HGDH, and ITGA9, which were directly bound with the LCN2 gene
3D structure prediction
The 3D structure of the LCN2 protein was analyzed by AlphaFold. The AlphaFold method assigns a confidence pLDDT score to each residue ranging from 0 to 100. The average pLDDT scores across all residues demonstrate an overall confidence in the entire protein chain. These 3D structure results show very high confidence (pLDDT > 90), while the other components are represented as unresolved loops with a low (70 > PLDDT > 50) and very low score (pLDDT50) and consist mostly of α-helical domains shown in Fig. 7.

AlphaFold 3D structure prediction of LCN2 gene. The AlphaFold method assigns a confidence pLDDT score to each individual residue ranging from 0 to 100. This 3D structure results reveal the very high confidence (pLDDT > 90), while the remaining components are illustrated as unresolved loops with the low (70 > pLDDT > 50) and extremely low scores (pLDDT50) and are primarily made up of α-helical domains
Discussion
In recent years SNPs served as promising markers for identifying loci linked to complex diseases and for pharmacogenetic applications. By studying the effects of functionally encoding SNPs on disease-related proteins, new drugs can be developed to correct the effects of these mutations in the population [30]. Many genes associated with disease have large databases containing deleterious SNPs, which has been a major concern in recent years [31]. Examining the presence of functional exonic SNPs within disease-associated proteins aims to enable the development of new treatments that mitigate the effects of these mutations in the population [4]. When occurring in genes, SNPs can affect mRNA splicing, nucleo-cytoplasmic export, stability, and translation. When present within the coding sequence and resulting in an amino acid change (known as a non-synonymous SNP or mutation), they can alter the protein’s activity [32]. Polymorphism in the LCN2 gene has been found to be associated with different diseases like cardiovascular disease, chronic damage to the renal system, colorectal and pancreatic cancer. In previous studies in animal models indicate that LCN2 plays significant roles in various physiological and pathological processes, including cell differentiation, apoptosis, organogenesis, inflammation, kidney damage, and liver injury. Additionally, LCN2 is suggested to be involved in cancer progression and metastasis [33]. A recent study has suggested, for the first time, that association of single nucleotide polymorphisms (SNPs) in the LCN2 gene may influence blood pressure without causing hypertension, yet still increase the risk of cardiovascular disease due to the continuous relationship between blood pressure and cardiovascular risk. Specifically, the SNP rs3814526 is associated with elevated blood pressure, indicating that lipocalin-2 may impact hypertension through inflammatory pathways [34].
Using several in silico methods, our study predicted the most deleterious nsSNPs structure and function of LCN2. The secondary structural predictions were analyzed by SOPMA and PSIPRED, while the protein–protein interaction and gene–gene interaction were analyzed by STRING and GeneMANIA. Finally, nsSNPs were submitted to AlphaFold for 3D structure prediction. Our study found that 6 functional SNPs rs11556770, rs139418967, rs142623708, rs200107414, rs201365744, and rs368926734 that have deleterious effects as determined by the conservation of amino acids, structural analysis, relative solvent accessibility, secondary structure prediction, and assessment of gene–gene and protein–protein interaction within the LCN2 gene. According to the I-MUTANT server, 5 amino acid changes were unstable and decreased the protein stability. In the MUpro server, all amino acids changes lead to decreased stability. The stability of proteins plays a pivotal role in shaping their conformational structure and functionality. Alterations in protein stability can influence misfolding, degradation, or the formation of abnormal protein aggregates [35]. Changes to amino acids that are involved in biological processes have a significant impact on protein function, as these amino acids are typically highly conserved [36]. The conservation analysis result showed that all 6 amino acids which are Q39H, L6P, M71I, Y52C, Y76H, and Y135 were found to be highly conserved. The exposed variations were found on the protein's surface, which could result in loss of interactions and structural changes, notably in the transmembrane domain [37]. PSIPRED analysis of LCN2 results revealed that the strand was the common secondary structure followed by coil and helix. SOPMA secondary structure found deleterious SNPs majorly in random coils and alpha helixes rather than beta twists, and extended strands.
