
Abstract
Background
Breast cancer is a common cause of cancer death among women with a complex and heterogeneous picture in histological, molecular and clinical features. The aim of this study was to identify hub gene and their target microRNAs in related pathways for breast cancer.
Methods
We selected screening methods for differentially expressed mRNAs and miRNAs using expression profile data of breast cancer from the cancer genome atlas. Using some databases for annotation, the functional and pathway enrichment for differential expression genes was performed. We selected genes and miRNAs with differential expression pattern. Then we determined target genes for differential expression miRNAs (DEMIs) and intersection between them was selected as differentially expressed miRNA–target genes for breast cancer. In the next step, we constructed miRNA–mRNA regulatory network and protein–protein interaction (PPI) network for more information.
Results
Top 10 DEMIs were identified from miRNA profile. Then, we selected 354 genes as target gene for 10 DEMIs. The miRNA–mRNA and PPI network were constructed, and 10 hub genes and 5 miRNAs identified that some of them are new for breast cancer. Also, miRNA–target genes with differential expressions in this study were all mainly involved in signaling pathways and developmental process.
Conclusion
This study identified some candidate biomarkers for breast cancer that they have a potential role in pathways related to breast. These findings can be used for research, early diagnosis and therapeutic goals.
Similar content being viewed by others

Introduction
Breast cancer is a common cause of cancer death that starts in the breast tissue and is associated with overgrowth and abnormal growth of cells in various breast tissues [1, 2]. Breast cancer is one of the most frequent cancers among females, and some serious health problems are related to breast cancer all over the world. This disorder is the second leading cause of cancer death among women and is a heterogeneous complex disorder with different histological, molecular and clinical phenotypes [3, 4].
Although some improvement strategies such as surgical and chemotherapy techniques have been achieved in recent years, late diagnosis and resistance to treatment are serious problems which lead to a poor prognosis for some patients in this regard [5, 6].
Based on previous studies, there are some evidences about role of genes and microRNAs in molecular pathways that are involved in progression and development of breast cancer [5]. MiRNAs are a class of small noncoding regulatory RNAs that are involved in controlling gene expression at the posttranscriptional level, and they are particularly promising due to their molecular stability, their ease of detection by noninvasive methods and their ability to provide improved subtype classification. Evaluation of differential expression profiles for these genes and microRNAs can be used as valuable clues to discover new biomarkers which are more effective for early diagnosis and therapeutic strategies in breast cancer patients [5, 7].
An ideal strategy to obtain valuable genetic information is bioinformatics analysis for the comprehensive analysis of large databases such as The Cancer Genome Atlas (TCGA). TCGA is a genomics program related to cancer landmarks, which contains the molecular profile of more than 20,000 primary cancers and matched normal samples including 33 cancer types [8].
In the present study, we used sophisticated bioinformatics methods to find useful potential biomarkers in molecular data from breast cancer.
Methods
Patients and sampling
We downloaded clinical data from breast cancer patients from the TCGA database. Criteria for including patient data in the present study include histopathological diagnosis of breast cancer, demographic information such as age, sex, race, disease status, survival status, pathological stage, TNM classification, and survival time. In total, data from 1097 patients were included in this study.
DEG identification
We collected RNA-seq level 3 data from the TCGA database. Using Voom and TMM normalization methods, raw readings of RNA-seq data were normalized. All analyses were performed with R software. The limma package was used to demonstrate the differential expression of miRNAs (DEmiRNAs) and mRNAs (DEmRNAs) between primary tumors and normal tissue. Data obtained based on change|log2 fold (FC)|> 1 are filtered for DEmiRNAs and DEmRNAs. P value < 0.05 and false detection rate (FDR) < 0.05 were considered for significant thresholds.
DEGs pathway enrichment analysis
Gene ontology (GO) was used to analyze functional enrichment in biological processes, cellular components and molecular functions levels. Due to classified genes in related pathways, the analysis of DEGs signaling pathways was carried out based on KEGG (Kyoto Encyclopedia of Genes and Genomes) database (https://www.genome.jp/kegg/) and DAVID ( the database for annotation, visualization and integrated discovery) online database (https://david.ncifcrf.gov/). In this part, pathways with P value < 0.05 were screened.
