
Abstract
Background
Acute myeloid leukemia (AML) is a heterogeneous disorder encompassing a set of hematopoietic tumors that develop when the myeloid precursor cells undergo disproportionate clonal proliferation. Homeobox A 9 (HOXA9) is a pioneer transcription factor in AML pathogenesis along with its cofactor myeloid ecotropic integration site 1 (MEIS1). Our work aimed to evaluate the different expression levels of HOXA9 and MEIS1 genes and their diagnostic and prognostic significance in adult Egyptian patients with de novo AML. The study was carried out on 91 de novo AML Egyptian patients and 41 healthy individuals. Bone marrow samples were obtained from both patients and controls and then tested by reverse transcription-quantitative polymerase chain reaction to assess the mRNA expression in the studied genes.
Results
HOXA9 and MEIS1 gene expression levels were significantly elevated in AML patients compared to controls (p < 0.001). There was a statistically significant positive correlation between HOXA9 and MEIS1 gene expression in AML patients. However, there was no association between HOXA9 and MEIS1 gene expression levels and disease-free survival (DFS) and overall survival (OS) (p = 0.264 and 0.351, respectively).
Conclusion
HOXA9 and MEIS1 genes are highly expressed in Egyptian AML patients, suggesting their interesting pathogenic role in AML. They could be used as markers for the diagnosis of AML, but not for the disease prognosis.
Similar content being viewed by others

Background
Acute myeloid leukemia (AML) is a malignant hematological disorder in which immature clonal myeloid cells excessively proliferate and show arrested differentiation [1]. There is a continuous increase in AML incidence. The risk of AML is higher in males than in females [2]. AML is the most common type of acute leukemia in adults and is associated with the highest leukemia-related mortality rate [3].
A deep understanding of AML biology has shown some progress, but it is sluggish to formulate this knowledge into reliable therapy protocols. Therefore, a comprehensive study of molecular mechanisms associated with the proliferation, carcinogenesis, and recurrence of AML is crucial for developing effective therapeutic and prognostic strategies [4].
The transcription factor genes necessary for normal blood cell development are the most frequently targeted sites of genetic alterations in leukemia. Among these transcription factors are the homeobox A9 (HOXA9) and the myeloid ecotropic integration site 1 (MEIS1) genes. They control progenitor cell regeneration during hematopoiesis and leukemogenesis [5, 6]. The expression of HOXA9 and MEIS1 genes leads to the transition into the cluster of differentiation 34 (CD34) stage of initial differentiation of progenitors [7].
AML is commonly featured by HOXA9 and MEIS1 gene expression, which are dominant collaborating oncoproteins leading to AML development [8, 9]. Despite blocking the differentiation of non-leukemogenic myeloid progenitors by the overexpressed HOXA9 gene [10], MEIS1 expression is mandatory to produce AML-initiating progenitors [11]. Thus, investigating the relationship between the transcription level of these genes and AML could help to identify a new targeted therapy for the disease.
The aim of the study was to evaluate HOXA9 and MEIS1 gene expression levels and their diagnostic and prognostic roles in de novo AML adult Egyptian patients.
Methods
Study group
Ninety-one de novo AML adult Egyptian patients and 41 healthy bone marrow donors were enrolled in the study.
The study was conducted between 2015 and 2018 at the Clinical Pathology Department at the National Cancer Institute, Cairo University, Egypt. The study was approved by the ethical scientific committee of Benha University and followed the Helsinki declaration guidelines.
AML was diagnosed and classified by the French–American–British (FAB) criteria [12]. The Medical Research Council Classification (MRC-C) was used to classify the cytogenetic risk in AML patients [13].
The inclusion criterion was adult patients with de novo AML. Patients ˂18 years, patients with secondary AML and patients with promyelocytic leukemia, and patients presented with AML with myelodysplasia were excluded from the study. Informed written consent was obtained from each patient included in the study.
All patients underwent a detailed history taking, complete clinical assessment, complete blood picture, and BM aspiration. The patients also underwent immunophenotyping (IPT), conventional karyotyping, fluorescence in situ hybridization (FISH), and reverse transcriptase PCR (RT-qPCR) for the genetic translocation t(8;21)(q22;q22); inv(16)(p13;1q22); or t(16;16)(p13.1;q22). Analysis of FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD) mutations was also performed and detected by fragment analysis.
Patients and controls were tested by RT-qPCR to determine the expression of messenger ribonucleic acid (mRNA) of HOXA9 and MEIS1 genes within the bone marrow samples.
Chemotherapy was induced in AML patients using the standard intensive 3 + 7 regimen (30 mg/m2 doxorubicin on days 1–3 and 100 mg/m2 cytarabine on days 1–7). However, dose reduction may be given for patients more than 60 years old. According to the cytogenetic risk and availability of a matched BM donor, the patients undergoing complete remission (CR) have received consolidation therapy. The patients with favorable risk were given high doses of cytarabine-based consolidation chemotherapy, while the intermediate-risk patients received either high doses of cytarabine-based consolidation chemotherapy or allogeneic BMT. Allogeneic BMT was considered the treatment of choice for adverse-risk patients. There were 9 patients who underwent allogeneic BMT.
