
Abstract
Background
Congenital muscular dystrophies (CMD) and congenital myopathies (CM) are clinically and genetically heterogeneous groups of neuromuscular disorders resulting in prenatal or early-onset hypotonia, muscle weakness, myogenic pattern, and dystrophic or myopathic features on muscle biopsy. In this study, we provide a genetic and molecular characterization of CMD and CM in Moroccan patients.
Patients and methods
In this cohort, we investigated 23 Moroccan patients from 21 families who consented to genetic testing. Firstly, genetic analysis in the probands was conducted by next-generation sequencing (NGS) technology using two approaches: targeted NGS gene panel and clinical exome sequencing to study the mutational spectrum and to achieve an accurate diagnosis of these hereditary myopathies in Morocco.
Results
NGS data analysis revealed 16 pathogenic variants harbored in 17 unrelated patients that were genetically resolved. The phenotypic forms identified were in order: LAMA2-related CMD (52.94%), LMNA-CMD (23.53%), and RYR1-related congenital myopathy (17.65%). The congenital titinopathy group was less frequent (5.88%). Here, we identified two novel recessive variants in LAMA2 gene: c.2164G > A (p.Glu722Lys), and c.(6992 + 1_6993-1)_(7300 + 1_7301-1)del p.(Pro2332Glnfs*10). Additionally, we expanded the phenotypic spectrum of a known heterozygous LMNA c.1718C > T p.(Ser573Leu) variant, and we report it for the first time to a form of CMD.
Conclusions
The introduction of the NGS tool in clinical practice allowed us to improve the diagnosis and the management of these neuromuscular diseases and to highlight the importance of molecular genetic diagnosis of these disorders that are underestimated in the Moroccan population.
Similar content being viewed by others


Introduction
Congenital muscular dystrophies (CMD) and congenital myopathies (CM) are a group of genetically and clinically heterogeneous hereditary neuromuscular disorders with autosomal dominant and/or recessive inheritance. The frequency of various phenotypic forms of CMD and CM varies between studies, depending on the cohort size and the patients’ ethnic origins [1,2,3]. They are characterized mostly by hypotonia during neonatal or early infancy, stable or progressive muscle weakness of different limbs, contractures, motor developmental delay, and a myogenic pattern on electromyogram (EMG) [4, 5]. In the case of CMD, other organ systems, such as the brain and eyes, may be affected, as seen in Walker-Warburg syndromes (WWS) or muscle–eye–brain diseases [4]. CMD’s histopathological pattern shows a degenerative primary muscle with dystrophic features, associated with moderate to high creatine kinase (CK) serum levels. On the other hand, CM’s cases are characterized by architectural abnormalities in the muscle fibers with specific structural changes, associated with normal to moderate CK [6,7,8]. Nonetheless, both forms most often have a myogenic pattern, and some CM forms have a normal EMG [9, 10].
The classification of these forms has traditionally been based on a combination of three parameters: clinical presentation, muscle histopathology and immunohistochemistry, and genetic data [11, 12]. In the last few years, next-generation sequencing has accelerated the identification of new genes involved in these disorders, leading to an expansion of their spectrum. NGS has revealed that the forms of these diseases can have a substantial overlap between the causative genes and clinical and histopathologic features of congenital myopathies [13].
Physicians confront a great challenge to have an accurate diagnosis of these complex muscle diseases. This is not only due to their high genotype and phenotype variability but also to the overlap of clinical signs between congenital muscular dystrophy and congenital myopathy [14, 15]. The diagnosis is made even more difficult by the fact that congenital hypotonia is a typical early clinical symptom in CMD and CM, but it is not a specific sign to either [16, 17]. Although histopathology and immunohistochemistry are central in both the diagnosis and the accurate identification and classification of these diseases, they are not done routinely in Morocco, as it is often unavailable to patients. The diagnosis is thus based on a clinical evaluation, coupled with various paraclinical tests (CK level, brain MRI, ocular, and cardiac investigation). Because of this reality, and given its analytic accuracy, high throughput, and potential cost-effectiveness, the NGS approach has quickly become an indispensable tool and a gold standard for the early diagnosis of these neuromuscular disorders in Morocco [18,19,20].
This present study aims to highlight the major contributions of Next-Generation Sequencing technology, as a first-line strategy, in achieving an accurate and rapid diagnosis of these rare neuromuscular disorders, and in establishing, in the future, a better molecular epidemiological characterization of CMD and CM forms in the Moroccan population.
Patients and methods
Study subjects
This cohort included a total of 23 Moroccan patients from 21 families referred between 2017 and 2022 to our Department of Medical Genetics for molecular genetic analysis with a suspected diagnosis of early-onset myopathy, including congenital muscular dystrophy and congenital myopathy. All patients were clinically assessed by pediatric neurologists. Inclusion criteria consisted in patients with early-onset hypotonia, muscle weakness, and delayed motor development, but non-inclusion criteria were the differential diagnosis of congenital muscle diseases such as congenital myasthenic syndromes, metabolic disorders and severe infectious diseases. Patients with neurogenic patterns seen on EMG and patients diagnosed with spinal muscular atrophy (SMA) were excluded. Moreover, the availability of histopathological features in muscle biopsy was available in three cases, and radiological findings data (cMRI/CT scan) were available in 13 cases. Written informed consent was obtained from all of the minor patients’ parents or adult patients before DNA collection by referring physicians in order to implement the genetic analysis.
NGS technology was used as a powerful genetic tool for the investigation of these patients.
Next-generation sequencing workflow
Genomic DNA extraction
Genomic DNA (gDNA) was extracted from 200 µl of peripheral blood through manual extraction with a commercial Kit based on Silica Spin Column isolation technology (PureLink™ Genomic DNA Mini Kit, Invitrogen™), and by semi-automated sample preparation using KingFisher Duo Prime with an automatable magnetic bead-based sample preparation technology (MagMAX DNA Multi-Sample Ultra 2.0 Kit, Applied Biosystems™). These two extraction methods achieve high molecular gDNA, following standard protocols according to the manufacturer’s instructions. gDNA purity and quantity were determined for each sample using NanoDrop™ 2000 Spectrophotometer (Thermo Scientific™) followed by Qubit 3.0 Fluorometer with the Qubit dsDNA HS Assay Kit (Invitrogen; Thermo Fisher Scientific) to accurately measure DNA quantity.