Protein–protein interaction (PPI) network construction analysis
The miRNA–mRNA network and protein–protein interaction (PPI) network were formed by Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) online tool. CytoHubba package of Cytoscape 3.9.1 was used to analyze and predict interactions and represent nodes and edges (score > 0.4). The cutoff criteria for construction of PPI for degree, node score, K-score and maximal depth were 2, 0.2, 2 and 100, respectively.
Results
Study population
Demographics results of participants including age, sex, ethnicity and race are shown in Table1. Also, the pathological characteristics based on The TNM Classification are shown in Fig. 1.

Tumor classification numbers are reported as percentages (NA, not available)
Identification and selection of DEGs in breast tumors and normal samples
In the first stage, DEGs were selected with P < 1.07E-30 and − 7 <|logFC|< 7. A total of 1639 DEGs were identified when normal samples were compared with breast tumor samples.
In the second stage, we identified top 10 miRNAs with differentially expressed from miRNA profile including 4 upregulated miRNAs and 6 downregulated miRNAs (Table 2). In the next stage, we identified a list of gene targets for this top 10 DEMIs by using multiMir package in R software which are 3141 in number. Then, from the sharing between the data of the first stage and the target genes, we reached 354 common genes (Fig. 2).

List 1 of genes obtained from RNA-seq level 3 analysis from the TCGA database and list 2 of target genes for top 10 DEMIs obtained with multimir software; the number of common genes between list 1 and 2 was 354
Functional and pathway enrichment analysis of differentially expressed genes (DEGs)
Based on GO enrichment analysis, DEGs were classified into three groups including a cellular component group, a biological process group and a molecular function group, which are shown in Fig. 3A–D

Gene ontology (GO) and signaling pathway enrichment analysis of DEGs. GO enrichment analysis of DEGs showed A cellular component, B biological process, C molecular function and D Signaling pathways enrichment of DEGs
In the cellular component group, GO terms mainly included chromosome, chromosomal part and chromatin (3A). In the biological process analysis, GO terms were mainly involved in the regulation of cellular, biological and developmental process (3B). Evaluation for molecular function group showed that GO terms mainly involved for transcriptional factors activity, enzymes binding, core promotor proximal region DNA binding and core promotor proximal region sequences (3C). Also, evaluation for pathway enrichment showed that DEGs were mainly involved in cancer pathways (3D).
Protein–protein interaction (PPI) network and modular analysis for DEGs
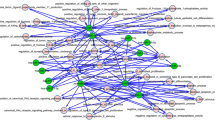
Withdrawing string network for 354 common genes (Fig. 4, node = 293, edge = 1454), we selected top 10 genes by using cytoHubba in Cytoscape. Also, among the 10 DEMIs, there were 5 DEMIs (hsa-miR-141-3p, hsa-miR-96-5p, hsa-miR-486-5p, hsa-miR-204-5p and hsa-miR-139-5p) related to top 10 genes. Therefore, PPI network consisted of 5 DEMIs as nodes and these 10 genes as edges. These 5 DEMIs (hsa-miR-141-3p, hsa-miR-96-5p, hsa-miR-486-5p, hsa-miR-204-5p and hsa-miR-139-5p) are related to 10 genes including JUN, CCNB1, AURKB, CCNA2, MKI67, FOXM1, ASPM, TTK, TOP2A and KIF23 (Fig. 5).

String network for 354 common genes

PPI network consisted of 5 DEMIs as nodes and these 10 top genes as edges
Evaluations for functional enrichment showed that they were mainly involved in signaling pathways and developmental process.
Discussion
Breast cancer is a heterogeneous complex disease with large heterogeneity in the genomic landscape with a high mortality rate. Examining the expression profile and related pathways can be a clue to early prognosis, diagnosis and treatment of breast cancer. The aim of the present study was to explore the expression profile and related pathways from breast cancer. Therefore, mRNA and miRNA expression profiles were used for differential expression mRNAs (DEMs) and miRNAs (DEMIs).
