
Abstract
Background
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease showing a rising prevalence globally. Genetic predisposition plays a key role in the development and progression of the disease pathogenicity.
Main body
This paper summarizes genetic associations based on their influence on several metabolic aspects such as lipid metabolism, glucose metabolism, hepatic iron accumulation and cholesterol metabolism toward the NAFLD pathogenicity. Furthermore, we present variations in some epigenetic characters and the microRNA profile with regard to NAFLD.
Conclusion
As reported in many studies, the PNPLA3 rs738409 variant seems to be significantly associated with NAFLD susceptibility. Other gene variants like TM6SF2 rs58542926, MBOAT7 rs641738 and GCKR variants also appear to be more prevalent among NAFLD patients. We believe these genetic variants may provide insights into new trends in developing noninvasive biomarkers and identify their suitability in clinical practice in the future.
Graphical abstract

Similar content being viewed by others

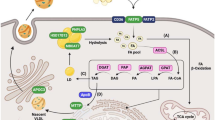
Background
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent chronic liver disease covering a wide spectrum of liver pathology (Fig. 1) characterized by accumulation of excess fat in the liver (> 5–10% by weight) in the absence of significant alcohol consumption [1].

NAFLD disease spectrum
NAFLD is considered the liver manifestation of metabolic syndrome [2]. As represented in Fig. 2, several factors could possibly increase the risk for NAFLD. Recent evidences suggest that there is a strong genetic contribution toward susceptibility to NAFLD and its severity [3]. A range of single nucleotide polymorphisms (SNPs) have been implicated in this regard, and in this paper, we aim to investigate the genetic background concerning various metabolic aspects of NAFLD by evaluating literature from several studies that reported genetic variants contributing to NAFLD pathogenicity (Fig. 3). PubMed and google scholar search terms “genetic background of NAFLD” and “genetic studies on NAFLD” were used in selecting high-quality articles for the analysis of our topic of interest.

Risk factors for NAFLD

Schematic representation showing genetic variants involved in different stages of NAFLD progression
Genetic polymorphisms incorporated in lipid metabolism related to NAFLD
PNPLA3 (patatin-like phospholipase domain-containing 3)
PNPLA3 encodes a protein with 481 amino acids that belonging to the patatin-like phospholipase domain-containing protein family, and its function remains unknown [4]. But the progenitor of the family, “patatin,” is a lipid acyl hydrolase (LAH) found in potato tubers. Patatin catalyzes the hydrolysis of monoacylglycerols (MAG) and expresses transesterification activity [5]. The human PNPLA family consists of nine members, and they show diverse enzymatic functions. PNPLA2 was previously identified as a major triglyceride hydrolase in the adipose tissue [6, 7], whereas other studies showed that PNPLA3 is a transmembrane polypeptide expressed in adipose tissue, liver and adrenal glands [8,9,10]. PNPLA3 is confined to the endoplasmic reticulum and on the lipid, droplet surfaces [11]. Several functional roles have been assigned to PNPLA3. These include hydrolase activity [9, 12, 13] a role in lipid droplet remodeling [4, 14] transacylase activity producing alkyl esters [13] and lysophosphatidic acid acyl transferase (LPAAT) activity producing phosphatidic acid triacylglycerol (Fig. 4). Phosphatidic acid is a common precursor for triacylglycerol synthesis [15].

Lysophosphatidic acid acyl transferase activity of PNPLA3 protein. GPAT, glycerol-3-phosphate acyltransferase; LPAAT, lysophosphatidic acid acyl transferase; PAP, phosphatidic acid phosphatase; DGAT, diacylglycerol acyltransferase
A SNP (rs738409) of cytosine to guanine substitution in PNPLA3 that changes codon 148 from isoleucine to guanine (I148M) has shown a strong association with NAFLD. Romeo and his colleagues who first reported an association of the PNPLA3 I148M variant with NAFLD pathogenicity identified this variation to be associated with hepatic fat levels and hepatic inflammation leading to the increased susceptibility to hepatic injury. Furthermore, they observed the carriers of the PNPLA3 I148M variant, to have elevated serum levels of liver-derived enzymes which are considered as the markers of liver inflammation [16]. He et al. (2009) have shown that the I148M substitution promoted triglyceride accumulation in hepatocytes thus interfering with hepatic triglyceride hydrolysis leading to hepatic steatosis [4]. Further, I148M variant was reported to be associated with higher LPAAT activity leading to steatosis [15].
Yamamoto and his colleagues suggested that PNPLA3 rs738409 variant is associated with NAFLD progression to cirrhosis and HCC in the Japanese population [17]. Tepper et al. (2018), who further validated this, showed a high frequency of the PNPLA3 rs738409 [C>G] variant among the Hmong population and that it predisposes to NAFLD/NASH [18]. Valenti et al. (2010) reported that PNPLA3 I148M in NAFLD patients is strongly associated with severe steatosis and with the presence of NASH and fibrosis in patients from Italy and the United Kingdom. This association was independent of age, basal metabolic rate (BMI) and diabetes. PNPLA3 rs738409 GG genotype was more prevalent among NAFLD patients, and it was found to influence high-density lipoprotein (HDL) cholesterol and alanine transaminase (ALT) levels [19]. Their study was extended to evaluate the predisposition of the PNPLA3 I148M variant to NASH and fibrosis in pediatric patients with NAFLD, and they observed that PNPLA3 rs738409 GG genotype had a very high risk of progressive liver disease in the pediatric cohort [20]. Previously, it was reported that children from different ethnicities including Asians, Americans and Hispanics are more predisposed to develop NAFLD [21].
Alisi et al. (2011) reviewed the genetic background of NAFLD and the metabolic syndrome. The report concludes that both conditions have common genetic origins. They further reported this to be valid for both adults and children [22]. Chan et al. (2017) conducted a retrospective cohort study to evaluate the risk of developing fatty liver in chronic hepatitis B virus (HBV) infected patients without significant alcohol intake. The study revealed that PNPLA3 rs738409 G allele was significantly associated with the susceptibility to the concurrent development of fatty liver in HBV patients. Furthermore, the study concluded that the concurrent fatty liver in HBV patients is a significant risk factor for progression to HCC in patients without significant alcohol intake [23].
Peroxisome-proliferator-activated receptors (PPARs)
PPARs are hormone receptors that bind to the promoters of the target genes in the ligand (hormone) bound state and activate transcription. There are three isotypes of PPARs, namely PPARα, PPARβ and PPARγ. All the members of PPAR are associated with lipid metabolism and transport. PPARα is expressed mostly in the adipose tissue and the liver. PPARα participates in fatty acid catabolism in the liver [24]. PPARβ mostly expressed in the gut, kidney, heart and the brain. PPARγ expressed mainly in the adipose tissue influences the fatty acid (FA) storage in the adipose tissue. Target genes of PPARγ directly participate in the lipogenic pathways. These include the genes expressing lipoprotein lipase (LPL), adipocyte FA binding protein, acyl-CoA and fatty acid transport protein (FATP) [24]. PPARγ also participates in the body’s fat cell differentiation program [25]. Two common variations of PPARγ, rs1801282 and rs3856806 are important in the lipid metabolism pathways.