GeneMANIA facilitates the identification of functional interactions between genes. GeneMANIA showed that interaction of 6 genes, MMP9, MMP2, LRP2, GID8, L2HGDH, and ITGA9, was directly bound with the LCN2 gene. Deleterious SNPs in the LCN2 gene may disrupt the interaction and function of other genes in the gene–gene interaction network. The LCN2 and MMP9 combination inhibits MMP9 autodegradation and increases MMP9 activity in vitro. The majority of LCN2's biological roles were discovered through studies done on mice. Nowadays, six potential LCN2 receptors have been found (NGALR, LRP2, LRP6, MCR4, MCR1, and MCR3), and their structures and affinities differ significantly. The mouse LRP6 protein, which serves as a co-receptor for Wnt and shares similar structural motifs as LRP2, has been shown to specifically interact with mouse LCN2. The study found that binding LCN2 to LRP6 efficiently inhibits Wnt/β-catenin signaling, as demonstrated by co-immunoprecipitation results [38]. In several studies, streptozotocin injection has been shown to elevate levels LCN2 in body fluids, such as urine, and in various body tissues, including the kidney. LCN2 is commonly used as a biomarker for both acute and chronic kidney injury [39,40,41,42].
The network of protein–protein interactions is critical for understanding the biological processes. Based on genomics data and fundamental assessment, functional and evolutionary aspects, these 7 proteins, CTLA4, LTF, SLC22A17, HAVCR1, MMP9, APP, and HAMP, have strong and direct interaction with LCN2 protein. Consequently, the variant protein containing damaging SNPs might engage with other proteins, leading to phenotypic alterations in protein expression (43). Recent study suggested that lipocalin-2 (LCN2) and hepcidin both contribute to iron homeostasis. LCN2 is a glycoprotein that transports hydrophobic ligands across cell membranes, regulates immunological responses, and keeps iron levels balanced. An engineered lipocalin generated from human LCN2 may bind the T cell co-receptor CTLA4 as a specified protein target with sub-nanomolar affinity [44]. Lactoferrin (LTF) and LCN2 both primarily operate in the sequestration of iron. Lactoferrin, a glycoprotein primarily known for its metal-binding abilities at mucosal surfaces, is also identified within neutrophil secondary granules and adorning neutrophil extracellular traps (NETs) [45]. Protein network research revealed that the LCN2-SLC22A17-MMP9 network has a role in TME through its interactions with fibronectin 1 and claudin 7, particularly in rectal tumors. LCN2, SLC22A17, and MMP9 expression and methylation status were consistent across all TCGA tumors, demonstrating that the LCN2-SLC22A17-MMP9 network was tightly controlled by DNA methylation within TME [46].
AlphaFold forecasts 3D protein structures and produces a predicted (pLDDT), which evaluates confidence for each residue. The LCN2 3D structure has high confidence (pLDDT > 90) and consists mostly of α-helical domains. This study examined the LCN2 gene polymorphism using various bioinformatics tools. From our study, 6 SNPs have been discovered to be both structurally and functionally detrimental, suggesting that they may impact the LCN2 protein's functions. The prediction of deleterious SNPs has been carried out using bioinformatics tools, but well-designed experimental and clinical analyses are necessary to investigate the impact of these nsSNPs on the structure and function of LCN2 protein.
Conclusion
Several online algorithmic tools relying on sequence and structural conservation were employed to pinpoint harmful nsSNPs within the LCN2 gene. Our study identified six nsSNPs as promising biomarkers for the LCN2 gene. Nevertheless, additional in vivo and in vitro investigations are essential to explore and confirm the involvement of the LCN2 nsSNPs in various diseases. Utilizing a variety of computational tools enhances the predictive capacity for assessing the impact of mutations on proteins and cost-effective screening approach to better inform diagnostic and experimental approaches. However, in silico tools alone are insufficient and their outcomes must be validated through additional biological evidence, serving as a basis for targeting pathogenic sites of the LCN2 protein.