Among top 10 DEMIs, 5 miRNAs including hsa-miR-141-3p, hsa-miR-96-5p, hsa-miR-486-5p, hsa-miR-204-5p and hsa-miR-139-5p were associated with 10 related genes including JUN, CCNB1, AURKB, CCNA2, MKI67, FOXM1, ASPM, TTK, TOP2A and KIF23. These miRNAs and genes were mainly involved in signaling pathways, cell cycle process such as cell division, and DNA rapier and DNA binding which are useful for the development early diagnosis and new therapeutic strategies. In this study, we found some microRNA related to hub genes that association between them and hub genes was studied in some previous studies. The association between these genes and miRNAs in breast cancer has not reported in previous studies. Therefore, they can be useful as biomarker.
In the current study, data analysis showed JUN and KIF23 genes are target genes for has-miR-139-5p with downregulated expression in tumor samples in comparison with normal samples. JUN regulates the activity of cyclinD1 and p53 and other genes involved in cellular growth and proliferation. Moreover, c-JUN plays an essential role in tumor progression [9, 10]. A study found that c-JUN overexpressed in invasive breast cancer [11] and increased microvessel density [10]. KIF23 is a crucial regulator of cytokinesis and microtubule-dependent molecular motors that move chromosomes during cell division and plays an important role in tumorigenesis and cancer progression [12, 13]. Zhi Li et al. found that KIF23 significantly upregulated and promoted EMT progression in TNBC [14]. miR-139-5p has a vital role in tumorigenesis and various types of cancers, including breast cancer. miR-139-5p had a significant role in breast cancer cell motility and invasion [15]. A study reported that miR-139-5p expression levels were significantly depleted in breast cancer samples. Yang Zhang et al. indicated that miR-139 inhibited JUN expression, and JUN could induce miR-139 expression. Aberration in this loop could develop gastric cancer. This result reveals miR-139-5p potential as a prognostic marker for breast cancer.
In this study, data analysis showed TTK gene as a target gene for hsa-miR-486-5p with a downregulated expression in tumor samples in comparison with normal sample. TTK is a component of the spindle of the checkpoint and required for chromosome alignment at the centromere during mitosis [16]. TTK expression level was elevated in different types of cancer, including lung, breast, bladder, prostate and anaplastic thyroid. Overexpression of TTK in cancer tissues correlates with poor prognosis in HER2-positive breast cancer [17,18,19]. miR-486-5p plays an important role in several cancers, and it is a tumor-specific miRNA. Gharehdaghci et al. reported that miR-486-5p expression decreased in breast cancer and affects cellular proliferation, invasion and apoptosis [20]. Therefore, hsa-miR-486-5p can be an ideal marker for more investigation for prognosis, diagnosis and therapeutic goals in breast cancer.
In the current study, the results showed AURKB and FXOM1 gene are target genes for has-miR-204-5p with a downregulated expression in tumor samples. Aurora kinase B (AURKB) is a conserved family of serine/threonine kinase that has an essential function during cell cycle, such as chromosome segregation, spindle organization and cytokinesis [21, 22]. AURKB is overexpressed in several cancers, such as triple-negative breast cancer. AURKB overexpression prevents cell apoptosis by impairing p53 function [23]. FOXM1 gene is another gene with differential expression in this study, plays a role in cell proliferation and DNA damage repair process and is crucial for G1/S transition [24]. Moreover, FOXM1 regulates cell cycle genes; thus, FOXM1 overexpression affects triple-negative proliferation [25]. Previous studies have reported that miR-204-5p acts as tumor inhibitor and downregulated in human cancers such as breast cancer, gastric cancer, papillary thyroid carcinoma and hepatocellular carcinoma. Also it has been confirmed that overexpression of miR-204-5p inhibits cell migration and metastasis in breast cancer [26].