Hui et al. (2008) initially reported a significant association between a PPARγ gene variant (rs3856806) and NAFLD through an adiponectin-related pathway [26]. PPARγ rs1801282 variant was reported to increase the resistance to oxidative stress, thus increasing susceptibility to develop NAFLD through a Chinese case–control study. Further, it was revealed that both the C/C genotype of PPARγ rs1801282 polymorphism and smoking were independent risk factors for NAFLD [27]. Gawrieh et al. (2011) investigated the association of PPARγ rs1801282 and PPARγ rs3856806 with NAFLD in a North American cohort. The study revealed two haplotypes with two major alleles (CC) and two minor alleles (GT) in the NAFLD cohort are associated with steatosis and fibrosis. Minor alleles (GT) are reported to show a protective form of NASH by lowering the risk for steatosis, inflammation and fibrosis. The study concluded that the two SNPs do not show sufficient individual effects, but an additive effect could possibly promote the risk of developing NAFLD and NASH [28]. Another genetic study on PPARγ rs1801282 polymorphism showed that the heterozygous genotype was significantly higher in NAFLD patients [29].
TM6SF2 (transmembrane 6 superfamily member 2)
Transmembrane 6 superfamily member 2 (TM6SF2) is identified as a casual gene associated with lipid traits. But its actual function remains unknown. TM6SF4 is expressed predominantly in the liver and intestine [30]. TM6SF2 is located in the 19q12 locus, and it encodes a protein of 351 amino acids [31]. Protein studies have revealed that TM6SF2 is comprised of 7–10 transmembrane domains, and it is localized in the endoplasmic reticulum (ER) and the ER-Golgi intermediate compartment of human liver cells [32]. TM6SF2 is a functional protein associated with hepatic triglyceride concentration. Functional studies have revealed that TM6SF2 inhibition is associated with reduced very-low-density lipoproteins (VLDL) secretion and elevated cellular triglyceride concentration leading to retention of TGs in hepatic lipid droplets causing a predisposition to fatty liver [30, 33]. But the overexpression of TM6SF2 is associated with reduced lipid droplet content [32]. Therefore, it is possible to suggest that TM6SF2 plays a role in NAFLD development. Another study demonstrated that TM6SF2 can influence total cholesterol levels and is associated with myocardial infarction [33].
Genome-wide association studies (GWAS) have revealed a TM6SF2 variant (rs58542926) associated with the elevated liver fat level. This variant is an adenine-to-guanine substitution in coding nucleotide 499, which replaces glutamate at residue 167 with lysine (c.499A>G; p.Glu167Lys). This variant was associated with decreased VLDL secretions from hepatocytes [30] and a higher risk of myocardial infarction [33]. An association of the p.Glu167Lys variant with fibrosis in patients with NAFLD [34] was also reported. This variant was highly prevalent in individuals with European ancestry; prevalence was moderate in African Americans and Hispanics and rare in Asians [30, 35]. Dongiovanni et al. (2015) studied suspicion of NASH in severely obese patients of European descent for the possible effect of the TM6SF2 p.Glu167Lys variant on liver diseases.. The TM6SF2 p.Glu167Lys variant was found to be associated with a higher prevalence of NASH and advanced fibrosis. Furthermore, the study revealed that the TM6SF2 p.Glu167Lys carriers are more susceptible to liver damage related to NAFLD. Nevertheless, obese carriers of TM6SF2 p.Glu167Lys were reported to be protected from cardiovascular risk. They suggest that inhibition of VLDL secretion from hepatocytes may protect against cardiovascular diseases, but on the other hand, it increases the risk of developing severe liver disease [36].
A systematic evaluation done by Holmen et al. (2014) reported that the TM6SF2 p.Glu167Lys C-allele carriage is a lipid-associated genetic variation influencing total cholesterol levels [33]. Also, strong associations were found between the variant and serum ALT levels [30, 33]. Further, the C>T minor allele in TM6SF2 p.Glu167Lys was reported to be associated with an increased risk of greater steatosis as well as with the severity of steatohepatitis. The variant genotype also has a strong association with an increased risk of advanced fibrosis independent of gender, age, BMI and diabetes [34]. Sookian et al. (2015) showed that the rs58542926 variant is associated with a higher risk of fatty liver. An allelic test on the study subjects has shown that the T allele was significantly associated with the disease progression. They also reported that TM6SF2 protein expression in the liver was remarkably decreased in NAFLD patients compared with the controls [37].
TRIB1 (tribbles-1)
TRIB1 protein function to regulate cell differentiation, cell division and in protein degradation processes [38]. TRIB1 polymorphisms are reported to be associated with lipid traits affecting lipid metabolism. Significant association of TRIB1 polymorphisms, rs17321515 (A>G) and rs2954029 (A>T) with serum TG levels [39, 40] and an association of TRIB1 rs17321515 with total cholesterol and LDL levels [41] has been reported. Liu et al. (2019) first reported an association of TRIB1 polymorphisms and the risk of NAFLD. A alleles of rs17321515 and rs2954029 were associated with the risk of NAFLD and rs17321515. A allele was associated with higher LDL levels in NAFLD [42]. Coronary heart disease (CHD) is a major complication observed in NAFLD; TRIB1 polymorphisms are implicated in CHD [43] and later studies identified AA and GA genotypes of rs17321515 as those associated with CHD in NAFLD [44]. Serum lipid levels were significantly increased in A allele carriers [44].
MBOAT7 (membrane-bound O-acyltransferase domain-containing 7)
In 2015, Buch et al. reported a gene variant in the membrane-bound O-acyltransferase domain-containing 7 (MBOAT7) at rs641738 that increased the risk of alcoholic cirrhosis [45]. Later, this variant was shown to be associated with susceptibility to the development and progression of NAFLD [46]. MBOAT7 catalyzes acyl-chain remodeling of phosphatidylinositols (PIs). Mainly, it catalyzes the transfer of polyunsaturated fatty acids like arachidonoyl-CoA to PIs like lysophosphatidylinositol, allowing maintaining the required level of desaturation in cell membranes. The rs641738 T allele reduces MBOAT7 expression affecting PI composition of the hepatocyte plasma membrane favoring hepatocellular fat accumulation and initiating inflammatory responses leading to NASH [46]. In an Italian NAFLD cohort, each MBOAT7 rs641738 T allele conferred an approximately 80% increased risk of HCC in patients without advanced liver fibrosis [47]. MBOAT7 variant has been thus implicated to predispose to HCC in non-cirrhotic patients, suggesting it to be a useful biomarker to identify such patients. Luukkonen et al. (2016) also confirmed that this variation was also associated with histological liver damage marked with hepatic phosphatidylinositol remodeling. In a cohort of 3854 patients of European descent, rs641738 was associated with increased hepatic fat content and development of NAFLD [48].
Genetic polymorphisms incorporated in glucose metabolism related to NAFLD
GCKR (glucokinase regulatory protein)
Glucokinase (GCK), the predominant hexokinase isoenzyme in the liver, is also expressed in the β-cells of the pancreas. It is highly sensitive to glucose and plays a key role in glucose metabolism. The activity of GCK is tightly regulated by GCK regulatory protein (GCKR). This protein–protein interaction shows a ligand-dependent and inhibitory GCK activity in response to plasma glucose fluctuations [49].
In 2011, Speolite et al. initially documented that GCKR variants were associated with changes in liver and serum lipid levels predisposing to liver fat deposition [50]. GCKR rs780094 is an intronic SNP which has shown associations with glucose levels. A GWAS done on the Finnish and Swedish populations [51] has shown that GCKR rs780094 polymorphism favors high TG levels and lower glucose levels. The same results were demonstrated in another study done on a European population [52]. Also, this variant showed an association with low insulin resistance and lower T2DM risk [51]. GCKR rs1260326, a non-synonymous variant with C to T substitution, substituting leucine for proline (P446L) was also associated with fasting plasma glucose and TG levels [51]. The T allele of rs1260326 C>T and rs780094 C>T variants also showed a clear association with NAFLD and NASH [53]. The rs1260326 T allele was significantly associated with a higher grade of hepatic steatosis in Indian patients but not in Malay and Chinese patients.. When the study was extended to observe the effect of both GCKR and PNPLA3 polymorphisms on the NAFLD risk, GCKR rs1260326, GCKR rs780094 and PNPLA3 rs738409 in combination led to a greater risk of developing NAFLD than either of the SNP alone [53].