Targeted gene panel approach using ion torrent platform
Seventeen unrelated patients were investigated by a multigene panel; six of them (P2 to P7) were analyzed by manual NGS workflow according to the manufacturer’s protocols and sequenced on the Ion PGM platform using Ion AmpliSeq On-Demand panel of 24 targeted genes, in which 17 genes (TCAP; LARGE1; ITGA7; LAMA2; COL6A1, COL6A2, COL6A3; DNM2; PMM2; DPM2; FKRP; FKTN; LMNA; POMT1; POMT2; POMGNT1, SEPN1) are involved in CMD, CM, and/or congenital disorder of glycosylation (CDG). Patient (P3) was already (diagnosed) in our lab and it was published [21].
The remaining 11 patients (P11 to P13, P15 to P22) were analyzed by automated NGS workflow using Ion Chef™ system and sequenced on the Ion GeneStudio™ S5 system. These 11 patients were diagnosed through sequencing of 26 targeted genes of myopathies, in which 18 genes (TCAP; RYR1; LARGE1; ITGA7; LAMA2; COL6A1, COL6A2, COL6A3; DNM2; PMM2; DPM2; FKRP; FKTN; LMNA; POMT1; POMT2; POMGNT1, SEPN1) are involved in CMD, CM, and/or CDG.
Eight targeted libraries per run were prepared on the Ion Chef™ system from 10 ng (0.67 ng/μl) of gDNA samples using Ion AmpliSeq™ Kit for Chef DL8 (Thermo Fisher Scientific), which was performed in accordance with the manufacturer’s instructions. Template preparation, chip loading, and sequencing of 275 base–read next-generation sequencing libraries were performed with the Ion 510™ & Ion 520™ & Ion 530™ Kit (Thermo Fisher Scientific) in accordance with the manufacturer’s protocol. Templated ISPs are enriched and loaded onto an Ion 510™ Chips v2 BC and sequenced on Ion GeneStudio™ S5 System with a read length set at 200 bp protocol and a number of flows at 550.
Raw data were processed for base calling, trimming, demultiplexing, alignment to the human reference genome assembly (Feb. 2009, GRCh37/hg19), and variant calling on the Ion Torrent Server (Thermo Fisher Scientific) using the Torrent Suite software version 5.12.3. The identified variations were annotated with Ion Reporter software 5.18.2.0 (Thermo Fisher Scientific).
Clinical exome sequencing approach using Illumina platform
Four unrelated patients (P1, P9, P10, and P23) from our series were able to benefit from clinical exome sequencing analysis. The clinical and molecular data of the patient (P9) was previously published by our team [22]. Clinical Exome Solution v2 kit (SOPHiA Genetics, Boston, USA) covers the coding regions of 4490 genes (target region of 12 Mb) related to rare and known inherited diseases. It was used for the enrichment of the conserved coding regions in these four probands’ DNA. Paired-end exome sequencing was performed on an Illumina NextSeq® 500 sequencer (Illumina Inc., San Diego, CA, USA) with a read length of 150 × 2 according to the manufacturer’s protocols.
Following sequencing, a custom cloud-based informatics pipeline was used to conduct alignment, variant identification, and annotation of sequencing data from FASTQ files using Sophia DDM® platform for genetic analysis of variants.
Variant filtering and pathogenicity interpretation of candidate variants
Common genetic variants or single nucleotide polymorphisms with a minor allele frequency (MAF) ≥ 1% were excluded by referring to public genetic databases, including dbSNP database (https://www.ncbi.nlm.nih.gov/snp/), 1000 Genomes Project (1000G; http://www.internationalgenome.org/), the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/). Moreover, MAF ≥ 2% in local resources (in-house database) was filtered out. The clinical significance of annotated variations was assessed with ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), Leiden Open Variation Database (LOVD; https://www.lovd.nl/), Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/omim), and PubMed (https://pubmed.ncbi.nlm.nih.gov/). The pathogenicity of the candidate variants was then assessed through a set of criteria, according to the American College of Medical Genetics and Genomics (ACMG) guidelines. The prediction of the pathogenicity of the identified variants was performed on MutationTaster (http://www.mutationtaster.org/), Sorting Intolerant From Tolerant (SIFT; https://sift.bii.a-star.edu.sg/), Polymorphism Phenotyping (PolyPhen v2; http://genetics.bwh.harvard.edu/pph2/), and Protein Variation Effect Analyzer (PROVEAN; http://provean.jcvi.org). The variants predicted to be benign or tolerated were filtered out.
All variants and regions included in the genes of interest with < 20X coverage depth were manually verified in the Integrative Genomics Viewer (IGV) version 2.5.0 (https://software.broadinstitute.org/software/igv/).
Confirmation of the identified variants
Sanger sequencing
Single nucleotide variants (SNV) and small insertion–deletions (Indel) identified in this study were confirmed by DNA Sanger sequencing in the probands using specific primer pairs (Additional file 1: Table S1). The segregation analysis was assessed on some families when the samples of the parents and/or siblings were available.
The Sanger sequencing was performed following the standard protocol of ABI Prism BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems®). They were sequenced on an ABI 3500 automated Genetic Analyzer (Applied Biosystems). The sequences were analyzed by the Sequencing Analysis software version 7.
Multiplex PCR and quantitative real-time PCR
Firstly, the large exon deletions in LAMA2 gene were confirmed by multiplex PCR (MPCR) performed on the genomic DNA of the patient (P11). Two pairs of primers were used to amplify exon 50 and exon 58 (internal control) of LAMA2 gene. The final volume of MPCR reaction was 20 µl using Platinum™ Multiplex PCR Master Mix (Applied Biosystems™). Migration of fragments amplified was performed on electrophoresis gel using 3% agarose and 1% nusieve and stained with SYBR™ Safe DNA Gel Stain (Invitrogen™).