In this study, results showed TOP2A and CCNB1 are target genes for has-miR-96-5p with upregulated expression in tumor samples in comparison with normal samples. TOP2A encodes a nuclear enzyme that regulates DNA topological state, and it is essential for chromosome condensation and chromatid separation. TOP2A is overexpressed in cancer types including breast, lung, ovarian and hepatic [27, 28]. TOP2A has located approximately 700kb telomeric to HER2. TOP2A gene is co-amplified in about one-third of all patients with HER2 amplification. TOP2A monitoring could predict breast cancer patients' overall survival and lower expression associated with better clinical outcomes [29]. CCNB1, a critical member of the cyclin family, plays a significant role in mitosis initiation and quality control mitosis steps [30]. Accumulating studies have demonstrated the role of CCNB1 in various cancers, including melanoma cancer, colorectal cancer, pancreatic cancer and breast cancer [31]. It was reported that CCNB1 activates p53 signaling pathways and promotes cell apoptosis, which may be an important prognostic marker for breast cancer patients [30]. Ding et al. have demonstrated that high expression of CCNB1 was significantly associated with poor hormone therapy outcomes and could be a biomarker for the prognosis of patients with ER-positive breast cancer. These reports agree with our demonstration that CCNB1 was associated with breast cancer [32, 33]. miR-96-5p is a oncogenic miRNA, and it is associated with several cancers including breast, prostate and bladder cancer [34]. Mou-chun Gong et al. found that TOP2A is a target for two microRNAs including miR-96-5p and miR-635. Based on the previous study, miR96-5p significantly upregulated in papillary thyroid cancer (PTC) [35]. miR-96-5p has a tumor-promoting role in breast cancer. A study reported that miR-96-5p was significantly overexpressed in breast cancer and promoted BC cell migration by targeting AK3 in MEK/ERK signaling [36]. These results showed miR-96-5p has potential to be a marker for prognosis, diagnosis and therapeutic goals in breast cancer.
In this study, results showed that ASPM, CCNA2, MKI76 and TOP2A are target genes for has-miR-141-3p with overexpression in tumor samples. ASPM regulates mitotic spindle organization, spindle orientation and cytokinesis [37]. The ASPM gene has an important role for making a protein that is involved in cell division. However, it appears to be particularly important for the division of cells in the developing brain. ASPM is overexpressed in various cancers, including hepatocellular, ovarian and prostate cancer. ASPM expression levels correlate with tumor grades, and overexpression of ASPM is associated with a worse prognosis [37]. Another study reported a correlation between CCNB2 and ASPM; thus, overexpression of CCNB2 and ASPM causes breast cancer development [38]. MKI67 is a nuclear protein and is closely related to cell mitosis and is widely used as a proliferation marker [39]. MKI67 is used as a prognostic marker in some cancers, including early breast cancer and non-small lung cancer. Moreover, the MKI67 expression level is related to pathological grade in tumors. Guangyu Sun et al. demonstrated that the expression of MKI67 gene was associated with metastasis and invasion of breast cancer [40]. CCNA2 is a regulator for cell cycle and plays a vital role in controlling the cell cycle at G1/S and G2/M steps [30]. Based on several studies, CCNA2 expression can be used as a prognostic biomarker for ER-positive for tamoxifen resistance in breast cancer [33]. MiR-141-3p is downregulated in breast cancer, and it has capability as a prognostic factor for overall survival in patients with breast cancer [41]. Shanping Sun et al. indicated that miR-141-3p downregulated in breast cancer and miR-141-3p introduced as hypoxia-responsive miRNA and it might be targeting AK3 gene in HMGB1/HIF1a signaling pathway [41].
Conclusion
There is a lack of reliable biomarkers for patient selection and therapeutic efficacy for patients with breast cancer. In this study through the formation of the protein–protein interaction network with the STRING database and bioinformatics analysis, we identified 10 hub genes and 5 microRNA that had high degrees of confidence, indicating that all of them might play pivotal roles in breast cancer-related pathways. These findings can be helpful for more information about molecular mechanism involved in breast cancer, and they can provide new insight for more investigation and development of early diagnosis and therapeutic targets for breast cancer. More investigations in experimental and clinical samples are needed.