INSR (insulin receptor) gene
INSR gene coding for the insulin receptor is located on the short arm of chromosome 19 and is composed of 22 exons [54]. Insulin participates in the glucose metabolism pathways through INSR, with liver cells being a major target. Defects in INSR lead to IR, a major risk factor for the development and progression of NAFLD.
Impaired secretion of insulin was identified as the major predictor for glucose intolerance in NAFLD patients, and histological severity of NAFLD was directly associated with glucose intolerance independent of adiposity [55]. NAFLD patients had a high prevalence of IR compared to controls. IR in NAFLD patients was associated with higher aspartate transaminase (AST) and ALT levels [56]. IR was reported to be more severe in NASH than in simple fatty liver [57]. The proportion of CT at the INSR exon 2-2257 locus was significantly lower in NAFLD than in the controls when compared with the CC genotype [58] the study concluded the protective effect of the CT genotype against NAFLD.
Genetic studies to investigate INSR polymorphisms in developing NAFLD are rare. INSR Exon 17 is an important motif as it encodes the tyrosine kinase domain of the INSR protein [59]. Therefore, mutations in the exon 17 can directly lead to IR and rs1799817 polymorphism is located in it [60]. Nobakht et al. (2020) who investigated INS and INSR polymorphisms on NAFLD risk in an Iranian population observed a 90% lower risk for NAFLD in carriers of the INSR rs1799817 “TT” genotype when compared with the “CC” genotype and suggested that “TT” genotype has a protective effect for NAFLD risk [54].
PC-1 (plasma cell antigen-1)/ENPP1 (ectoenzyme nucleotide pyrophosphate phosphodiesterase 1)
ENPP1 is a membrane glycoprotein that binds with INSR and inhibits its effects on glucose metabolism. Thus, overexpression of ENPP1 can lead to IR [61]. PC-1/ENPP1 Lys121Gln polymorphism is a gain of function variant causing stronger interaction with INSR.
Dongiovanni et al. (2010) observed the PC-1/ENPP1 Lys121Gln polymorphism to be associated with increased fibrosis and fibrosis severity in NAFLD patients compared to healthy controls in a European cohort. This variant was also associated with diabetes and metabolic abnormalities in NAFLD patients, suggesting that these abnormalities may occur consequent to IR in them. The study concludes that PC-1/ENPP1 Lys121Gln polymorphism influences INSR activity thus predisposing to liver damage in NAFLD [62].
Genetic polymorphisms incorporated in hepatic iron accumulation related to NAFLD
HFE polymorphisms
The HFE gene shows two common missense mutations (C282Y, H63D) in patients suffering from hereditary hemochromatosis (HH). HH is an autosomal recessive genetic disorder that causes enhanced iron absorption and hepatic iron deposition that can increase the risk of developing cirrhosis and HCC [63, 64].
C282Y, a G to A transition at nucleotide 845 that changes the amino acid cysteine to tyrosine, and H63D a C to G transition at nucleotide 187 that results in a histidine to aspartic acid substitution are reported as the most prevalent genotypes associated with HH [65, 66]. In an Indian population study, C282Y mutation was not present among patients with liver disorders including NASH, but the H63D variant showed 14.8% prevalence among NASH patients, though they did not have iron overload. Two of the NASH patients with hepatic iron load were heterozygous for H63D, and one homozygous patient did not have hepatic iron overload. The study concludes that iron overload in NASH and other liver malignancies is a non-HFE type in Indians. [67]. Saremi et al. (2016) reported a significant association of HFE C282Y polymorphism with NAFLD in Iranian patients with T2DM [68]. Hepcidin is the key regulator of iron homeostasis of the body. Nelson et al. (2012) investigated whether the iron loading in NAFLD/NASH is influenced by the hepcidin regulation among HFE genotypes and observed a positive correlation between hepatic iron stores and decreased serum hepcidin levels in all the HFE genotypes tested (C282Y, H63D and wild type). The study suggests that hepcidin regulation in NAFLD is determined by the iron stores of the body, regardless of HFE genotypes. Furthermore, the study found a potential role of HFE H63D in NAFLD pathogenesis possibly through an iron-independent pathway [69]. A similar result was found in a Polish study where a higher serum iron level was identified as a risk factor for NAFLD pathogenicity, regardless of HFE mutations [70].
Genetic polymorphisms incorporated in cholesterol metabolism related to NAFLD
SREBF (sterol regulatory element-binding factor) polymorphisms
SREBPs are being identified as regulators of cholesterol and lipid metabolism, and there are three members in the human SREBP family named SREBP-1a, SREBP-1c and SREBP-2 [71]. The sterol regulatory element-binding factor (SREBF)-1c gene codes for a transcription factor which is involved in de novo lipogenesis and hepatic insulin sensitivity [72]. (SREBF)-1c rs11868035 A/G variant is located in the intron region. A allele of this variant together with BMI changes was associated with an increased risk for NAFLD [72]. Furthermore, GA/AA carriers of the NAFLD cohort are reported to show more severe steatosis, higher NAFLD activity score and a higher prevalence of NASH. Also, SREBF-1c rs11868035 SNP was associated with impaired glucose homeostasis in NAFLD patients [72]. In contrast, in a Han Chinese population, none of the four common SNPs (rs62064119, rs2297508, rs11868035 and rs13306741) in the SREBP-1c gene were associated with the NAFLD risk or with the total cholesterol levels [73]. SREBP-2 is encoded by a separate gene on human chromosome 22q13 and is closely associated with cholesterol synthesis [74, 75]. Studies have shown enhanced SREBP-2 expression and free cholesterol in NAFLD patients compared to healthy controls. This shows that free cholesterol plays an important role in NAFLD, and it is correlated with SREBP-2 expression [76]. Wang et al. (2014) showed that the SREBP-2 rs2228314 G > C polymorphism increases the risk of NAFLD in the Han Chinese population [77].
Miscellaneous
Steatohepatitic HCC (SH-HCC) is a histological subtype highly associated with metabolic syndrome [78]. Ando et al. (2015) conducted a genetic study with regard to the CTNNB1 (Catenin beta-1) gene mutations to interpret phenotypic characteristics of SH-HCC. Exon 3 of the CTNNB1 gene was previously known to be a mutational hotspot region according to the Catalogue of Somatic Mutations in Cancer (COSMIC). The study involved viable tumor tissues of 197 HCCs; 70 SH-HCCs and 127 conventional HCCs (C-HCCs). The mutational analysis revealed that 12 of 84 HCCs had missense mutations of the CTNNB1 gene with a single SH-HCC case and 11 C-HCC cases. The study concludes that CTNNB1 mutations were less frequent in SH-HCC than in C-HCC [79].
Epigenetics
Epigenetic changes that affect gene expression without altering the DNA sequence reveal new perspectives on the pathogenesis of NAFLD. The prevalence of unhealthy diets and physical inactivity has led to the development of NAFLD through epigenetic mechanisms.
Epigenetic alterations of tumor suppressor genes contribute to the HCC emergence. Nishida et al. (2016) examined the DNA methylation levels in HCC and their surrounding non-cancerous liver. The study concludes that the epigenetic alterations in the tumor suppressor genes that are leading to hepatocarcinogenesis could result from oxidative DNA damage in hepatocytes. Furthermore, they have identified that the serum alpha-fetoprotein (AFP) levels and degree of ballooning show independent associations with this oxidative DNA damage [80].