Afterward, this copy number variant was subsequently confirmed by quantitative real-time PCR (qPCR) in the proband and her parents using SYBR® Green dyes and loaded on a QuantStudio™ 7 Flex Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific). To quantify the region of interest, a pair of primers was designed in such a way that the forward primer (5ʹ-CGCTGGTACCCCAACATCTC-3ʹ) was hybridized within the exonic sequence (ex50) and the reverse primer (5ʹ-TCTTGGCTGCCAGACAATCAT-3ʹ) was incorporated within the intron 50–51 sequence with an amplicon length of 230 base pairs. As a reference gene, we used the KIF1A with the following primers: (ex12_F: 5ʹ-GGAGCAGACATAGCCCTGG-3ʹ and ex12_R: 5ʹ-CCTAATTCAAGCACGAGAGG-3ʹ).
Patient (P11), his parents, and unrelated normal samples were analyzed together with the calibrator sample. Each sample was run in triplicates. All data were calculated by the comparative Ct method to detect the relative copy number of exon 50 in LAMA2 gene using a sample of normal control DNA as a calibrator. The relative copy numbers were determined by the expression: 2−∆∆Ct, with ∆∆Ct as the difference in ΔCt between the target and reference samples. The first ΔCt is the difference in Ct between the target and reference genes.
The 2−∆∆Ct ratio was expected to be about 1 in normal controls, about 0.5 in carriers, and 0 in patients with homozygous deletion.
Conservation analysis of amino acids sequence
Protein multiple sequence alignments for variants harbored in P7, and P12 were performed by the Clustal Omega tool (https://www.ebi.ac.uk/Tools/msa/clustalo/). The input file sequences in format FASTA were obtained from the UniProt database (https://www.uniprot.org/).
Results
Clinical features of patients
Clinical and paraclinical details of patients are summarized in Table 1.
Molecular diagnostic
In this cohort, we report molecular data of 17 unrelated patients from 21 families. The proportion of resolved patients with DNA variations in the analyzed genes using two diagnostic approaches of NGS is (17/21; 80.95%), while the remaining 4 patients represent (4/21; 19.05%) of undiagnosed cases. Among 17 patients who were investigated by the targeted NGS gene panel, a confirmed molecular genetic diagnosis was established in 13 patients (P2 to P7, P11 to P13, and P18 to P21) representing (13/17; 76.47%), while genetic testing of known and available genes included in our panel was negative for 4 remaining patients (P15 to P17, and P22) representing (4/17; 23.53%). The coverage sequencing of the coding regions of interest represents 98% in these unresolved four patients, with a read deep > 10X. All other four patients analyzed by CES approach (P1, P9, P10, and P23) were genetically resolved by identified variants in LAMA2, TTN, and LMNA genes (Fig. 1).

Flowchart diagram to illustrate the analysis method used in this cohort
A total of 16 variants (two are novel) were identified in the 17 unrelated positive probands. The most frequently mutated gene analyzed by both NGS approaches was LAMA2 in nine unrelated patients (P1 to P7, P11, and P23), followed by LMNA in four patients (P10, P12, P18, and P21), RYR1 in three unrelated patients (P13, P19, and P20), and then TTN in one patient (P9).
There are four diagnostic groups of patients that bear mutations in four different genes: (i) Laminin subunit alpha 2-related congenital muscular dystrophy (LAMA2-related CMD) is the most prevalently mutated form observed in nine probands (9/17; 52.94%). All had a phenotype ranged from severe to moderate form of CMD. The identified variants were three splice sites, two missenses, two frameshifts, and one-nonsense variations. (ii) Lamin-related congenital muscular dystrophy (LMNA-related CMD) was detected in four probands (4/17; 23.53%). The variants were three missenses and one in-frame variant. (iii) Ryanodine receptor type 1-related congenital myopathy (RYR1-related CM) was observed in three probands (3/17; 17.65%) with three missense variants. (iv) Titin-related congenital myopathy (TTN-related CM) was observed in a female child (1/17; 5.88%) diagnosed with Early-Onset Myopathy with Fatal Cardiomyopathy and caused by a frameshift variant. Molecular findings of patients are summarized in Table 2.
Through this study, we identified two novel variants in LAMA2 gene. The first missense c.2164G > A p.(Glu722Lys) variant identified in P7 (IV:8) is located in coding exon 15 of the LAMA2 gene, resulting in a G to A substitution at nucleotide position 2164. This sequence change replaces glutamic acid with lysine at codon 722 of the Laminin-α2 protein. Glutamic acid (E) is highly conserved among different species. This variant has not been previously reported in databases and was not found in 138 Moroccan clinical exomes (in-house database). Segregation analysis revealed the presence of this variant in the proband’s sister P8 (IV:7) with a similar phenotype (Fig. 2). The sample DNA of the proband’s mother was not available. It was classified as likely pathogenic according to ACMG/AMP criteria.

Analysis of a novel LAMA2 c.2164G > A p.(Glu722Lys) variant. A Pedigree of the studied family. Affected individuals are denoted by filled symbols (IV:7; IV:8), while unaffected individuals are denoted by unfilled squares and circles. The black arrow indicates the proband P7. Asterisks indicate tested individuals. B IGV screenshot of a next-generation sequencing panel showing the homozygous DNA variation of exon 15 in LAMA2 gene identified in the proband P7 (IV:8). C, D Representative Sanger sequencing chromatograms with c.2164G > A variant position in the patient and his sister compared with a reference sequence. E Residue conservation analysis of the p.Glu722Lys variant in LAMA2 in orthologous proteins. The LAMA2 p.Glu722Lys variant is shown with a black box identifying the corresponding amino acid position. «sp» for UniProtKB/Swiss-Prot, «tr» for UniProtKB/TrEMBL, *(Asterisk) positions with a single, fully conserved residue,: (colon) positions with conservation between amino acid groups of similar properties
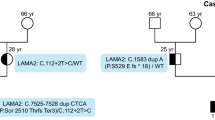
The second novel LAMA2 variant is a large homozygous deletion of exons 50–51, NM_000426.4(LAMA2):c.(6992 + 1_6993-1)_(7300 + 1_7301-1)del p.(Pro2332Glnfs*10), identified in P11 using IGV v2.5.0 (Fig. 3). It has not been reported in databases or found in personal data (in-house database). It results in a frameshift variant starting from amino acid position 2332 and introduces a premature termination signal at codon position 2341. It may lead to the synthesis of a truncated laminin-alpha 2 protein lacking 782 C-terminal amino acid residues. qPCR analysis of exon 50 validated the molecular genetic diagnosis in the proband (IV:1) with no copy of this exon and showed that his parents (III:2, III:7) are heterozygous with one copy (Fig. 4).

