
Abstract
Background
Von Hippel-Lindau (VHL) syndrome is an autosomal dominantly inherited disorder that predisposes to multiple neoplasms. Patients may develop hemangioblastomas of the central nervous system and retina, multiple cysts in the pancreas and kidneys, renal carcinoma, and pheochromocytomas, among other lesions. This disease is caused by germline genetic variants in the VHL gene. The regulation of the alpha subunit of hypoxia-inducible factor-1 is the key tumor suppressor function of the VHL protein. To date, more than seven hundred variants have been reported in VHL gene. This study aimed to investigate the molecular etiology of VHL syndrome in Cuban patients.
Results
DNA samples from twenty-two individuals were analyzed by Sanger sequencing or enzymatic restriction. The analysis identified four novel pathogenic variants for the Cuban population: c.463 + 2T > C, C162W, R167W, and S183X, in addition to D121G and R161X, previously described in another work. The diagnosis was confirmed in seven patients with clinical manifestations and family history. Two at-risk family members without clinical signs were positive for presymptomatic diagnosis.
Conclusions
The spectrum of germinal point mutations of VHL gene in Cuban patients was updated. The presence of genetic variants was ruled out in eight asymptomatic relatives, which is a psychological relief for these individuals. The results allow for offering other at-risk relatives the presymptomatic diagnosis and the possibility of receiving genetic counseling.
Similar content being viewed by others


Background
Von Hippel-Lindau syndrome (OMIM #193300) is a heritable multisystem cancer disease. It is characterized by the appearance of highly vascularized tumors, both benign and malignant, in multiple locations. Patients may develop hemangioblastomas of the central nervous system and retina, and visceral tumors such as clear cell renal carcinoma, pheochromocytomas, epididymal cystadenomas, pancreatic neuroendocrine tumors, and other lesions such as cysts in the pancreas and kidneys [1, 2].
It is rare to find all manifestations in the same patient; in familial cases, up to 50% of patients have only one manifestation of the syndrome [3]. The disease has a highly variable inter- and intra-familial expressivity. The location and dynamics of tumor development, the severity and diversity of manifestations, and the age of first symptoms vary considerably [2]. Both genetic and environmental modifying factors have been reported to influence expressivity, as with other familial cancer syndromes [4]. Penetrance varies with age, and the average age at diagnosis is 29 years for cerebellar hemangioblastoma and 44.8 years for clear cell renal carcinoma [5].
VHL syndrome has been subdivided according to the presence or absence of pheochromocytomas. Type 1-affected patients within a family have a minimal risk of developing pheochromocytomas but are at elevated risk for other lesions. In Type 2, on the other hand, there is a substantial risk of developing pheochromocytomas. This type is further subdivided into Type 2A, without the manifestation of renal cell carcinoma, which has the best prognosis; Type 2B, which has a high incidence of renal cell carcinoma, is the rarest form but the only one in which all manifestations of VHL syndrome are present; and Type 2C, where pheochromocytomas are the only clinical manifestation [6].
The disease originates from germline variants in the VHL tumor suppressor gene (OMIM *608537), which is located on 3p25.26 [7]. Patients are heterozygotes for VHL with one wild type and one defective allele. Somatic inactivation of the second functional allele in susceptible cells, and therefore loss of VHL function, leads to pathological features of disease [8].
To date, more than 700 variants in VHL gene have been registered in HGMD [9]. Approximately 89% of the index cases analyzed have point variants, while the remainder have large or complete deletions [10]. It is proposed that 20% of pathogenic alterations of the VHL gene sequence occur de novo [2].
Germline variants in VHL gene are also associated with other clinical entities and can be detected in patients with nonsyndromic familial pheochromocytoma (OMIM #171300), while variants in homozygous or compound heterozygous state can cause familial erythrocytosis-2, (OMIM #263400), without manifestation of Von Hippel-Lindau syndrome.
The best understood function of VHL protein is its role in processes that sense oxygen levels. Symptoms associated with VHL syndrome and sporadic renal cell carcinoma, such as increased angiogenesis and polycythemia, suggest a correlation between the function of VHL protein and the adaptive response to oxygen deprivation [10].