(See Additional file 1 for summary of the NAFLD-associated genetic variants).
MicroRNA
miR-122
MicroRNA 122 (miR-122) was identified as a dominant hepatocyte-specific miRNA accounting for 70% of the liver’s total miRNAs. [81]. Pivot roles for endogenous miR-122 were identified as tumor suppression and hepatocyte survival [82]. Also, Esau et al. (2006) showed that miR-122 is a key regulator of cholesterol and fatty acid metabolism in adult liver using mice models [83]. Furthermore, pathogenic repression of miR-122 has been observed in liver diseases such as NASH, cirrhosis and HCCs [84,85,86]
Tsai et al. (2012) observed key clinical phenotypes of human liver diseases in mice with a targeted deletion of Mir122a. These mice developed steatohepatitis, fibrosis and HCC together with disrupted livers which closely resembled disruptions found in human HCC. This study confirms the low expression of miR-122 in chronic liver diseases and HCCs, suggesting the restoration of miR-122 may be a therapy for such diseases [87]. Cheung et al. (2008) tabulated potential targets of miR-122 and pattern of expression regarding human NASH by studying patients with metabolic syndrome with/without suspected NAFLD. Significantly decreased liver tissue miR-122 levels were seen in NASH, and they also observed silencing of miR-122 can activate some lipogenic genes which are expressed in human NASH in vitro. [84]. In contrast to the situation in the liver tissue, Cermelli et al. (2011) reported that serum levels of miR-122 were significantly higher in NAFLD patients when compared with controls. Furthermore, the study shows a positive correlation between miR-122 and disease severity from simple steatosis to steatohepatitis and also with ALT and AST levels.. miR-122 appeared better than ALT, in detecting early disease stages in NAFLD conferring it as a suitable prognostic biomarker [88]. In a case–control study in biopsy-proven NAFLD, Pirola et al. (2015) showed that serum miR-122 level was upregulated in 7.2 folds in both simple steatosis and NASH and a systematic downregulation in miR-122 in the liver tissue in NASH [89]. Schütte et al. (2015) suggest that the high level of circulating miR-122 may be influenced by inflammation or apoptosis of hepatocytes in conditions like NAFLD, indicating miR-122 as a noninvasive biomarker for such HCC-related risk conditions [90]. Zhang et al. (2017) did a weighted gene co-expression network analysis (WGCNA) to identify potential key miRNAs and genes associated with the prognosis of HCC. The study concludes that the hsa-miR-363-5p may be a potential prognostic marker for HCC as its low expression was closely related to better survival of HCC [91]. Gene expression is regulated by upstream regulators (UR) like miRNAs, growth factors, transcription factors and cytokines [92]. Seshachalam et al. (2018) revealed that the miR-1249 is a major activated UR in 112 differentially expressed genes which are specific to NAFLD-HCC. Furthermore, five other miRNAs were also activated as URs in NAFLD-HCC (miR-7159-5p, miR-766-5p, miR-7056-5p, miR-6777-5p and miR-1249-5p). Also, it was found that the miR-4661-5p prominently inhibited UR in NAFLD-HCC [92].
(See Additional file 2 for summary of the included studies representing the association between miR-122 expression and NAFLD).
Association of genetic variations with NAFLD through different ethnicities
Danford et al. (2018) suggested that genetic factors play an important role in ethnic differences for NAFLD susceptibility among individuals [93]. In a large ethnically diverse cohort including African Americans, Japanese Americans, Latinos, native Hawaiians and Whites, PNPLA3 rs738409 variant showed a similar risk allele association with NAFLD, across all five ethnicities studied, while Latinos showed the strongest among all. Hispanics showed high frequency for this variant, while African Americans showed a lower frequency for the risk allele [94]. Further, Han et al. (2021) reported that this variant has shown increased susceptibility to NAFLD among ethnicities such as Hispanics, African Americans, East Asians, and South Asians [95]. Interestingly, Asians represent a distinct phenotype as “lean NAFLD” showing a BMI lower than the generally accepted obese range [96]. PNPLA3 rs738409 variant has been reported to show associations with this lean NAFLD phenotype found among Asians [95, 97]). TM6SF2 rs58542926 is another variant associated with lipid traits in NAFLD. Hispanics and Europeans were reported to show similar low frequencies for this variant [94, 98]. Also, this variant was reported to be lower in frequency in Chinese compared to Caucasians [99]. Another important variant associated with NAFLD severity is MBOAT7 rs641738. This variant was reported to show associations with NAFLD among Caucasians but not among Hispanics and African Americans [100]. Interestingly, among Hispanic obese children, this variant was reported to show a protective role against NAFLD [101]. Two of the reported variants in the GCKR (rs780094 and rs1260326) gene were significant genetic determinants of NAFLD, particularly among African Americans and Latinos [94]. Understanding such ethnic variabilities for predisposing to NAFLD is important as it directly affects the generalizability of research data.
Association of NAFLD with COVID-19
A significant association between liver injury and the severity of COVID-19 infection has been reported in multiple studies recently [102,103,104]. Vrsaljko et al. (2022) showed NAFLD patients had a longer duration of hospitalization in COVID-19 infection together with significant elevations in liver-associated markers and more frequent pulmonary thrombosis [105]. Further, NAFLD patients with advanced fibrosis were reported to have a higher risk of developing severe COVID-19 [106, 107]. Another recent meta-analysis showed that NAFLD is an independent risk factor for severe COVID-19 in younger patients [108]. The presence of cirrhosis was reported to show higher mortality in COVID-19 patients compared to those without underline cirrhosis (p < 0.001) [109]. Similar observations were made by Sarin et al. (2020), reporting that obese cirrhotics were more susceptible for liver injury than normal weight cirrhotics in COVID-19 (p = 0.02) [110]. Also, obese patients with advanced NAFLD stages, such as in NASH, have been reported to show high likelihood in predisposing to COVID-19 [111]. A Chinese hospital-based study demonstrated that COVID-19 patients with NAFLD had significantly longer viral shredding time compared to non NAFLD patients (p < 0.0001). Further, the study reported that COVID-19 patients with NAFLD background had abnormal liver function with high risk of NAFLD progression [112]. In contrast, some reports conclude that the presence of NAFLD does not affect the severity of COVID-19 infection [113, 114]. Multiple studies have shown an important association between metabolic associated fatty liver disease (MAFLD) and COVID-19 severity [110,115].
Future perspectives
Due to the complexity beh ind the NAFLD pathogenesis, identification of favorable drug targets is still emerging. On this regard, defining pathological drivers for NAFLD could be a preferred approach. Toxic alterations in the liver’s metabolic homeostasis are the key inducer of hepatic injury in non-alcoholic backgrounds. Genetics plays an important role in this regard as many studies including GWAS have defined loci which can induce metabolic dysfunction. In this review, we showed that such variations could be associated either with multiple stages or only with a specific stage of NAFLD. This is promising as those loci could be used as genetic biomarkers with both diagnostic and prognostic properties toward NAFLD. But to standardize such attempts, future studies should enable translational research with reproducible results with larger sample sizes. The recently proposed early diagnostic MAFLD criteria perform better than NAFLD identification criteria [116,117]. Basically, it combines metabolic syndrome evidences with hepatic steatosis features [118]. A recent Chinese community-based study has further investigated the genetic contribution together with such metabolic dysfunction features toward MAFLD development [119]. Such attempts that combine the metabolic and genetic signatures in defining MAFLD/NAFLD should be encouraged. Although liver biopsy remained the gold standard for diagnosis of NAFLD, it has major limitations due to the invasiveness of the procedure, cost and 10% false negativity [120]. Abdominal ultrasound is an effective NAFLD surveillance strategy, but still has questionable cost-effectiveness [121]. Therefore, genetic biomarkers with metabolic dysfunction features are promising due to noninvasiveness and cost-effectiveness. Future studies should validate such genetic and metabolic data through molecular assays to use them in developing favorable drug targets.