Representation of the next-generation sequencing data coverage depth detected using the Integrative Genomics Viewer (IGV). This snapshot shows the homozygous deletion of exons 50–51 (indicated with red arrows) in the LAMA2 gene identified in patient P11

Molecular analysis of a novel copy number variation in LAMA2 gene. A Pedigree of the studied family. The black arrow indicates the proband P11 (IV:1) and asterisks indicate tested individuals. B Representative agarose gel picture of multiplex PCR products migration using ChemiPRO Chemiluminescence Imaging System (Cleaver Scientific) with GenePIX software V1.6.3.8. Original agarose gel is presented in (Additional file 2: Fig. S1). Lanes 1 and 3 indicate the presence of two bands in the normal controls. Lane 2 indicates the absence of the band of targeted amplicon (exon 50) and the presence of the band of control amplicon (exon 58) in the patient (IV:1). Lane 4 indicates the absence of the bands in the negative control. C Real-time amplification plots of targeted LAMA2 gene. D Real-time amplification plots of endogenous KIF1A gene. E Bar graph showed the copy number results calculated from delta-delta Ct
The known de novo heterozygous LMNA missense c.1718C > T p.(Ser573Leu) variant harbored by patient P12 (IV:3) is located in a highly conserved amino acid position in orthologous proteins. Both parents (III:1, III:2) were normal for this variant. The LMNA gene is completely sequenced at 100% with a minimal read deep of 33x. The coverage sequencing of the coding regions of interest represents 97% in this patient with a read deep > 10X. This variant has never been reported in Moroccan exomes data (in-house database), and it is associated with our patient for the first time with congenital muscular dystrophy (Fig. 5).

Analysis of a known LMNA c.1718C > T p.(Ser573Leu) variant associated with another phenotype was never reported. A Pedigree of the studied family. The affected individual P12 is denoted by filled square (IV:3), while unaffected individuals are denoted by unfilled squares and circles. Asterisks indicate tested individuals. B IGV screenshot of a next-generation sequencing panel showing the heterozygous DNA variation of exon 11 in LMNA identified in the patient (P12). C–E Representative Sanger sequencing chromatograms with c.1718C > T variant position. The patient (IV:3) is heterozygous and their parents (III:1, III:2) are healthy for the variant. F Residue conservation analysis of the p.Ser573Leu variant in LMNA in different species. The LMNA p.Ser573Leu variant is shown with a black box identifying the corresponding amino acid position. «sp» for UniProtKB/Swiss-Prot, «tr» for UniProtKB/TrEMBL, * (Asterisk) positions with a single, fully conserved residue,: (colon) positions with conservation between amino acid groups of similar properties
The accession numbers from ClinVar database, SCV002515382, and SCV002524097 were assigned to our novel missense and CNV LAMA2 variants, respectively. Moreover, the SCV002547317 was assigned to the known LMNA variant involved in CMD phenotype.
Discussion
The diagnosis of inherited skeletal muscle disorders especially early-onset myopathies represents a great challenge not only for their high clinical and pathological heterogeneity but also because of the large number and great diversity of inheritance patterns seen [18, 42].
It should be noted that congenital hypotonia is a nonspecific clinical criterion that can occur in association with multiple other conditions. Generally, the proportion of newborns/infants with central hypotonia is much more than the peripheral hypotonia types. In several studies, the frequency of peripheral causes of neonatal hypotonia (neuromuscular disorders) was estimated to range between 9% and 46.9% of diagnosed patients with hypotonia [17, 43,44,45,46,47]. Moreover, patients with neonatal hypotonia related to CM or CMD/CM represent approximately 18.2% to 61.5% of peripheral hypotonia [17, 43, 45,46,47].
Usually, clinicians find major difficulties in targeting the gene responsible for the disease. Despite the lack of clinical information in several patients, NGS technology overcomes these difficulties through rapid screening of several genes to achieve a correct and definitive molecular genetic diagnosis for these heterogeneous diseases [48].
In the last update, the 2021 version of the GeneTable of neuromuscular disorders includes about 36 genes associated with all congenital muscular dystrophy phenotypes and ≈45 genes with all congenital myopathy forms [49].
In this cohort, four different conditions/classes of congenital muscle diseases have been described. The predominance of AR forms is probably due to the prevalence of consanguinity, which represents 15.25% in Morocco [50]. The identified variants were classified as pathogenic or likely pathogenic.
LAMA2-related congenital muscular dystrophy
In this study, the most prevalent pathology was laminin-α2 chain-related congenital muscular dystrophy, also known as Merosin-Deficient Congenital Muscular Dystrophy type 1A (MDC1A) phenotype. It represents 52.94% of positive patients. Similarly, MDC1A is the most severe and common form of CMD in many European countries with an average frequency of 30–40% among CMD patients [2, 51]. Moreover, a recent study revealed that MDC1A is the most common CMDs in Qatar, accounting for 48.5% [52]. The prevalence of MDC1A is estimated at 1–9/1000000 (ORPHA: 258).
In all our MDC1A patients, the initial clinical symptoms were identified at birth, as in previous studies. Generally, MDC1A is caused by variations in the LAMA2 gene, which has been associated with the complete absence of the laminin-α2 chain and characterized by early-onset symptoms [53]. In the literature, frequency of seizures was estimated to 30% of patients [54]. Two siblings of our LAMA2 mutated patients (P7 and P8) had developed epileptic seizures but were controlled by treatment. Specific abnormal cerebral white matter signals are regularly observed by 1 year of age on an MRI scan [55].
MDC1A results from different and numerous pathogenic variants that are scattered along the 65 exons of LAMA2 gene. More than 500 variants are reported in The Human Gene Mutation Database (accessed on 12 December 2022; HGMD Professional 2021.4). Interestingly, missense substitutions were associated with some reported patients with milder CMD and partial LAMA2 deficiency.