Availability of data and materials
All data generated or analyzed during the study are included in this manuscript and supplementary files, all of which were submitted.
References
Adorno G, Lopez E, Burg MA, Loerzel V, Killian M, Dailey AB et al (2018) Positive aspects of having had cancer: a mixed-methods analysis of responses from the American Cancer Society Study of Cancer Survivors-II (SCS-II). Psychooncology 27(5):1412–1425
Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G (2021) Epidemiology and risk factors
Ferlay JS, Foucher EL (2013) Cancerinci denceandmortalitypatternsinEurope: estimatesfor40countriesin 2012. EurJCancer 49(6):1374
Sun Y-S, Zhao Z, Yang Z-N, Xu F, Lu H-J, Zhu Z-Y et al (2017) Risk factors and preventions of breast cancer. Int J Biol Sci 13(11):1387
Xia L, Su X, Shen J, Meng Q, Yan J, Zhang C et al (2018) ANLN functions as a key candidate gene in cervical cancer as determined by integrated bioinformatic analysis. Cancer Manag Res 10:663
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339(6127):1546–1558
Zhang G-M, Goyal H, Song L-L (2018) Bioinformatics analysis of differentially expressed miRNA-related mRNAs and their prognostic value in breast carcinoma Corrigendum in/10.3892/or. 2018.6505. Oncol Rep 39(6):2865–2872
Silva TC, Colaprico A, Olsen C, D'Angelo F, Bontempi G, Ceccarelli M, et al (2016) TCGA Workflow: analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research, 5
Zhang L, Sun S, Wang Y, Mo Y, Xiong F, Zhang S et al (2020) Gossypol induces apoptosis of multiple myeloma cells through the JUN-JNK pathway. Am J Cancer Res 10(3):870
Jiao X, Katiyar S, Willmarth NE, Liu M, Ma X, Flomenberg N et al (2010) c-Jun induces mammary epithelial cellular invasion and breast cancer stem cell expansion. J Biol Chem 285(11):8218–8226
Vleugel MM, Greijer AE, Bos R, van der Wall E, van Diest PJ (2006) c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum Pathol 37(6):668–674
Liu Y, Chen H, Dong P, Xie G, Zhou Y, Ma Y et al (2020) KIF23 activated Wnt/β-catenin signaling pathway through direct interaction with Amer1 in gastric cancer. Aging (Albany NY) 12(9):8372
Sun X, Jin Z, Song X, Wang J, Li Y, Qian X et al (2015) Evaluation of KIF23 variant 1 expression and relevance as a novel prognostic factor in patients with hepatocellular carcinoma. BMC Cancer 15(1):1–9
Li Z, Yang H-Y, Zhang X-L, Zhang X, Huang Y-Z, Dai X-Y et al (2022) Kinesin family member 23, regulated by FOXM1, promotes triple negative breast cancer progression via activating Wnt/β-catenin pathway. J Exp Clin Cancer Res 41(1):1–15
Krishnan K, Steptoe AL, Martin HC, Pattabiraman DR, Nones K, Waddell N et al (2013) miR-139-5p is a regulator of metastatic pathways in breast cancer. RNA 19(12):1767–1780
Zhang H, Yao W, Zhang M, Lu Y, Tang J, Jiang M et al (2021) TTK inhibitor promotes radiosensitivity of liver cancer cells through p21. Biochem Biophys Res Commun 550:84–91
Tang J, Lu M, Cui Q, Zhang D, Kong D, Liao X et al (2019) Overexpression of ASPM, CDC20, and TTK confer a poorer prognosis in breast cancer identified by gene co-expression network analysis. Front Oncol 9:310
Zhou Q, Ren J, Hou J, Wang G, Ju L, Xiao Y et al (2019) Co-expression network analysis identified candidate biomarkers in association with progression and prognosis of breast cancer. J Cancer Res Clin Oncol 145(9):2383–2396
Xie Y, Wang A, Lin J, Wu L, Zhang H, Yang X et al (2017) Mps1/TTK: a novel target and biomarker for cancer. J Drug Target 25(2):112–118