Conclusion
The genetic background of NAFLD is being widely investigated to identify pathogenic gene variants that may predispose to such conditions. Various studies were designed addressing candidate genes which may have some role in NAFLD pathogenicity. In this review, genes having potential roles in processes like lipid metabolism, glucose metabolism, hepatic iron accumulation, cholesterol metabolism and epigenetic characters were addressed in relation to NAFLD pathogenicity. PNPLA3 rs738409 variant seems to be significantly associated with NAFLD disease susceptibility as studies on this gene variant were replicated in many populations. Also, other gene variants like TM6SF2 rs58542926, MBOAT7 rs641738 and GCKR variants appear to be more prevalent in the NAFLD susceptibility. Circulating miR-122 was also reported to be upregulated in NASH validating the circulating miR profile as a prognosis biomarker. Patient characteristics and environmental factors to influence the outcome of the genetic effects. Therefore, factors like age, sex, BMI, co-morbidities such as diabetes mellitus and dietary factors should also be considered when comparing patient and control populations. Liver biopsy, a diagnostic method commonly used to detect NAFLD, is an invasive procedure. Therefore, studies revealing gene variants associated with NAFLD are important in developing noninvasive biomarkers for disease prediction, detection and monitoring prognosis.
Availability of data and materials
Not applicable.
Abbreviations
- NAFLD:
-
Non-alcoholic fatty liver disease
- NAFL:
-
Non-alcoholic fatty liver
- NASH:
-
Non-alcoholic steatohepatitis
- LC:
-
Liver cirrhosis
- HCC:
-
Hepatocellular carcinoma
- T2DM:
-
Type 2 diabetes mellitus
- IR:
-
Insulin resistance
- SNPs:
-
Single nucleotide polymorphisms
- PNPLA3 :
-
Patatin-like phospholipase domain-containing 3
- LAH:
-
Lipid acyl hydrolase
- MAG:
-
Monoacylglycerols
- LPAAT:
-
Lysophosphatidic acid acyl transferase
- BMI:
-
Basal metabolic rate
- HDL:
-
High-density lipoproteins
- ALT:
-
Alanine transaminase
- HBV:
-
Hepatitis B virus
- PPARs:
-
Peroxisome-proliferator-activated-receptors
- FA:
-
Fatty acid
- LPL:
-
Lipoprotein lipase
- FATP:
-
Fatty acid transport protein
- LDL:
-
Low density lipoprotein
- TG:
-
Triglyceride
- TM6SF2 :
-
Transmembrane 6 superfamily member 2
- ER:
-
Endoplasmic reticulum
- VLDL:
-
Very-low-density lipoproteins
- GWAS:
-
Genome-wide association studies
- CHD:
-
Coronary heart disease
- MBOAT7 :
-
Membrane bound O-acyltransferase domain-containing 7
- PIs:
-
Phosphatidylinositols
- GCKR:
-
GCK regulatory protein
- GCK:
-
Glucokinase
- INSR :
-
Insulin receptor
- AST:
-
Aspartate transaminase
- PC-1:
-
Plasma cell antigen-1
- ENPP1:
-
Ectoenzyme nucleotide pyrophosphate phosphodiesterase 1
- HH:
-
Hereditary hemochromatosis
- SREBF:
-
Sterol regulatory element-binding factor
- SH-HCC:
-
Steatohepatitic HCC
- CTNNB1 :
-
Catenin beta-1
- COSMIC:
-
Catalogue of Somatic Mutations in Cancer
- C-HCCs:
-
Conventional HCCs
- AFP:
-
Alpha fetoprotein
- miR:
-
Micro RNA
- WGCNA:
-
Weighted gene co-expression network analysis
- UR:
-
Upstream regulators
- MAFLD:
-
Metabolic-associated fatty liver disease
References
Lindenmeyer CC, McCulloug AJ (2018) The natural history of nonalcoholic fatty liver disease—an evolving view. Clin Liver Dis 22:11–21
Lee L, Sanders RA (2012) Metabolic syndrome. Pediatr Rev 33:459–468
Hernaez R (2012) Genetic factors associated with the presence and progression of nonalcoholic fatty liver disease: a narrative review. Gastroenterol Hepatol 35:32–41
He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV et al (2009) A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem 285:6706–6715
Macrae AR, Visicchio JE, Lanot A (1998) Application of potato lipid acyl hydrolase for the synthesis of monoacylglycerols. J Am Oil Chem’ Soc 75:1489–1494
Kienesberger PC, Oberer M, Lass A, Zechner R (2008) Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. J Lipid Res J 50(Suppl):S63–S68
Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ (2006) Characterization of the human patatin-like phospholipase family. J Lipid Res J 47:1940–1949
Baulande S, Lasnier F, Lucas M, Pairault J (2001) Adiponutrin, a transmembrane protein corresponding to a novel dietary- and obesity-linked mRNA specifically expressed in the adipose lineage. J Biol Chem 276:33336–33344
Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D et al (2005) Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J Lipid Res 46:2477–2487
Kershaw EE, Hamm JK, Verhagen LA, Peroni O, Katic M, Flier JS (2006) Adipose triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin. Diabetes 55:148–157
Trépo E, Romeo S, Zucman-Rossi J, Nahon P (2016) PNPLA3 gene in liver diseases. J Hepatol 65:399–412
Huang Y, Cohen JC, Hobbs HH (2011) Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem 286:37085–37093
Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW (2004) Identification, cloning, expression, and purification of three novel human calcium independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem 279:48968–48975
Ruhanen H, Perttilä J, Hölttä-Vuori M, Zhou Y, Yki-Järvinen H, Ikonen E et al (2014) PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res J 55:739–746
Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY et al (2012) Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab 15:691–702
Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA et al (2008) Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40:1461–1465
Yamamoto K, Kogiso T, Taniai M, Hashimoto E, Tokushige K (2018) Differences in the genetic backgrounds of patients with alcoholic liver disease and non-alcoholic fatty liver disease. JGH Open 3:17–24
Tepper CG, Dang JHT, Stewart SL, Fang DM, Wong KA, Liu SY et al (2018) High frequency of the PNPLA3 rs738409 [G] single-nucleotide polymorphism in Hmong individuals as a potential basis for a predisposition to chronic liver disease. Cancer 124:1583–1589
Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P et al (2010) Homozygosity for the PNPLA3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 51:1209–1217
Valenti L, Alisi A, Galmozzi E, Bartuli A, Del Menico B, Alterio A et al (2010) I148M patatin-like phospholipase domaincontaining 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology 52:1274–1280
Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C (2006) Prevalence of fatty liver in children and adolescents. Pediatrics 118:1388–1393
Alisi A, Cianfarani S, Manco M, Agostoni C, Nobili V (2011) 2011 Non-alcoholic fatty liver disease and metabolic syndrome in adolescents: pathogenetic role of genetic background and intrauterine environment. Ann Med 44:29–40
Chan AWH, Wong GLH, Chan HY, Tong JHM, Yu YH, Choi PCL et al (2017) Concurrent fatty liver increases risk of hepatocellular carcinoma among patients with chronic hepatitis B. J Gastroenterol Hepatol (Aust) 32:667–676
Kersten S, Desvergne B, Wahli W (2000) Roles of PPARs in health and disease. Nature 405:421–424
Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS et al (1999) PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 4:611–617