All patients of our series who carried LAMA2 variants had a severe phenotype of LAMA2. The frameshift c.1377delC variant harbored in P6 was previously reported in a Moroccan patient and published as 1426delC following the old nomenclature [25]. It was not found in ClinVar database but was listed in the Moroccan Genetic Disease Database (local database of Pasteur Institute of Morocco, Laboratory of Human Molecular Genetics).
The splicing c.8244 + 1G > A variant described in P4 and P23 was previously identified at homozygous or compound heterozygous state in the Tunisian and Algerian populations, and other countries [24, 27,28,29]. We assume that it could be a recurrent variant, especially in the Maghreb. Our hypothesis remains valid as long as the allele frequency of this variant is low and estimated at 1/120032 chromosomes (0.0008%) in Exome Aggregation Consortium (ExAC) and 2/250124 chromosomes (0.0008%) in the Genome Aggregation Database (gnomAD_exome), but higher at 7/264690 chromosomes (0.0026%) in Trans-Omics for Precision Medicine (TOPMed) (accessed on 12 December 2022).
LMNA-related congenital muscular dystrophy
The second frequent clinical phenotype in our series is LMNA-related CMD or L-CMD caused by variations in LMNA gene encoding two splicing products (lamin A/C). To date over 100 cases of L-CMD have been reported in the literature with an autosomal dominant inheritance with a prevalence estimated at < 1/1000000 (ORPHA:157,973, last update: November 2020). In addition, more than 700 LMNA variants have been identified (accessed on 12 December 2022; HGMD Professional 2021.4). Patients with L-CMD are described as having severe weakness in the first year, with marked weakness in the neck extensors, resulting in the "dropped head syndrome."
In our study, L-CMD represents the second most common condition with (4 patients/17; 23.53%) of positive patients. In comparison with previous studies, the proportion of patients with LMNA variants was 12.5% in CMD Chinese patients [56], and 8.8% in the UK population [2].
The heterozygous missense c.1718C > T variant harbored in patient P12 was previously reported at a heterozygous state in other several clinical phenotypes like dilated cardiomyopathy, ventricular tachycardia, and ventricular ectopy, familial partial lipodystrophy subtype 2, Charcot-Marie-Tooth disease, and in a patient with a limb-girdle muscular dystrophy form with no cardiac involvement [34,35,36,37]. Moreover, it was found in heterozygous individuals affected by Emery-Dreifuss muscular dystrophy type 2 [57], but it has also been reported in the homozygous state in a patient with progeroid features with no cardiac findings [38]. We identified for the first time a de novo heterozygous missense c.1718C > T variant in congenital muscular dystrophy without cardiac involvement, and we assume that it is a strong candidate variant responsible for CMD in our patient.
It was suggested that additional factors, either genetic or environmental, may contribute to the precise tissue involvement [36]. The association between LMNA gene mutations and different phenotypes is complex and poorly understood and can present with a wide range of severity [58]. Several hypotheses have been proposed, including mechanical shearing, differential gene regulation by interaction with nuclear chromatin, and interaction between mutant lamins A/C and other nuclear proteins [59].
RYR1-related congenital myopathy
RYR1-related myopathies are a group of congenital muscle diseases due to variations in the RYR1 gene that encodes the skeletal muscle sarcoplasmic reticulum calcium release channel (Ryanodine receptor type 1). Main histological subtypes of RYR1-RM include central core disease (CCD), multiminicore disease (MmD), core–rod myopathy (CRM), centronuclear myopathy (CNM), and congenital fiber-type disproportion (CFTD). A range of RYR1-RM clinical phenotypes has also emerged more recently and includes King Denborough syndrome (AD, MIM#619,542), RYR1 rhabdomyolysis-myalgia syndrome, atypical periodic paralysis, Malignant hyperthermia susceptibility 1 (AD, MIM#145,600), Congenital myopathy 1A, autosomal dominant, with susceptibility to malignant hyperthermia (AD, MIM#117,000), Congenital myopathy 1B, autosomal recessive (AR, MIM#255,320), and late-onset axial myopathy [60,61,62,63].
To date, more than 800 RYR1 variants have been identified (accessed on 12 December 2022; HGMD Professional 2021.4) and can be inherited in both autosomal dominant and recessive manners. The majority of variants are categorized as “variants of uncertain significance” [64]. Symptoms of RYR1-related diseases are often present from birth or appear in early infancy and the disease course is often non-progressive or very slowly progressive.
In this study, RYR1-related congenital myopathies represent the third most common class with 3 mutated patients (3 patients/17; 17.65%). In comparison, Gonzalez-Quereda and their colleagues found that 15.7% of patients with variants occurred in RYR1 gene as the most common causative gene in a large cohort of Spanish patients [19].
After Sanger sequencing confirmation, the little brother P14 of the proband P13 harbored the same homozygous pathogenic missense c.1201C > T p.(Arg401Cys) variant in exon 12 with a similar clinical phenotype of congenital myopathy. Their parents were heterozygous for this familial variant. The same variant has been previously reported in individuals with malignant hyperthermia at a heterozygous state [39]. However, it was reported in a patient with centronuclear myopathy [40], and in another patient with severe congenital RYR1-associated myopathy in a recessive manner [41].
The known heterozygous missense c.7880 T > G p.(Val2627Gly) variant in exon 49 presented in the P19 and the homozygous missense c.13892A > G p.(Tyr4631Cys) variant in exon 95 harbored in the P20 are causative of RYR1-related congenital myopathy.
The association with malignant hyperthermia in genetically susceptible individuals may reveal and confirmed after a clinical reaction by exposure to volatile anesthetics or succinylcholine [65].
Congenital titinopathy
TTN is a huge gene with 364 exons. It encodes titin, the largest described protein in humans [66]. Given the size of titin, it is not unexpected that recessive prenatal or infant onset forms of titinopathy have been reported in several clinically different diseases affecting skeletal and/or cardiac muscle, including early-onset myopathy with fatal cardiomyopathy (EOMFC) [67], centronuclear myopathy [68], core myopathy with heart disease [69], and arthrogryposis multiplex congenita with myopathy [70]. In contrast to many other neuromuscular disorders, cardiac involvement is a substantial source of morbidity and mortality [71], while in most cases of congenital myopathy, cardiac involvement is not a major concern [72]. Nevertheless, the TTN gene is among the most frequent genes related to severe cardiac involvement in the literature [73]. To date, more than 1000 variants are reported in The Human Gene Mutation Database (accessed on 12 December 2022; HGMD Professional 2021.4).