Regulation of the alpha subunit of hypoxia-inducible factor-1 (HIF1A) is the key tumor suppressor function of VHL protein. A large number of disease-associated variants have been shown to significantly impair the interaction between VHL protein and HIF-alpha. This interaction is mediated by the formation of a complex called VCB-CR, which, in addition to VHL protein, includes elongin C and B, cullin-2, and E3 ubiquitin-protein ligase RBX1 [11].
The small size of the VHL gene, with a coding sequence of only 642 base pairs distributed in three exons, allowed its early molecular characterization. This work aimed to investigate the molecular etiology of VHL syndrome in Cuban patients with a clinical diagnosis of VHL syndrome, as well as in patients with isolated clinical manifestations, without family history of the disease. The analysis of at-risk relatives of index cases with molecular diagnosis was also carried out.
Methods
Subjects
A total of twenty-two individuals were studied between 2014 and 2022. Of these, seven patients had clinical manifestations of VHL syndrome and family history, ten were asymptomatic relatives at risk for the disease, and another five patients had pheochromocytoma or hemangioblastoma, with no family history of the disease.
The patients were seen in the multidisciplinary consultation for the diagnosis and treatment of patients with VHL syndrome, of the Clinical Surgical Hospital Hermanos Ameijeiras (HCQHA), of Havana, Cuba. Molecular analysis of the VHL gene was performed at the Department of Molecular Biology, National Center for Medical Genetics (CNGM), of Cuba.
DNA analysis
After informed consent, peripheral blood samples were collected. Genomic DNA isolation was performed by conventional saline precipitation method or automated using magnetic bead technology (QIAsymphony DNA Midi Kit and QIAsymphony SP, QIAGEN).
Sanger sequencing or RFLP-PCR was used in the analysis process. The primer sets for PCR are described in Table 1. These oligonucleotides were the same used for DNA sequencing.
The GenomeLab GeXP (Beckman Coulter) sequencer was used for bidirectional sequencing of PCR products. The sequences obtained for each sample were compared with the reference sequence of the human VHL gene (GenBank: NG_008212). For the nomenclature of the variants detected, the VHL variant 1 transcript (GenBank: NM_000551) and the variant 1 isoform of VHL protein (GenBank: NP_000542) were used as references. We used the in-silico tool Human Splicing Finder (RRID:SCR_005181) to measure the functional impact of a detected splice site variant.
PCR–RFLP analysis was used to genotype the D121G and R161X variants [12], which were detected in the first three families with VHL syndrome studied in Cuba.
Results
DNA sequencing identified four pathogenic variants not previously detected in the Cuban patients: c.463 + 2T > C, in intron 2, and C162W, R167W, and S183X, in the third exon. These are in addition to the previously detected D121G and R167X mutations in exons 2 and 3, respectively. No variant has been found in exon 1. Details of the variants identified so far in Cuba are shown in Table 2. All are registered in the ClinVar [13] and VHLdb [14] databases.
To date, mutations in the VHL gene have been detected in seven Cuban families (see Additional file 1: Figs. 1–7). The same sequence change, D121G, is present in two families with no established linkage. In this study, the clinical diagnosis could be confirmed in seven of the 12 (58.3%) patients with some clinical manifestation and in 100% of the cases with symptoms and a family history of the disease.
A total of three patients were positive for R167W and another two had S183X. The c.463 + 2T > C and C162W variants have only been detected in the index cases, the only individuals in their respective families with genetic testing performed. One subject tested positive for the presymptomatic diagnosis of variant D121G and another for R161X. In addition, eight asymptomatic members were negative for the variants in VHL gene.
Discussion
The clinical diagnosis, molecular confirmation, and management of patients with manifestations of VHL syndrome have been ensured by the joint work of the specialized Clinical Genetics consultation of the HCQHA and the molecular genetics laboratory of the CNGM.
Until now, the molecular study has identified six variants in Cuban patients. All of them are described in the literature and registered in different variant databases. Although studies with larger case series have found a greater number of variants in exons 1 and 3 [10, 15], no patients with pathogenic mutations in the first exon have been detected in Cuba. However, four of the six nucleotide sequence changes were identified in exon 3.