Gharehdaghchi Z, Baradaran B, Salehzadeh A, Kazemi T (2020) miR-486-5p regulates cell proliferation and migration in breast cancer. Meta Gene 23:100643
Zhu Q, Sun Y, Zhou Q, He Q, Qian H (2018) Identification of key genes and pathways by bioinformatics analysis with TCGA RNA sequencing data in hepatocellular carcinoma. Mol Clin Oncol 9(6):597–606
Huang C, Luo H, Huang Y, Fang C, Zhao L, Li P et al (2021) AURKB, CHEK1 and NEK2 as the potential target proteins of scutellaria barbata on hepatocellular carcinoma: an integrated bioinformatics analysis. Int J Gen Med 14:3295
Naorem LD, Muthaiyan M, Venkatesan A (2019) Integrated network analysis and machine learning approach for the identification of key genes of triple-negative breast cancer. J Cell Biochem 120(4):6154–6167
Ziegler Y, Guillen VS, Kim SH, Katzenellenbogen JA, Katzenellenbogen BS (2021) Transcription regulation and genome rewiring governing sensitivity and resistance to FOXM1 inhibition in breast cancer. Cancers 13(24):6282
Li S, Liu N, Piao J, Meng F, Li Y (2020) CCNB1 expedites the progression of cervical squamous cell carcinoma via the regulation by FOXM1. Onco Targets Ther 13:12383
Liang WH, Li N, Yuan ZQ, Qian XL, Wang ZH (2019) DSCAM-AS1 promotes tumor growth of breast cancer by reducing miR-204-5p and up-regulating RRM2. Mol Carcinog 58(4):461–473
Gao Y, Zhao H, Ren M, Chen Q, Li J, Li Z et al (2020) TOP2A promotes tumorigenesis of high-grade serous ovarian cancer by regulating the TGF-β/Smad pathway. J Cancer 11(14):4181
Qi L, Zhou B, Chen J, Hu W, Bai R, Ye C et al (2019) Significant prognostic values of differentially expressed-aberrantly methylated hub genes in breast cancer. J Cancer 10(26):6618
Fountzilas G, Valavanis C, Kotoula V, Eleftheraki AG, Kalogeras KT, Tzaida O et al (2012) HER2 and TOP2A in high-risk early breast cancer patients treated with adjuvant epirubicin-based dose-dense sequential chemotherapy. J Transl Med 10(1):1–21
Liu F, Wu Y, Mi Y, Gu L, Sang M, Geng C (2019) Identification of core genes and potential molecular mechanisms in breast cancer using bioinformatics analysis. Pathol Res Pract 215(7):152436
Lu Y, Yang G, Xiao Y, Zhang T, Su F, Chang R et al (2020) Upregulated cyclins may be novel genes for triple-negative breast cancer based on bioinformatic analysis. Breast Cancer 27(5):903–911
Wei L-M, Li X-Y, Wang Z-M, Wang Y-K, Yao G, Fan J-H et al (2021) Identification of hub genes in triple-negative breast cancer by integrated bioinformatics analysis. Gland Surg 10(2):799
Deng J-L, Xu Y-h, Wang G (2019) Identification of potential crucial genes and key pathways in breast cancer using bioinformatic analysis. Front Genet 10:695
Wei S, Zheng Y, Jiang Y, Li X, Geng J, Shen Y et al (2019) The circRNA circPTPRA suppresses epithelial-mesenchymal transitioning and metastasis of NSCLC cells by sponging miR-96-5p. EBioMedicine 44:182–193
Gong M-C, Chen W-Q, Jin Z-Q, Lyu J, Meng L-H (2021) Prognostic value and significant pathway exploration associated with TOP2A involved in papillary thyroid cancer. Int J Gen Med 14:3485
Wy Q, Sc F, Yq S, Gq J (2020) MiR-96-5p promotes breast cancer migration by activating MEK/ERK signaling. J Gene Med 22(8):e3188
Shubbar E, Kovács A, Hajizadeh S, Parris TZ, Nemes S, Gunnarsdóttir K et al (2013) Elevated cyclin B2 expression in invasive breast carcinoma is associated with unfavorable clinical outcome. BMC Cancer 13(1):1–10
Shi H, Zhang L, Qu Y, Hou L, Wang L, Zheng M (2017) Prognostic genes of breast cancer revealed by gene co-expression network analysis. Oncol Lett 14(4):4535–4542