Our homozygous pathogenic frameshift c.106541delA p.(Asp35514Valfs*32) variant harbored in patient P9 with an EOMFC phenotype was previously published [22].
For the patients (P15, P16, P17, and P22), further sequencing including a large gene panel is slated to be carried out to investigate these four genetically unresolved patients. They may be due to variants in other myopathic or neuromuscular genes that were not covered by our custom gene panel.
Conclusions
We would like to emphasize the importance of family segregation studies as they offer useful information on the clinical significance of the variants and give further data about their association with the disease.
We assume that several Moroccan patients with neuromuscular disorders are undiagnosed due to difficulties for patients accessing specialized centers of genetic pediatric neurology in our context, in addition, to the limited means of genetic investigation, offered by only a few centers in our country. That is why, we investigated molecular analysis and application of a targeted NGS-based method and clinical exome sequencing in Moroccan patients with early-onset myopathies.
In summary, the variation of these genetic results of congenital myopathies and congenital muscular dystrophies emphasizes the crucial role of NGS technology in accurate genetic diagnosis, notably, for some cases with an atypical phenotype that does not fit with the classical description of these diseases. The NGS-based molecular diagnosis allows us to significantly increase the rate of positive cases for heterogeneous genetic diseases and expand the mutation spectrum to better understand the genotype–phenotype correlations of congenital muscle diseases. The results of this study will contribute to the achievement of an epidemiological characterization of early-onset myopathy, highlighting the importance of these neuromuscular diseases that are underestimated in the Moroccan population, and improvement of the diagnosis and management of these genetic disorders with potentially significant improvement in prognosis of patients.
Availability of data and materials
The principal data generated and/or analyzed during the current study are included in the published article. The datasets used in this study are available from the corresponding author on reasonable request.
Abbreviations
- CDG:
-
Congenital disorder of glycosylation
- CES:
-
Clinical exome sequencing
- CK:
-
Creatine kinase
- CM:
-
Congenital myopathy
- CMD:
-
Congenital muscular dystrophy
- CNV:
-
Copy number variation
- EMG:
-
Electromyogram
- EOMFC:
-
Early-onset myopathy with fatal cardiomyopathy
- MAF:
-
Minor allele frequency
- MDC1A:
-
Merosin-deficient congenital muscular dystrophy type 1A
- MPCR:
-
Multiplex PCR
- MRI:
-
Magnetic resonance imaging
- NGS:
-
Next-generation sequencing
- qPCR:
-
Quantitative PCR
- SNV:
-
Single nucleotide variant
- VUS:
-
Variant of uncertain significance
References
Peat RA, Smith JM, Compton AG, Baker NL, Pace RA, Burkin DJ et al (2008) Diagnosis and etiology of congenital muscular dystrophy. Neurology 71(5):312–321
Sframeli M, Sarkozy A, Bertoli M, Astrea G, Hudson J, Scoto M et al (2017) Congenital muscular dystrophies in the UK population: clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul Disord 27(9):793–803
Clement EM, Feng L, Mein R, Sewry CA, Robb SA, Manzur AY et al (2012) Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord 22(6):522–527
Lisi MT, Cohn RD (2007) Congenital muscular dystrophies: new aspects of an expanding group of disorders. Biochim Biophys Acta 1772(2):159–172
Colombo I, Scoto M, Manzur AY, Robb SA, Maggi L, Gowda V et al (2015) Congenital myopathies: natural history of a large pediatric cohort. Neurology 84(1):28–35
Emery AE (2002) The muscular dystrophies. Lancet 359(9307):687–695
Sewry CA (2008) Pathological defects in congenital myopathies. J Muscle Res Cell Motil 29(6–8):231–238
North KN, Wang CH, Clarke N, Jungbluth H, Vainzof M, Dowling JJ et al (2014) Approach to the diagnosis of congenital myopathies. Neuromuscul Disord 24(2):97–116
Quijano-Roy S, Renault F, Romero N, Guicheney P, Fardeau M, Estournet B (2004) EMG and nerve conduction studies in children with congenital muscular dystrophy. Muscle Nerve 29(2):292–299
Wallgren-Pettersson C, Sainio K, Salmi T (1989) Electromyography in congenital nemaline myopathy. Muscle Nerve 12(7):587–593
Fu XN, Xiong H (2017) Genetic and clinical advances of congenital muscular dystrophy. Chin Med J 130(21):2624–2631
Mercuri E, Muntoni F (2012) The ever-expanding spectrum of congenital muscular dystrophies. Ann Neurol 72(1):9–17
Pelin K, Wallgren-Pettersson C (2019) Update on the genetics of congenital myopathies. Semin Pediatr Neurol 29:12–22
Ravenscroft G, Bryson-Richardson RJ, Nowak KJ, Laing NG (2018) Recent advances in understanding congenital myopathies. F1000Res 7
Fardeau M, Desguerre I (2013) Diagnostic workup for neuromuscular diseases. Handb Clin Neurol 113:1291–1297
Prasad AN, Prasad C (2011) Genetic evaluation of the floppy infant. Semin Fetal Neonatal Med 16(2):99–108
Wang Y, Peng W, Guo HY, Li H, Tian J, Shi YJ et al (2016) Next-generation sequencing-based molecular diagnosis of neonatal hypotonia in Chinese Population. Sci Rep 6:29088
Chae JH, Vasta V, Cho A, Lim BC, Zhang Q, Eun SH et al (2015) Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. J Med Genet 52(3):208–216
Gonzalez-Quereda L, Rodriguez MJ, Diaz-Manera J, Alonso-Perez J, Gallardo E, Nascimento A et al (2020) Targeted next-generation sequencing in a large cohort of genetically undiagnosed patients with neuromuscular disorders in Spain. Genes 11:5
Herman I, Lopez MA, Marafi D, Pehlivan D, Calame DG, Abid F et al (2021) Clinical exome sequencing in the diagnosis of pediatric neuromuscular disease. Muscle Nerve 63(3):304–310
El Kadiri Y, Ratbi I, Laarabi FZ, Kriouile Y, Sefiani A, Lyahyai J (2021) Identification of a novel LAMA2 c.2217G > A, p.(Trp739*) mutation in a Moroccan patient with congenital muscular dystrophy: a case report. BMC Med Genom 14(1):113