In this work, we detected two missense variants (C162W and R167W), one nonsense (S183X) and one splice-site mutation (c.463 + 2T > C).
The c.463 + 2T > C variant has been found both in the germline and at the somatic level and is classified as pathogenic [16, 17]. Patients with VHL syndrome, familial erythrocytosis-2, or renal cell carcinoma have also been identified as carriers of c.463 + 2T > C. This sequence change affects the donor splice site in intron two. This is a highly conserved site and computational prediction of its effect using the Human Splicing Finder program indicates that this alteration probably affects the splicing mechanism. The computational tools also predict that another variant, identified more frequently at this donor site, c.463 + 1G > T, produces the same effect [15, 18]. Nucleotide substitutions within the consensus splice site are a relatively common cause of aberrant intron deletion mechanisms [19]. The splice donor site affected by c.463 + 2T > C is located upstream of the exon 3 sequence encoding the elongin C-binding domain of VHL protein. This domain is required for the stability of the protein and its tumor suppressor activity [20]. Variants that destabilize elongin C-binding domain have been reported to be pathogenic [19, 21]. This suggests that this region is critical for VHL protein function and other variants that alter this region may also be pathogenic.
The C162W variant has been identified in different populations. It has been found both in the germline and at the somatic level, and is classified by different investigators as pathogenic [19, 22]. Patients with VHL syndrome, familial erythrocytosis-2, or renal cell carcinoma carry this variant. This sequence change replaces cysteine with tryptophan at codon 162 of VHL protein. The cysteine residue is highly conserved evolutionarily, and there are large physicochemical differences between cysteine (polar) and tryptophan (apolar). An experimental study has shown that this missense change disrupts VHL protein-dependent degradation in the proteasome, failing to induce ubiquitination of target proteins [19]. Schoenfeld et al. suggest that C162 is a critical residue for binding to elongin C [20], so it is likely that the stability and functionality of the VCB-CR complex are affected.
The R167W variant is commonly described in studies of the VHL gene. It has been found both in the germline and at the somatic level, and is classified by several investigators as pathogenic [23, 24]. Patients with VHL syndrome, renal cell carcinoma, pheochromocytomas, or familial erythrocytosis-2 carry this variant. This sequence change replaces arginine with tryptophan at codon 167 of VHL protein. The arginine residue 167 is highly conserved evolutionarily, and there are moderate physicochemical differences between arginine (positively charged) and tryptophan (apolar). Codon 167 is located in the α-domain of VHL protein, and residue R167 has been reported to be important in maintaining protein structure [25]. Functional studies show that VHL protein with the R167W variant does not bind to elongin C, nor elongin B, resulting in an unstable protein that is rapidly degraded by the proteasome [20]. Other experimental studies have shown that VHL protein with this variant does not fully stabilize the Jade-1 protein, which is an important factor in the suppression of renal tumors [26]. Recently Buart et al. demonstrated that VHL protein with this variant interferes with the plasticity of renal carcinomas and may affect cell behavior by exacerbating their phenotypic change [27]. Other variants in this codon: R167G, R167Q, and R167L, associated with VHL syndrome, have been registered in variant databases, confirming the functional importance of this region of the protein.
The S183X variant has also been found both in the germline and at the somatic level, and is classified as pathogenic [15, 28]. Patients with VHL syndrome, renal cell carcinoma, or kidney cancer have been reported to carry this variant. Although there are no functional studies for this variant, it has been reported to segregate with the disease in families affected with VHL syndrome. This allelic variant produces a truncated protein with loss of the last 31 amino acids, starting from the serine residue at position 183. The 1999 study by Stebbins et al. established that the leucine residues at positions 184 and 188 (L184 and L188) are involved in the packing of the α-domain helices, and that residue L184 is an important contact between VHL protein and elongin C [25]. These amino acids with determinant roles in protein stability are lost in the S183X variant, so VHL protein can be expected to result in an unstable protein and to be rapidly degraded by the proteasome.