Availability of data and materials
All data analyzed during this study are included in this study.
Abbreviations
- AAA:
-
Abdominal aortic aneurysms
- CST3:
-
Cystatin-C
- CTLA4:
-
Cytotoxic T-lymphocyte protein 4
- CVD:
-
Cardiovascular disease
- FN1:
-
Fibronectin
- GID8:
-
GID complex subunit 8 homolog
- GWA:
-
Genome-wide association
- HAMP:
-
Hepcidin-20
- HAVCR1:
-
Hepatitis A virus cellular receptor 1
- ITGA9:
-
Integrin subunit alpha 9
- L2HGDH:
-
L-2-hydroxyglutarate dehydrogenase
- LCN2:
-
Lipocalin-2
- LRP2:
-
LDL receptor-related protein 2
- LTF:
-
Lactotransferrin
- LYZ:
-
Lysozyme
- MAPP:
-
Multivariate Analysis of Protein Polymorphism
- MMP9:
-
Matrix mettaloproteinase-9
- MR:
-
Mineralocorticoid receptor
- NCBI:
-
National Centre For Biotechnology Information
- NGAL:
-
Neutrophil gelatinase-associated lipocalin
- nsSNPs:
-
Non-synonymous single nucleotide polymorphism
- P13:
-
Peptidase inhibitor 3
- POLYPHEN-2:
-
Polymorphism phenotyping2
- PPI:
-
Protein–protein interactions
- PROVEAN:
-
Protein Variation Effect Analyzer
- S100A8:
-
S100 calcium binding protein A8
- S100P:
-
S100 calcium binding protein P
- Serpinb5:
-
Serpin family B member 5
- SIFT:
-
Sorting intolerant from tolerant
- SLC22A17:
-
Solute carrier family 22 member 17
- SNAP2:
-
Screening for non-acceptable polymorphism
- SNP:
-
Single nucleotide polymorphism
- TCN1:
-
Transcobalamin1
- TME:
-
Tumor microenvironment
- WFDC2:
-
WAP four-disulfide core domain 2
References
Navapour L, Mogharrab N (2021) In silico screening and analysis of nonsynonymous SNPs in human CYP1A2 to assess possible associations with pathogenicity and cancer susceptibility. Sci Rep 11(1):1–15. https://doi.org/10.1038/s41598-021-83696-x
Hewagama A, Richardson B (2009) The genetics and epigenetics of autoimmune diseases. J Autoimmun 33(1):3–11
Akter M, Khan SF, Sajib AA, Rima FS (2022) A comprehensive in silico analysis of the deleterious nonsynonymous SNPs of human FOXP2 protein. PLoS ONE 17(8):1–14. https://doi.org/10.1371/journal.pone.0272625
Ajith A, Subbiah U (2023) In silico screening of non-synonymous SNPs in human TUFT1 gene. J Genet Eng Biotechnol 21(1):95
Zhang M, Huang C, Wang Z, Lv H, Li X (2020) In silico analysis of non-synonymous single nucleotide polymorphisms (nsSNPs) in the human GJA3 gene associated with congenital cataract. BMC Mol Cell Biol 21(1):1–13
Asaf S (2023) Lipocalin 2—not only a biomarker: a study of current literature and systematic findings of ongoing clinical trials. Immunol Res 71(3):287–313
Al Jaberi S, Cohen A, D’Souza C, Abdulrazzaq YM, Ojha S, Bastaki S et al (2021) Lipocalin-2: structure, function, distribution and role in metabolic disorders. Biomed Pharmacother 142:112002