El Kadiri Y, Ratbi I, Sefiani A, Lyahyai J (2022) Clinical and molecular genetic analysis of early-onset myopathy with fatal cardiomyopathy: novel biallelic M-line TTN mutation and review of the literature. Gene Reports 27:101587
Tai H, Cui L, Guan Y, Liu M, Li X, Shen D et al (2017) Correlation of Creatine kinase levels with clinical features and survival in amyotrophic lateral sclerosis. Front Neurol 8:322
Oliveira J, Gruber A, Cardoso M, Taipa R, Fineza I, Goncalves A et al (2018) LAMA2 gene mutation update: toward a more comprehensive picture of the laminin-alpha2 variome and its related phenotypes. Hum Mutat 39(10):1314–1337
Allamand V, Guicheney P (2002) Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for alpha2 chain of laminin). Eur J Hum Genet 10(2):91–94
Oliveira J, Santos R, Soares-Silva I, Jorge P, Vieira E, Oliveira ME et al (2008) LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet 74(6):502–512
Louhichi N, Richard P, Triki CH, Meziou M, Ayadi H, Guicheney P et al (2006) Novel mutations in LAMA2 gene responsible for a severe phenotype of congenital muscular dystrophy in two Tunisian families. Arch Inst Pasteur Tunis 83(1–4):19–23
Siala O, Louhichi N, Triki C, Moriniere M, Rebai A, Richard P et al (2007) Severe MDC1A congenital muscular dystrophy due to a splicing mutation in the LAMA2 gene resulting in exon skipping and significant decrease of mRNA level. Genet Test 11(3):199–207
Sonia Nouioua AC, Benahmed M, Mathieu C, Krahn M, Hamadouche T, MeriemTazir (2017) Description d’une famille algérienne associant une dysferlinopathie et une merosinopathie primaires. Revue Neurologique 173:S68–S69
Xiong H, Tan D, Wang S, Song S, Yang H, Gao K et al (2015) Genotype/phenotype analysis in Chinese laminin-alpha2 deficient congenital muscular dystrophy patients. Clin Genet 87(3):233–243
Tan D, Yang H, Yuan Y, Bonnemann C, Chang X, Wang S et al (2015) Phenotype-genotype analysis of chinese patients with early-onset LMNA-related muscular dystrophy. PLoS ONE 10(6):e0129699
Tekin H, Yilmaz S, Tekgul H, Gokben S, Aktan G (2020) Dropped head related lamin A/C associated congenital muscular dystrophy case; previously defined as emerydreifuss muscular dystrophy. Turk J Pediatr 62(1):130–135
Bonne G, Mercuri E, Muchir A, Urtizberea A, Becane HM, Recan D et al (2000) Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol 48(2):170–180
Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A et al (2008) Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol 52(15):1250–1260
Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E et al (2003) Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 41(5):771–780
Lanktree M, Cao H, Rabkin SW, Hanna A, Hegele RA (2007) Novel LMNA mutations seen in patients with familial partial lipodystrophy subtype 2 (FPLD2; MIM 151660). Clin Genet 71(2):183–186
Benedetti S, Menditto I, Degano M, Rodolico C, Merlini L, D’Amico A et al (2007) Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology 69(12):1285–1292
Van Esch H, Agarwal AK, Debeer P, Fryns JP, Garg A (2006) A homozygous mutation in the lamin A/C gene associated with a novel syndrome of arthropathy, tendinous calcinosis, and progeroid features. J Clin Endocrinol Metab 91(2):517–521
Miller DM, Daly C, Aboelsaod EM, Gardner L, Hobson SJ, Riasat K et al (2018) Genetic epidemiology of malignant hyperthermia in the UK. Br J Anaesth 121(4):944–952
Abath Neto O, Moreno CAM, Malfatti E, Donkervoort S, Bohm J, Guimaraes JB et al (2017) Common and variable clinical, histological, and imaging findings of recessive RYR1-related centronuclear myopathy patients. Neuromuscul Disord 27(11):975–985
Bharucha-Goebel DX, Santi M, Medne L, Zukosky K, Dastgir J, Shieh PB et al (2013) Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum. Neurology 80(17):1584–1589
Tian X, Liang WC, Feng Y, Wang J, Zhang VW, Chou CH et al (2015) Expanding genotype/phenotype of neuromuscular diseases by comprehensive target capture/NGS. Neurol Genet 1(2):e14
Paro-Panjan D, Neubauer D (2004) Congenital hypotonia: is there an algorithm? J Child Neurol 19(6):439–442
Shahabi NS, Fakhraee H, Kazemian M, Afjeh A, Fallahi M, Shariati M et al (2017) Frequency and causes of hypotonia in neonatal period with the gestational age of more than 36 weeks in NICU of Mofid children hospital, Tehran, Iran During 2012–2014. Iran J Child Neurol 11(1):43–49
Laugel V, Cossee M, Matis J, de Saint-Martin A, Echaniz-Laguna A, Mandel JL et al (2008) Diagnostic approach to neonatal hypotonia: retrospective study on 144 neonates. Eur J Pediatr 167(5):517–523
Richer LP, Shevell MI, Miller SP (2001) Diagnostic profile of neonatal hypotonia: an 11-year study. Pediatr Neurol 25(1):32–37
Birdi K, Prasad AN, Prasad C, Chodirker B, Chudley AE (2005) The floppy infant: retrospective analysis of clinical experience (1990–2000) in a tertiary care facility. J Child Neurol 20(10):803–808
Oliveira J, Goncalves A, Taipa R, Melo-Pires M, Oliveira ME, Costa JL et al (2016) New massive parallel sequencing approach improves the genetic characterization of congenital myopathies. J Hum Genet 61(6):497–505
Benarroch L, Bonne G, Rivier F, Hamroun D (2020) The 2021 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul Disord 30(12):1008–1048
Jaouad IC, Elalaoui SC, Sbiti A, Elkerh F, Belmahi L, Sefiani A (2009) Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders. J Biosoc Sci 41(5):575–581
Graziano A, Bianco F, D’Amico A, Moroni I, Messina S, Bruno C et al (2015) Prevalence of congenital muscular dystrophy in Italy: a population study. Neurology 84(9):904–911
Abdel Aleem A, Elsaid MF, Chalhoub N, Chakroun A, Mohamed KAS, AlShami R et al (2020) Clinical and genomic characteristics of LAMA2 related congenital muscular dystrophy in a patients’ cohort from Qatar A population specific founder variant. Neuromuscul Disord 30(6):457–471