Hui Y, Yu-Yuan L, Yu-Qiang N, Wei-Hong S, Yan-Lei D, Xiao-Bo L et al (2008) Effect of peroxisome proliferator-activated receptors-γ and co-activator-1α genetic polymorphisms on plasma adiponectin levels and susceptibility of non-alcoholic fatty liver disease in Chinese people. Liver Int 28:385–392
Yang Z, Wen J, Li Q, Tao X, Ye Z, He M et al (2012) PPARG gene Pro12Ala variant contributes to the development of non-alcoholic fatty liver in middle-aged and older Chinese population. Mol Cell Endocrinol 48:255–259
Gawrieh S, Marion MC, Komorowski R, Wallace J, Charlton M, Kissebah A et al (2011) Genetic variation in the peroxisome proliferator activated receptor-gamma gene is associated with histologically advanced NAFLD. Dig Dis Sci 57:952–957
Gupta AC, Chaudhory AK, Sukriti PC, Sakhuja P, Singh Y et al (2011) Peroxisome proliferators-activated receptor γ2 Pro12Ala variant is associated with body mass index in non-alcoholic fatty liver disease patients. Hepatol Int 5:575–580
Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A et al (2014) Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 46:352–356
Carim-Todd L, Escarceller M, Estivill X, Sumoy L (2000) Cloning of the novel gene TM6SF1 reveals conservation of clusters of paralogous genes between human chromosomes 15q24→q26 and 19p13.3→p12. Cytogenet Genome Res 90:255–260
Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A et al (2014) TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci USA 111:8913–8918
Holmen OL, Zhang H, Fan Y, Hovelson DH, Schmidt EM, Zhou W et al (2014) Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet 46:345–351
Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JBS et al (2014) TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun 5:1–6
Wong VW-S, Wong GL-H, Tse C-H, Chan HL-Y (2014) Prevalence of TM6SF2 variant and nonalcoholic fatty liver disease in Chinese. J Hepatol 61:708–709
Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E et al (2015) Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 61:506–514
Sookoian S, Castaño GO, Scian R, Mallardi P, Fernández Gianotti T, Burgueño AL et al (2015) Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 61:515–525
Lohan F, Keeshan K (2013) The functionally diverse roles of tribbles. Biochem Soc Trans 41:1096–1100
Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ et al (2008) Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet 40:189–197
Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R et al (2008) Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 40:161–169
Tai ES, Sim XL, Ong TH, Wong TY, Saw SM, Aung T et al (2009) Polymorphisms at newly identified lipid-associated loci are associated with blood lipids and cardiovascular disease in an Asian Malay population. J Lipid Res J 50:514–520
Liu Q, Xue F, Meng J, Liu S-S, Chen L-Z, Gao H et al (2019) TRIB1 rs17321515 and rs2954029 gene polymorphisms increase the risk of non-alcoholic fatty liver disease in Chinese Han population. Lipids Health Dis 18:61
Wang L, Jing J, Fu Q et al (2015) Association study of genetic variants at newly identified lipid gene TRIB1 with coronary heart disease in Chinese Han population. Lipids Health Dis 14:46
Liu Q, Liu SS, Zhao ZZ, Zhao BT, Du SX, Jin WW et al (2019) TRIB1 rs17321515 gene polymorphism increases the risk of coronary heart disease in general population and non-alcoholic fatty liver disease patients in Chinese Han population. Lipids Health Dis 18:165
Buch S, Stickel F, Trépo E, Way M, Herrmann A, Nischalke HD et al (2015) A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet 47:1443–1448
Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R et al (2016) The MBOAT7-TMC4 Variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 150:1219–1230
Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, Miele L et al (2017) MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci Rep 7:2–11
Luukkonen PK, Zhou Y, Hyötyläinen T, Leivonen M, Arola J, Orho-Melander M et al (2016) The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J Hepatol 65:263–1265
Iynedjian PB (2009) Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 66:27–42
Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, NASH CRN; GIANT Consortium; MAGIC Investigators, Voight BF, Carr JJ, Feitosa MF, Harris TB, Fox CS, Smith AV, Kao WH, Hirschhorn JN, Borecki IB, GOLD Consortium et al (2011) Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 7:e1001324
Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H et al (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336
Orho-Melander M, Melander O, Guiducci C, Perez-Martinez P, Corella D, Roos C et al (2008) Common missense variant in the glucokinase regulatory protein gene is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes 57:3112–3121
Tan H-L, Zain SM, Mohamed R, Rampal S, Chin K-F, Basu RC et al (2013) 2013 Association of glucokinase regulatory gene polymorphisms with risk and severity of non-alcoholic fatty liver disease: an interaction study with adiponutrin gene. J Gastroenterol Hepatol 49:1056–1064
Nobakht H, Mahmoudi T, Sabzikarian M, Tabaeian SP, Rezamand G, Asadi A et al (2020) Insulin and insulin receptor gene polymorphisms and susceptibility to nonalcoholic fatty liver disease. Arq Gastroenterol 57:203–208
Ohmi S, Ono M, Takata H, Hirano S, Funakoshi S, Nishi Y et al (2017) Analysis of factors influencing glucose tolerance in Japanese patients with non-alcoholic fatty liver disease. Diabetol Metab Syndr 9:65
Li M, Zhang S, Wu Y, Ye J, Cao X, Liu J et al (2015) Prevalence of insulin resistance in subjects with nonalcoholic fatty liver disease and its predictors in a Chinese population. Dig Dis Sci 60:2170–2176
Gholam P, Flancbaum L, Machan JT, Charney DA, Kotler DP (2007) Nonalcoholic fatty liver disease in severely obese subjects. Am J Gastroenterol 102:399–408
Wang L, Mi J, Wu JX, Zhao XY, Cheng H, Ding XY et al (2004) Study on the relationship between polymorphism of insulin-receptor gene EXON2-2257 and insulin resistance in Chinese people. Zhonghua Liu Xing Bing Xue Za Zhi 25:49–53
Seino S, Seino M, Bell GI (1990) Human insulin receptor gene: partial sequence and amplification of exons by polymerase chain reaction. Diabetes 39:123–128
Moller DE, Yokota A, White MF, Pazianos AG, Flier JS (1990) A naturally occurring mutation of insulin receptor alanine 1134 impairs tyrosine kinase function and is associated with dominantly inherited insulin resistance. J Biol Chem 265:14979–14985
Grarup N, Urhammer SA, Ek J, Albrechtsen A, Glümer C, Borch-Johnsen K et al (2006) Studies of the relationship between the ENPP1 K121Q polymorphism and type 2 diabetes, insulin resistance and obesity in 7,333 Danish white subjects. Diabetologia 49:2097–2104
Dongiovanni P, Valenti L, Rametta R, Daly AK, Nobili V, Mozzi E et al (2010) Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 59:267–273
Deugnier YM, Loréal O, Turlin B, Guyader D, Jouanolle H, Moirand R et al (1992) Liver pathology in genetic hemochromatosis: a review of 135 homozygous cases and their bioclinical correlations. Gastroenterology 102:2050–2059
Niederau C, Fischer R, Pürschel A, Stremmel W, Häussinger D, Strohmeyer G (1996) Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 110:1107–1119
Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A et al (1996) A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 13:399–4085
Barton JC, Shih WW, Sawada-Hirai R, Acton RT, Harmon L, Rivers C et al (1997) Genetic and clinical description of hemochromatosis probands and heterozygotes: evidence that multiple genes linked to the major histocompatibility complex are responsible for hemochromatosis. Blood Cells Mol Dis 23:135–145
Dhillon BK, Das R, Garewal G, Chawla Y, Dhiman RK, Das A et al (2007) Frequency of primary iron overload and HFE gene mutations (C282Y, H63D and S65C) in chronic liver disease patients in north India. World J Gastroenterol 13:2956–2959
Saremi L, Lotfipanah S, Mohammadi M, Hosseinzadeh H, Sayad A, Saltanatpour Z (2016) Association of HFE gene mutations with nonalcoholic fatty liver disease in the Iranian population. Cell Mol Biol (Noisy-le-grand) 62:123–128
Nelson JE, Brunt EM, Kowdley KV, Nonalcoholic Steatohepatitis Clinical Research Network (2012) Lower serum hepcidin and greater parenchymal iron in nonalcoholic fatty liver disease patients with C282Y HFE mutations. Hepatology 56:1730–1740
Raszeja-Wyszomirska J, Kurzawski G, Lawniczak M, Miezynska-Kurtycz J, Lubinski J (2010) Nonalcoholic fatty liver disease and HFE gene mutations: a Polish study. World J Gastroenterol 16:2531–2536
Willger SD, Puttikamonkul S, Kim K-H, Burritt JB, Grahl N, Metzler LJ et al (2008) A sterol-regulatory element binding protein is required for cell polarity, hypoxia adaptation, azole drug resistance, and virulence in Aspergillus fumigatus. PLoS Pathog 4:e1000200
Musso G, Bo S, Cassader M, Franco De Michieli FD, Gambino R (2013) Impact of sterol regulatory element-binding factor-1c polymorphism on incidence of nonalcoholic fatty liver disease and on the severity of liver disease and of glucose and lipid dysmetabolism. Am J Clin Nutr 98:895–906
Peng XE, Chen FL, Liu W, Hu Z, Lin X (2016) Lack of association between SREBF-1c gene polymorphisms and risk of non-alcoholic fatty liver disease in a Chinese Han population. Sci Rep 6:32110
Hua X, Wu J, Goldstein JL, Brown MS, Hobbs HH (1995) Structure of the human gene encoding sterol regulatory element binding protein-1 (SREBF1) and localization of SREBF1 and SREBF2 to chromosomes 17p11.2 and 22q13. Genomics 25:667–673
Bauer S, Wanninger J, Schmidhofer S, Weigert J, Neumeier M, Dorn C et al (2011) Sterol regulatory element-binding protein 2 (SREBP2) activation after excess triglyceride storage induces chemerin in hypertrophic adipocytes. Endocrinology 152:26–35
Caballero F, Fernández A, Lacy AMD, Fernández-Checa JC, Caballería J, García-Ruiz C (2009) Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol 50:789–796
Wang Y, Tong J, Chang B, Wang B-F, Zhang D, Wang B-Y (2014) Relationship of SREBP-2 rs2228314 G>C polymorphism with nonalcoholic fatty liver disease in a Han Chinese population. Genet Test Mol Biomark 18:653–657
Salomao M, Remotti H, Vaughan R, Siegel AB, Lefkowitch JH, Moreira RK (2012) The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum Pathol 43:737–746
Ando S, Shibahara J, Hayashi A, Fukayama M (2015) β-catenin alteration is rare in hepatocellular carcinoma with steatohepatitic features: immunohistochemical and mutational study. Virchows Arch 467:535–542
Nishida N, Yada N, Hagiwara S, Sakurai T, Kitano M, Kudo M (2016) Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J Gastroenterol Hepatol 31:1646–1653
Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA et al (2004) miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and MayDownregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol 1:106–113
Sekine S, Ogawa R, Ito R, Hiraoka N, McManus MT, Kanai Y et al (2009) Disruption of Dicer1 induces dysregulated fetal gene expression and promotes hepatocarcinogenesis. Gastroenterology 136:2304–2315
Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M et al (2006) miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 3:87–98
Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW et al (2008) Nonalcoholic steatohepatitis is associated with altered hepatic microRNA expression. Hepatology 48:1810–1820
Burchard J, Zhang C, Liu AM, Poon RTP, Lee NPY, Wong K-F et al (2010) microRNA-122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol Syst Biol 6:402
Kutay H, Bai S, Datta J, Motiwala T, Pogribny I, Frankel W et al (2006) Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem 99:671–678
Tsai WC, Da HS, Hsu CS, Lai TC, Chen SJ, Shen R et al (2012) MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Investig 122:2884–2897
Cermelli S, Ruggieri A, Marrero JA, Ioannou GN, Beretta L (2011) Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 6:e23937
Pirola CJ, Gianotti TF, Castaño GO, Mallardi P, Martino JS, Ledesma MMGL et al (2015) Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut 64:800–812
Schütte K, Schulz C, Link A, Malfertheiner P (2015) Current biomarkers for hepatocellular carcinoma: surveillance, diagnosis and prediction of prognosis. World J Hepatol 7:139–149
Zhang J, Fan J, Zhou C, Qi Y (2017) miR-363-5p as potential prognostic marker for hepatocellular carcinoma indicated by weighted co-expression network analysis of miRNAs and mRNA. BMC Gastroenterol 17:1–9
Seshachalam VP, Sekar K, Hui KM (2018) Insights into the etiology-associated gene regulatory networks in hepatocellular carcinoma from The Cancer Genome Atlas. J Gastroenterol Hepatol 33:2037–2047
Danford CJ, Yao ZM, Jiang ZG (2018) Non-alcoholic fatty liver disease: a narrative review of genetics. J Biomed Res 32:389–400
Wang J, Conti DV, Bogumil D, Sheng X, Noureddin M, Wilkens LR et al (2021) Association of genetic risk score with NAFLD in an ethnically diverse cohort. Hepatol Commun 5:1689–1703
Han MAT, Yu Q, Tafesh Z, Pyrsopoulos N (2021) Diversity in NAFLD: a review of manifestations of nonalcoholic fatty liver disease in different ethnicities globally. J Clin Transl Hepatol 9:71–80
Sookoian S, Pirola CJ (2017) Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol Hepatol 23:1–12
Wei JL, Leung JC, Loong TC, Wong GL, Yeung DK, Chan RS et al (2015) Prevalence and severity of nonalcoholic fatty liver disease in non-obese patients: a population study using proton-magnetic resonance spectroscopy. Am J Gastroenterol 110:1306–1314
Kallwitz ER, Tayo BO, Kuniholm MH, Cai J, Daviglus M, Cooper RS et al (2019) American ancestry is a risk factor for suspected nonalcoholic fatty liver disease in Hispanic/Latino adults. Clin Gastroenterol Hepatol 17:2301–2309
Zhou J, Zhou F, Wang W, Zhang XJ, Ji YX, Zhang P et al (2020) Epidemiological features of NAFLD from 1999 to 2018 in China. Hepatology 71:1851–1864
Umano GR, Caprio S, Di Sessa A, Chalasani N, Dykas DJ, Pierpont B, Bale AE, Santoro N (2018) The rs626283 variant in the MBOAT7 gene is associated with insulin resistance and fatty liver in Caucasian obese youth. Am J Gastroenterol 113:376–383
Mansoor S, Maheshwari A, Di Guglielmo M, Furuya K, Wang M, Crowgey E et al (2021) The PNPLA3 rs738409 variant but not MBOAT7 rs641738 is a risk factor for nonalcoholic fatty liver disease in Obese U.S. children of Hispanic ethnicity. Pediatr Gastroenterol Hepatol Nutr 24:455–469