Molter CW, Muszynski EF, Tao Y, Trivedi T, Clouvel A, Ehrlicher AJ (2022) Prostate cancer cells of increasing metastatic potential exhibit diverse contractile forces, cell stiffness, and motility in a microenvironment stiffness-dependent manner. Front Cell Dev Biol 10:932510
Jang H (2022) LCN2 deficiency ameliorates doxorubicin-induced cardiomyopathy in mice. Biochemical and biophysical research communications. Biochem Biophys Res Commun 588:8–14
Marques FZ, Prestes PR, Byars SG, Ritchie SC, Würtz P, Patel SK et al (2017) Experimental and human evidence for lipocalin-2 (neutrophil gelatinase-associated lipocalin [NGAL]) in the development of cardiac hypertrophy and heart failure. J Am Heart Assoc 6(6):e005971
Zhang H (2021) Lipocalin 2: could it be a new biomarker in pediatric pulmonary hypertension associated with congenital heart disease? Rev Cardiovasc Med 22.2:531–536
Xu G (2012) Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J Biol Chem 287(7):4808–4817
Ong KL, Tso AW, Cherny SS, Sham PC, Lam TH, Lam KS, Cheung BM (2011) Role of genetic variants in the gene encoding lipocalin-2 in the development of elevated blood pressure. Clin Exp Hypertens 33:484–491
Arifuzzaman M, Mitra S, Hamza A, Das R, Absar N, Dash R (2018) In silico analysis of non synonymous single nucleotide polymorphisms (nsSNPs) of SMPX gene in hearing impairment. bioRxiv. 461764.
Thakur R, Shankar J (2016) In silico analysis revealed high-risk single nucleotide polymorphisms in human Pentraxin-3 gene and their impact on innate immune response against microbial pathogens. Front Microbiol 7:1–12
Singh S (2021) Computational prediction of the effects of non-synonymous single nucleotide polymorphisms on the GPI-anchor transamidase subunit GPI8p of Plasmodium falciparum. Comput Biol Chem 92:107461
Elkhattabi L, Morjane I, Charoute H, Amghar S, Bouafi H, Elkarhat Z et al (2019) In silico analysis of coding/noncoding SNPs of human RETN gene and characterization of their impact on resistin stability and structure. J Diabetes Res 2019:1
AbdulAzeez S, Borgio JF (2016) In-silico computing of the most deleterious nsSNPs in HBA1 gene. PLoS ONE 11(1):1–13
Cheng J, Randall A, Baldi P (2006) Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct Funct Genet 62(4):1125–1132
Kellogg EH, Leaver-Fay A, Baker D (2011) Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins Struct Funct Bioinforma 79(3):830–838
Narayana Swamy A, Valasala H, Kamma S (2015) In silico evaluation of nonsynonymous single nucleotide polymorphisms in the ADIPOQ gene associated with diabetes, obesity, and inflammation. Avicenna J Med Biotechnol 7(3):121–127
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T et al (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44(W1):W344–W350
Klausen MS, Jespersen MC, Nielsen H, Jensen KK. NetSurfP-2.0 : improved prediction of protein structural features by integrated deep learning. 2018;(1).