No variants were detected after sequencing of the VHL gene in five of the patients who presented clinical manifestations of VHL with no family history of the disease. Two of these presented with pheochromocytoma alone, and the other three with hemangioblastoma of the retina or cerebellum, in one case also accompanied by ovarian cyst. It should be noted that susceptibility to develop isolated pheochromocytoma may be caused by germline variants in several genes (OMIM #171300), not analyzed in this investigation.
On the other hand, as mentioned above, up to 11% of the variants identified in patients with VHL syndrome are large deletions or duplications [10], which cannot be detected with the techniques employed in this study. However, patients with isolated manifestations may be sporadic cases and have no germline involvement. To elucidate this, additional testing should be performed for large rearrangements in the VHL gene.
The phenotype–genotype relationship behaved, in most cases, as reported in previous studies. Amino acid changes that result in a VHL protein that is normal concerning HIF regulation are more likely to be associated with VHL Type 2 [29, 30]. As in families 2 and 3, in which D121G is segregated, functional studies showed that the VHL protein with this mutation retained many of its properties, including its in vitro interaction with HIF and the regulation of gene expression controlled by this transcription factor, as well as its ubiquitination (although at a lower level) and its interaction with elongin C [31, 32].
Amino acid changes that destabilize the packing of the α-helical domains; decrease the stability of the interface between the α and β domains; interfere with binding to elongin C and HIF-alpha; or alter the hydrophobic residues of the central core of VHL protein, can result in loss of HIF regulation and are more likely to result in VHL Type 1 [29]. The family 7, where C162W segregates, appears to be an example. An exception to this rule is the R167W variant, segregating in family 6, which as previously described results in an unstable protein that is rapidly degraded by the proteasome [20]. However, this variant has been described mainly in families with VHL Type 2 [33, 34], which was in agreement with our results.
Deletions, reading frame shifts, variants resulting in protein truncation, and splice site variants generally lead to VHL Type 1 manifestation [29, 30]. This is also true for families 1 and 4, where R161X and S183X segregate, respectively. In the case of the variant c.463 + 2T > C (family 5, VHL type 2), there was no coincidence with the expected findings for the splice site variants. As described by other authors [16, 35], the manifestations of patients with this variant do not include pheochromocytomas. The same occurs in patients with other intronic variants that affect the splicing mechanism between exons 2 and 3 [36]. One possibility would be that these individuals had not yet developed pheochromocytomas at the time of study or that a modifying factor is influencing the different phenotypic expression among these patients.
Conclusions
So far, six germline mutations have been detected in Cuban patients with clinical diagnosis of VHL syndrome. Of these, c.463 + 2T > C, C162W, R167W, and S183X were find for the first time in Cuban patients. This has contributed to establishing the etiology of the disease in seven families, which constitutes a valuable support to genetic counseling and better management of patients. At the same time, it has been possible to rule out the risk of developing the syndrome in several asymptomatic relatives, with the consequent psychological benefit for these individuals.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- VHL:
-
Von Hippel-Lindau
- HIF1A:
-
Alpha subunit of hypoxia-inducible factor-1
- VCB-CR:
-
Complex formed by VHL protein, elongin C and B, cullin-2, and ring-box 1 protein
- PCR:
-
Polymerase chain reaction
- PCR–RFLP:
-
Restriction Fragment Length Polymorphism analysis of PCR-amplified fragments
References
Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM et al (2003) von Hippel-Lindau disease. Lancet 361(9374):2059–2067
Schmid S, Gillessen S, Binet I, Brandle M, Engeler D, Greiner J et al (2014) Management of von hippel-lindau disease: an interdisciplinary review. Oncol Res Treat 37(12):761–771
Crespigio J, Berbel LCL, Dias MA, Berbel RF, Pereira SS, Pignatelli D et al (2017) Von Hippel-Lindau disease: a single gene, several hereditary tumors. J Endocrinol Invest 41(1):21–31
Binderup MLM (2018) von Hippel-Lindau disease: diagnosis and factors influencing disease outcome. Dan Med J 65(3):B5461
Maher ER, Neumann HP, Richard S (2011) von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 19(6):617–623
Varshney N, Kebede AA, Owusu-Dapaah H, Lather J, Kaushik M, Bhullar JS (2017) A review of von Hippel-Lindau syndrome. J Kidney Cancer VHL 4(3):20–29
Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML et al (1993) Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260(5112):1317–1320
Robinson CM, Ohh M (2014) The multifaceted von Hippel-Lindau tumour suppressor protein. FEBS Lett 588(16):2704–2711
The Human Gene Mutation Database (2022) Institute of Medical Genetics. Cardiff University. https://www.hgmd.cf.ac.uk/ac/gene.php?gene=VHL. Accessed 13 August 2022
Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP et al (2010) Genetic analysis of von Hippel-Lindau disease. Hum Mutat 31(5):521–537
Gossage L, Eisen T, Maher ER (2015) VHL, the story of a tumour suppressor gene. Nat Rev Cancer 15(1):55–64
Esperon AA, Noa IV, Reyes L (2013) Introduction of a PCR-RFLP method for the detection of two mutations in VHL gene. Medisur 11(3):361–367
ClinVar (2022) National Center for Biotechnology Information. National Institutes of Health. https://www.ncbi.nlm.nih.gov/clinvar/. Accessed 13 Aug 2022.