Shi Y, Li Y, Yan C, Su H, Ying K (2019) Identification of key genes and evaluation of clinical outcomes in lung squamous cell carcinoma using integrated bioinformatics analysis. Oncol Lett 18(6):5859–5870
Sun G, Wang S, Wang Y (2019) Expressions of Topo IIα and Ki67 in breast cancer and its clinicopathologic features and prognosis. Pak J Med Sci 35(3):715
Sun S, Ma J, Xie P, Wu Z, Tian X (2020) Hypoxia-responsive miR-141–3p is involved in the progression of breast cancer via mediating the HMGB1/HIF-1α signaling pathway. J Gene Med 22(10):e3230
Acknowledgements
The authors also gratefully acknowledge technical and financial support provided by the Ahvaz Jundishapur University of Medical Sciences
Funding
The study was funded by the Ahvaz Jundishapur University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
MR, SMR and AP performed data collection and analysis. ET performed project administration. NG and GS performed writing and original draft. SH, ET, AS and SM performed review and editing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Ahvaz Jundishapur University of Medical Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Effect of ASPM expression level & cancer type on BRCA patient survival.
Additional file 2
. Effect of AURKB expression level & cancer type on BRCA patient survival.
Additional file 3
. Effect of CCNA2 expression level & cancer type on BRCA patient survival.
Additional file 4
. Effect of CCNB1 expression level & cancer type on BRCA patient survival.
Additional file 5
. Effect of FOXM1 expression level & cancer type on BRCA patient survival.
Additional file 6
. Egyptian Journal of Medical Human Genetics.
Additional file 7
. Effect of JUN expression level & cancer type on BRCA patient survival.
Additional file 8
. Effect of KIF23 expression level & cancer type on BRCA patient survival.
Additional file 9
. Effect of MKI67 expression level & cancer type on BRCA patient survival
Additional file 10
. Effect of TOP2A expression level & cancer type on BRCA patient survival.
Additional file 11
. Effect of TTK expression level & cancer type on BRCA patient survival.
Additional file 12
. Expression of ASPM in BRCA based on sample types.
Additional file 13
. Expression of AURKB in BRCA based on sample types.
Additional file 14
. Expression of CCNA2 in BRCA based on sample types.
Additional file 15
. Expression of CCNB1 in BRCA based on sample types.
Additional file 16
. Expression of FOXM1 in BRCA based on sample types.
Additional file 17
. Expression of JUN in BRCA based on sample types.
Additional file 18
. Expression of KIF23 in BRCA based on sample types.
Additional file 19
. Expression of MKI67 in BRCA based on sample types.
Additional file 20
. Expression of TOP2A in BRCA based on sample types.
Additional file 21
. Expression of TTK in BRCA based on sample types.
Additional file 22
. List 1 and List 2 genes contain DEGs obtained from first stage analysis and gene targets for top 10 DEMIs
Additional file 23
. Effect of TOP2A expression level & cancer type on BRCA patient survival.
Additional file 24
. Common genes between list 1 and 2
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Rezaeijo, S.M., Rezaei, M., Poursheikhani, A. et al. Integrative bioinformatics analysis of miRNA and mRNA expression profiles identified some potential biomarkers for breast cancer. Egypt J Med Hum Genet 24, 62 (2023). https://doi.org/10.1186/s43042-023-00443-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-023-00443-9