Parohan M, Yaghoubi S, Seraji A (2020) Liver injury is associated with severe coronavirus disease 2019 (COVID-19) infection: a systematic review and meta-analysis of retrospective studies. Hepatol Res 50:924–935
Zhang C, Shi L, Wang FS (2020) Liver injury in COVID-19: management and challenges. Lancet Gastroenterol Hepatol 5:428–430
Satapathy SK, Kuntzen C, Qiu H, Jiang Y, Bodenheimer HC, Roth NC, Bernstein DE, Northwell Health COVID-19 Research Consortium et al (2021) Severity of liver test abnormalities in coronavirus disease 2019 depends on comorbidities and predicts early in-hospital mortality. Eur J Gastroenterol Hepatol 33:e320–e328
Vrsaljko N, Samadan L, Viskovic K, Mehmedović A, Budimir J, Vince A et al (2022) Association of nonalcoholic fatty liver disease with COVID-19 severity and pulmonary thrombosis: CovidFAT, a prospective, observational cohort study. Open Forum Infect Dis 9(4):ofac073
Yao R, Zhu L, Wang J et al (2021) Risk of severe illness of COVID-19 patients with NAFLD and increased NAFLD fibrosis scores. J Clin Lab Anal 35:e23880
Yoo HW, Jin HY, Yon DK, Effenberger M, Shin YH, Kim SY et al (2021) Non-alcoholic fatty liver disease and COVID-19 susceptibility and outcomes: a Korean Nationwide Cohort. J Korean Med Sci 36(41):e291
Wang Y, Duan G, Yang H (2023) NAFLD was independently associated with severe COVID-19 among younger patients rather than older patients: a meta-analysis. J Hepatol 78(4):e136–e139
Marjot T, Moon AM, Cook JA, Abd-Elsalam S, Aloman C, Armstrong MJ et al (2021) Outcomes following SARS-CoV-2 infection in patients with chronic liver disease: an international registry study. J Hepatol 74(3):567–577
Sarin SK, Choudhury A, Lau GK, Zheng MH, Ji D, Abd-Elsalam S, APASL COVID Task Force et al (2020) APASL COVID liver injury spectrum study (APCOLIS Study-NCT 04345640) pre-existing liver disease is associated with poor outcome in patients with SARS CoV2 infection; the APCOLIS study (APASL COVID-19 liver injury spectrum study). Hepatol Int 14:690–700
Fondevila MF, Mercado-Gómez M, Rodríguez A, Gonzalez-Rellan MJ, Iruzubieta P, Valentí V et al (2021) Obese patients with NASH have increased hepatic expression of SARS-CoV-2 critical entry points. J Hepatol 74:469–471
Ji D, Qin E, Xu J, Zhang D, Cheng G, Wang Y et al (2020) Non-alcoholic fatty liver diseases in patients with COVID-19: a retrospective study. J Hepatol 73(2):451–453
Nath P, Kumar R, Mallick B et al (2022) Effect of nonalcoholic fatty liver disease (NAFLD) on COVID-19: a single-center study of 3983 patients with review of literature. Cureus 14(7):e26683. https://doi.org/10.7759/cureus.26683
Mushtaq K, Khan MU, Iqbal F, Alsoub DH, Chaudhry HS, Ata F et al (2021) NAFLD is a predictor of liver injury in COVID-19 hospitalized patients but not of mortality, disease severity on the presentation or progression—the debate continues. J Hepatol 74:482–484
Zhou YJ, Zheng KI, Wang XB, Yan HD, Sun QF, Pan KH et al (2020) Younger patients with MAFLD are at increased risk of severe COVID-19 illness: a multicenter preliminary analysis. J Hepatol 73:719–721
Lin S, Huang J, Wang M, Kumar R, Liu Y, Liu S et al (2020) Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int 40:2082–2089
Niriella MA, Ediriweera DS, Kasturiratne A, De Silva ST, Dassanayaka AS, De Silva AP et al (2021) Outcomes of NAFLD and MAFLD: results from a community-based, prospective cohort study. PLoS ONE 16:e0245762
Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, RomeroGomez M et al (2020) A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol 73(1):202–209
Xia M, Zeng H, Wang S, Tang H, Gao X (2021) Insights into contribution of genetic variants towards the susceptibility of MAFLD revealed by the NMR-based lipoprotein profiling. J Hepatol 74:974–977
Andreana L, Isgrò G, Pleguezuelo M, Germani G, Burroughs AK (2009) Surveillance and diagnosis of hepatocellular carcinoma in patients with cirrhosis. World J Hepatol 1(1):48–56
Geh D, Rana FA, Reeves HL (2019) Weighing the benefits of hepatocellular carcinoma surveillance against potential harms. J Hepatocell Carcinoma 6:23–30
Acknowledgements
Not applicable.
Authors' information
Rohan Chaminda Siriwardana: Professor in Hepatobiliary and liver transplant at the Department of Surgery, Faculty of Medicine, University of Kelaniya, Sri Lanka. His research interests are Hepatocellular carcinoma, liver transplantation and cryptogenic cirrhosis.
Madunil Anuk Niriella: Professor in Gastroenterology at the Department of Medicine, Faculty of Medicine, University of Kelaniya, Sri Lanka. His major research specialities are on Epidemiology and genetics of NAFLD, chronic liver disease epidemiology and Cirrhosis and complications. He was a fellow of American Gastroenterology Association and American Society of Gastrointestinal Endoscopy American Association for the Study of Liver Disease.
Some of the research publications of RCS and MAN related to NAFLD:
Liver Transplantation for Non-Alcoholic Steatohepatitis Related Cirrhosis. Siriwardana RC, Niriella MA, Dassnayake AS, Gunathillake B, de Silva HJ.Prog Transplant. 2017 Mar;27(1):107–108. https://doi.org/10.1177/1526924816677996.
Non-alcoholic fatty liver disease among potential live liver donors–a preliminary experience from Sri Lanka. Silva H, Siriwardana RC, Niriella MA, Dassanayake AS, Liayange CA, Gunathilake B, De Silva HJ.Indian J Gastroenterol. 2014 Nov;33(6):573–4. https://doi.org/10.1007/s12664-014-0478-7. Epub 2014 Jun 3.PMID: 24882252.
Cryptogenic cirrhosis is the leading cause for listing for liver transplantation in Sri Lanka. Siriwardana RC, Niriella MA, Liyanage CA, Wijesuriya SR, Gunathilaka B, Dassanayake AS, De Silva HJ.Indian J Gastroenterol. 2013 Nov;32(6):397–9. https://doi.org/10.1007/s12664-013-0376-4. Epub 2013 Sep 3.PMID: 23999684.
Genetic variants of NAFLD in an urban Sri Lankan community. Niriella, M.A.; Kasturiratne, A.; Akiyama, K.; Takeuchi, F.; Isono, M.; Dassanayake, A.S.; de Silva, A.P.; Wickremasinghe, A.R.; Kato, N.; de Silva, H.J. Journal of Gastroenterology and Hepatology. 2013; 28(Suppl 3):184.
Association of Serum Ferritin with Diabetes and Alcohol in Patients with Non-Viral Liver Disease-Related Hepatocellular Carcinoma. Siriwardana RC, Niriella MA, Dassanayake A, Ediriweera D, Gunetilleke B, Sivasundaram T, de Silva J.Liver Cancer. 2017 Nov;6(4):307–312. https://doi.org/10.1159/000477266. Epub 2017 Aug 29.PMID: 29234634.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SMS wrote the manuscript. ASH, RCS, KHT, MAN and SDS reviewed the manuscript and provided critical comments. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Summery of the NAFLD-associated genetic variants.
Additional file 2:
Summery of the included studies representing the association between miR-122 expression and NAFLD.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Samarasinghe, S.M., Hewage, A.S., Siriwardana, R.C. et al. Genetic and metabolic aspects of non-alcoholic fatty liver disease (NAFLD) pathogenicity. Egypt J Med Hum Genet 24, 53 (2023). https://doi.org/10.1186/s43042-023-00433-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-023-00433-x