VHLdb (2022) Database of von Hippel-Lindau protein interactors and mutations. University of Padua. http://vhldb.bio.unipd.it/mutations. Accessed 5 Aug 2022
Dandanell M, Friis-Hansen L, Sunde L, Nielsen FC, Hansen TV (2012) Identification of 3 novel VHL germ-line mutations in Danish VHL patients. BMC Med Genet 13:54
Hes FJ, van der Luijt RB, Janssen AL, Zewald RA, de Jong GJ, Lenders JW et al (2007) Frequency of Von Hippel-Lindau germline mutations in classic and non-classic Von Hippel-Lindau disease identified by DNA sequencing, Southern blot analysis and multiplex ligation-dependent probe amplification. Clin Genet 72(2):122–129
Charbotel B, Gad S, Caiola D, Beroud C, Fevotte J, Bergeret A et al (2007) Trichloroethylene exposure and somatic mutations of the VHL gene in patients with Renal Cell Carcinoma. J Occup Med Toxicol 2:13
Siu WK, Ma RC, Lam CW, Mak CM, Yuen YP, Lo FM et al (2011) Molecular basis of von Hippel-Lindau syndrome in Chinese patients. Chin Med J (Engl) 124(2):237–241
Iturrioz X, Parker PJ (2007) PKCzetaII is a target for degradation through the tumour suppressor protein pVHL. FEBS Lett 581(7):1397–1402
Schoenfeld AR, Davidowitz EJ, Burk RD (2000) Elongin BC complex prevents degradation of von Hippel-Lindau tumor suppressor gene products. Proc Natl Acad Sci 97(15):8507–8512
Couve S, Ladroue C, Laine E, Mahtouk K, Guegan J, Gad S et al (2014) Genetic evidence of a precisely tuned dysregulation in the hypoxia signaling pathway during oncogenesis. Cancer Res 74(22):6554–6564
Hong B, Ma K, Zhou J, Zhang J, Wang J, Liu S et al (2019) Frequent mutations of VHL gene and the clinical phenotypes in the largest Chinese cohort with von Hippel-Lindau disease. Front Genet 10:867
Wang Y, Liang G, Tian J, Wang X, Chen A, Liang T et al (2018) Pedigree analysis, diagnosis and treatment in Von Hippel-Lindau syndrome: a report of three cases. Oncol Lett 15(4):4882–4890
Sivaskandarajah GA, Arnason TG (2018) Unsuspected von Hippel-Lindau syndrome in acute-onset resistant hypertension. BMJ Case Reports. https://doi.org/10.1136/bcr-2018-225162
Stebbins CE, Kaelin WG Jr, Pavletich NP (1999) Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science 284(5413):455–461
Zhou MI, Wang H, Foy RL, Ross JJ, Cohen HT (2004) Tumor suppressor von Hippel-Lindau (VHL) stabilization of Jade-1 protein occurs through plant homeodomains and is VHL mutation dependent. Cancer Res 64(4):1278–1286
Buart S, Terry S, Diop MK, Dessen P, Couvé S, Abdou A et al (2021) The most common VHL point mutation R167Q in hereditary VHL disease interferes with cell plasticity regulation. Cancers (Basel) 13(15):3897
Reich M, Jaegle S, Neumann-Haefelin E, Klingler JH, Evers C, Daniel M et al (2021) Genotype-phenotype correlation in von Hippel-Lindau disease. Acta Ophthalmol 99(8):e1492–e1500
van Leeuwaarde RS, Ahmad S, Links TP, Giles RH (2000) Von Hippel-Lindau Syndrome. In: GeneReviews. University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK1463. Accessed 29 September 2020
Wittstrom E, Nordling M, Andreasson S (2014) Genotype-phenotype correlations, and retinal function and structure in von Hippel-Lindau disease. Ophthalmic Genet 35(2):91–106