Oliveira JPFJ, Santos M et al (2012) LAMA2 muscular dystrophy. In: Adam MP, Ardinger HH, Pagon RA et al (eds) GeneReviews®. University of Washington, Seattle
Natera-de Benito D, Muchart J, Itzep D, Ortez C, Gonzalez-Quereda L, Gallano P et al (2020) Epilepsy in LAMA2-related muscular dystrophy: An electro-clinico-radiological characterization. Epilepsia 61(5):971–983
Ge L, Liu A, Gao K, Du R, Ding J, Mao B et al (2018) Deletion of exon 4 in LAMA2 is the most frequent mutation in Chinese patients with laminin alpha2-related muscular dystrophy. Sci Rep 8(1):14989
Ge L, Zhang C, Wang Z, Chan SHS, Zhu W, Han C et al (2019) Congenital muscular dystrophies in China. Clin Genet 96(3):207–215
Bernasconi P, Carboni N, Ricci G, Siciliano G, Politano L, Maggi L et al (2018) Elevated TGF beta2 serum levels in Emery-Dreifuss Muscular Dystrophy: Implications for myocyte and tenocyte differentiation and fibrogenic processes. Nucleus 9(1):292–304
Francisco ARG, Santos Goncalves I, Veiga F, Mendes Pedro M, Pinto FJ, Brito D (2017) Complex phenotype linked to a mutation in exon 11 of the lamin A/C gene: hypertrophic cardiomyopathy, atrioventricular block, severe dyslipidemia and diabetes. Rev Port Cardiol 36(9):669
Worman HJ, Courvalin JC (2004) How do mutations in lamins A and C cause disease? J Clin Invest 113(3):349–351
Lawal TA, Todd JJ, Meilleur KG (2018) Ryanodine receptor 1-related myopathies: diagnostic and therapeutic approaches. Neurotherapeutics 15(4):885–899
Witting N, Werlauff U, Duno M, Vissing J (2017) Phenotypes, genotypes, and prevalence of congenital myopathies older than 5 years in Denmark. Neurol Genet 3(2):e140
Dowling JJ, Lillis S, Amburgey K, Zhou H, Al-Sarraj S, Buk SJ et al (2011) King-Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord 21(6):420–427
Matthews E, Neuwirth C, Jaffer F, Scalco RS, Fialho D, Parton M et al (2018) Atypical periodic paralysis and myalgia: a novel RYR1 phenotype. Neurology 90(5):e412–e418
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424
Hopkins PM (2011) Malignant hyperthermia: pharmacology of triggering. Br J Anaesth 107(1):48–56
Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M et al (2001) The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 89(11):1065–1072
Carmignac V, Salih MA, Quijano-Roy S, Marchand S, Al Rayess MM, Mukhtar MM et al (2007) C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol 61(4):340–351
Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, Schmitz-Abe K, DeChene ET, Swanson LC et al (2013) Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 81(14):1205–1214
Chauveau C, Bonnemann CG, Julien C, Kho AL, Marks H, Talim B et al (2014) Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet 23(4):980–991
Fernandez-Marmiesse A, Carrascosa-Romero MC, Alfaro Ponce B, Nascimento A, Ortez C, Romero N et al (2017) Homozygous truncating mutation in prenatally expressed skeletal isoform of TTN gene results in arthrogryposis multiplex congenita and myopathy without cardiac involvement. Neuromuscul Disord 27(2):188–192
Feingold B, Mahle WT, Auerbach S, Clemens P, Domenighetti AA, Jefferies JL et al (2017) Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation 136(13):e200–e231
Claeys KG (2020) Congenital myopathies: an update. Dev Med Child Neurol 62(3):297–302
Oates EC, Jones KJ, Donkervoort S, Charlton A, Brammah S, Smith JE 3rd et al (2018) Congenital Titinopathy: comprehensive characterization and pathogenic insights. Ann Neurol 83(6):1105–1124
Acknowledgements
We thank the families for their participation in this study, and we are grateful to Dr. Selouani and Pr. Alaoui for improving the English language of the manuscript. Youssef El Kadiri was supported by the excellence research Grant from the National Centre for Scientific and Technical Research (CNRST), Rabat, Morocco.
Funding
This research did not receive any specific Grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
YEK contributed to data curation, investigation, methodology, visualization, validation, formal analysis, and writing—original draft. IR contributed to investigation, methodology, and writing—review and editing. MO, SCE, ICJ, and MS performed investigation. AZ performed data curation. NB performed investigation. AS contributed to investigation, conceptualization and supervision, and writing—review and editing. JL performed conceptualization, supervision, and writing—review and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The whole pre-analytic and post-analytic steps described in this work (genetic consultations, blood sampling, molecular analysis, genetic reports and genetic counseling consultations) were performed for the patients by the Department of Medical Genetics of the National Institute of Health as medical services in agreement with the tenets of the Declaration of Helsinki. All methods were carried out in accordance with relevant guidelines and regulations. Written informed consent was obtained from all of the minor patients’ parents or adult patients before DNA collection by referring clinicians in order to implement the genetic analysis. All ethical issues of the National Institute of Health in Rabat, Morocco, that included the Department of Medical Genetics are in charge of an Intramural Advisory Committee. This committee gave its approval to publish clinical and molecular data of the patients described in the paper in anonymous state.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Set of primers used for confirmation of DNA variations.
Additional file 2: Fig. S1.
Original agarose gel of Figure 4B.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
El Kadiri, Y., Ratbi, I., Ouhenach, M. et al. Early diagnosis of congenital muscular pathologies using next-generation sequencing: experiences from a tertiary center in Morocco. Egypt J Med Hum Genet 24, 36 (2023). https://doi.org/10.1186/s43042-023-00416-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-023-00416-y