Buchan DWA, Jones DT (2019) The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res 47(W1):W402–W407
Geourjon C, Deléage G (1995) Sopma: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 11(6):681–684
Ul Ain Farooq Q (2021) Protein-protein interactions: methods, databases, and applications in virus-host study. World J Virol 10(6):288
Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447(7145):661–678
Warde-Farley D (2010) The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38:214–220
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596(7873):583–589. https://doi.org/10.1038/s41586-021-03819-2
Javed R (2010) Current research status, databases and application of single nucleotide polymorphism. Pak J Biol Sci: PJBS 13:657–663
Rozario LT, Sharker T, Nila TA (2021) In silico analysis of deleterious SNPs of human MTUS1 gene and their impacts on subsequent protein structure and function. PLoS ONE 16.6:e0252932
Robert F, Pelletier J (2018) Exploring the impact of single-nucleotide polymorphisms on translation. Front Genet 9:507
Candido S, Abrams SL, Steelman LS, Lertpiriyapong K, Fitzgerald TL, Martelli AM, McCubrey JA (2016) Roles of NGAL and MMP-9 in the tumormicroenvironment and sensitivity to targeted therapy. Biochimica et Biophysica Acta BBA-Molecular Cell Research. 1863(3):438–448
Ong K-L, Tso AWK, Cherny SS, Sham P-C, Lam T-H, Lam KSL, Cheung BM (2011) Role of genetic variants in the gene encoding lipocalin-2 in the development of elevated blood pressure. Clin Exp Hypertens 33(7):484–491
Witham S (2011) A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins: Struct Funct Bioinform 79:2444–2454
Irfan M, Iqbal T, Hashmi S, Ghani U, Bhatti A (2022) Insilico prediction and functional analysis of nonsynonymous SNPs in human CTLA4 gene. Sci Rep 12(1):1–11. https://doi.org/10.1038/s41598-022-24699-0
Ajith A (2023) In silico prediction of deleterious non-synonymous SNPs in STAT3. Asian Biomed 17:185–199
Li D (2020) Lipocalin-2—the myth of its expression and function. Basic Clin Pharmacol Toxicol 127(2):142–151
Bhusal A, Rahman MH, Lee IK, Suk K (2019) Role of hippocampal lipocalin-2 in experimental diabetic encephalopathy. Front Endocrinol 2019(10):25
Arellano-Buendía AS, García-Arroyo FE, Cristobal-Garcíá M, Loredo-Mendoza ML, Tapia-Rodríguez E, Sanchez-Lozadá LG, Osorio-Alonso HD (2014) Urinary excretion of neutrophil gelatinase-associated lipocalin in diabetic rats. Oxid Med Cell Longev 1:961
Korrapati MC, Shaner BE, Neely BA, Alge JL, Arthur JM, Schnellmann RG (2012) Diabetes-induced renal injury in rats is attenuated by suramin. J Pharmacol Exp Ther 343(1):34–43
Liu F, Yang H, Chen H, Zhang M, Ma Q (2015) High expression of neutrophil gelatinase-associated lipocalin (NGAL) in the kidney proximal tubules of diabetic rats. Adv Med Sci 60(1):133–138
Ali Mohamoud HS (2014) First comprehensive in silico analysis of the functional and structural consequences of SNPs in human GalNAc-T1 gene. Comput Math Methods Med 1:904052
Schönfeld D (2009) An engineered lipocalin specific for CTLA-4 reveals a combining site with structural and conformational features similar to antibodies. Proc Natl Acad Sci 106(20):8198–8203
Sheldon JR (2022) Lipocalin-2 is an essential component of the innate immune response to Acinetobacterbaumannii infection”. PLoS Pathog 18(9):e1010809
Candido S (2022) Bioinformatic analysis of the LCN2-SLC22A17-MMP9 network in cancer: the role of DNA methylation in the modulation of tumor microenvironment. Front Cell Dev Biol 10:945586
Acknowledgements
The authors express gratitude to the DST-FIST (Ref. No.SR/FST/College-23/2017), Government of India, New Delhi, for the utilizing the funded research equipment facilities at Sree Balaji Dental College and Hospital, Pallikaranai, Chennai, Tamil Nadu, India.
Funding
This research did not receive any specific grant from funding agencies in the public or commercial sectors.
Author information
Authors and Affiliations
Contributions
Kaniha Sivakumar and Usha Subbiah contributed to conception and design and editing and review. Kaniha Sivakumar was involved in literature search and manuscript preparation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sivakumar, K., Subbiah, U. Computational analysis of non-synonymous SNPs in the human LCN2 gene. Egypt J Med Hum Genet 25, 94 (2024). https://doi.org/10.1186/s43042-024-00565-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-024-00565-8