Hansen WJ, Ohh M, Moslehi J, Kondo K, Kaelin WG, Welch WJ (2002) Diverse effects of mutations in Exon II of the von Hippel-Lindau (VHL) tumor suppressor gene on the interaction of pVHL with the cytosolic chaperonin and pVHL-dependent ubiquitin ligase activity. Mol Cell Biol 22(6):1947–1960
Ruiz-Llorente S, Bravo J, Cebrian A, Cascon A, Pollan M, Telleria D et al (2004) Genetic characterization and structural analysis of VHL Spanish families to define genotype-phenotype correlations. Hum Mutat 23(2):160–169
Ciotti P, Garuti A, Gulli R, Ballestrero A, Bellone E, Mandich P (2009) Germline mutations in the von Hippel-Lindau gene in Italian patients. Eur J Med Genet 52(5):311–314
Zhang J, Ma J, Du X, Wu D, Ai H, Bai J et al (2015) Clinical and genetic investigation of a multi-generational Chinese family afflicted with von Hippel-Lindau disease. Chin Med J (Engl) 128(1):32–38
Glavac D, Neumann HP, Wittke C, Jaenig H, Masek O, Streicher T et al (1996) Mutations in the VHL tumor suppressor gene and associated lesions in families with von Hippel-Lindau disease from central Europe. Hum Genet 98(3):271–280
Liu Z, Zhou J, Li L, Yi Z, Lu R, Li C et al (2020) Intronic mutation of the VHL gene associated with central nervous system hemangioblastomas in two Chinese families with Von Hippel-Lindau disease: case report. BMC Med Genet 21(1):191
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conception and design of the study. Material preparation, data collection, and analysis were performed by AAEA and ILR. Patient interview and genetic counseling were performed by IVNH. TCM supervised the study. The first draft of the manuscript was written by AAEA, and all authors commented on earlier versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval was obtained from the Ethics Committee and the Scientific Council of the National Center of Medical Genetics. The procedures used in this study adhere to the tenets of the Declaration of Helsinki. Informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
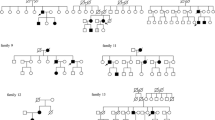
Additional file 1: Figure 1
Pedigree of family 1 where the R161X (c.481C > T) mutation is being segregated (VHL Type 1). Figure 2 Pedigree of family 2 where the D121G (c.362A > G) mutation is being segregated (VHL Type 2). Figure 3 Pedigree of family 3 where the D121G (c.362A > G) mutation is being segregated (VHL Type 2). Figure 4 Pedigree of family 4 where the S183X (c.548C > A) mutation is being segregated (VHL Type 1). Figure 5 Pedigree of family 5 where the c.463+2T > C mutation is being segregated (VHL Type 2). Figure 6 Pedigree of family 6 where the R167W (c.499C > T) mutation is being segregated (VHL Type 2). Figure 7 Pedigree of family 7 where the C162W (c.486C > G) mutation is being segregated (VHL Type 1).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Esperón Álvarez, A.A., Noa Hechavarría, I.V., López Reyes, I. et al. Germline variants in the Von Hippel-Lindau tumor suppressor gene in Cuban patients. Egypt J Med Hum Genet 25, 35 (2024). https://doi.org/10.1186/s43042-024-00506-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-024-00506-5