CR was defined as regaining the BM’s normal cellularity with fewer than 5% leukemic blasts, an absolute neutrophil count above 1.0*109/L, and a platelet count above 100*109/L. Resistance was defined as failure to fulfill CR, CR with incomplete recovery, or partial remission (PR) in a patient alive ≥ 7 days after completion of initial therapy, with evidence of persistent leukemia in peripheral blood and/or bone marrow examination [14, 15]. Disease-free survival (DFS) was defined as the duration between gaining CR and relapse or death. Overall survival (OS) was defined as the time from involvement in the study to death from any cause; patients not known to have died at the last follow-up are censored on the date they were last known to be alive [16].
RNA extraction and cDNA synthesis
The BM samples (1 ml) were collected from all patients and controls on potassium ethylenediaminetetraacetic acid (K-EDTA) containing tubes. Ribonucleic acid (RNA) extraction from BM cells was performed using QIAamp® RNA Mini Kit (QIAGEN, Austin, Texas, USA, Catalog no. 52304) according to the manufacturer's instructions.
The concentration and purity of the extracted RNA were assessed by spectrophotometer NanoDrop (Quawell, Q-500, Scribner, USA) and then stored at -80° C till used. The extracted RNA was reverse-transcribed to complementary DNA (cDNA) using the available cDNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific, USA; Catalog no. 4368814) as recommended by the manufacturer's instructions. The synthesized cDNA was stored at -20° C until performing quantitative real-time PCR.
Gene expression analysis
We used the quantitative real-time PCR to determine the level of expression of HOXA9, MEIS1 genes, and the reference gene (β-Actin). The RT-qPCR reactions were analyzed according to the manufacturer's instructions using TaqMan® Universal PCR Master Mix II (Catalog no 4440043, Thermo fisher scientific, Applied Biosystems, USA) and TaqMan® Gene Expression Assays for HOXA9 mRNA (Assay ID: Hs04931836_s1; Catalog no 4331182, Thermo Fisher Scientific, USA), MEIS1 mRNA (Assay ID: Hs00180020_m1; Catalog no 4331182, Thermo Fisher Scientific, USA), and β-Actin (Assay ID: Hs03929097_g1; Catalog no 4331182, Thermo Fisher Scientific, USA). The PCR conditions were as follows: polymerase activation (95 °C for 10 min), then denaturation (40 cycles of 95 °C for 30 s), followed by annealing and extension (60 °C for 60 s). The fluorescence was measured using Step One plus™ Real-Time PCR System (Applied Biosystems, USA). The data were presented by cycle threshold (Ct). The Ct was obtained for HOXA9 or MEIS1 as the target gene (Ct-target) and for the reference gene β-Actin (Ct-reference). Data were analyzed using relative quantitation (RQ). RQ and results were expressed as 2−∆∆Ct according to Schmittgen and Livak [17].
Statistical methods
The obtained data were statistically analyzed using version 24 of the SPSS statistical software (SPSS Inc., Chicago, IL, USA). Categorical data analysis was performed using the chi-square and Fisher's exact test. The Mann–Whitney test compared quantitative data between groups. The performance of the tested genes as AML biomarkers was tested using receiver operating characteristic (ROC) analysis. The correlation between HOXA9 and MEIS1 gene expression was analyzed using the Spearman-rho test. Cumulative DFS and OS were compared by Kaplan–Meier survival analysis and the log-rank test. p values < 0.05 were considered statistically significant. Patients were classified according to HOXA9 and MEIS1 gene expression into low and overexpression groups based on the cutoff values obtained by ROC curve analysis.
Results
Patients’ characteristics
The main demographic, clinical, and laboratory characteristics of the studied patients are summarized in Table 1.
Determination of delta Ct cutoff value
ROC analysis was applied to determine the best cutoff value for HOXA9 and MEIS1 gene expression for the diagnosis of AML. The curve value showed that the best cutoff for HOXA9 mRNA expression was 1.06, with an AUC of 0.910. The sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and accuracy were 87.9%, 92.9%, 98.8%, 54.2%, and 88.6%, respectively, while for MEIS1 mRNA expression, the best cutoff value was 1.3, with an AUC of 0.831. The sensitivity, specificity, positive predictive value (PPV), negative (NPV) predictive value, and accuracy were 82.4%, 100%, 100%, 46.7%, and 84.8%, respectively. This indicated that HOXA9 and MEIS1 mRNA expression levels could be used in AML diagnosis (Fig. 1 and Additional file 1: Table S1).

ROC analysis of HOXA9 and MEIS1 gene expression for discriminating adult AML patients
The expression levels of HOXA9 and MEIS1 mRNA in AML and control group
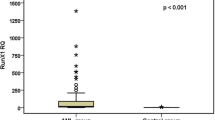
The relative expression levels of HOXA9 mRNA and MEIS1 mRNA in newly diagnosed AML patients were significantly higher than in the healthy controls (p < 0.001). A broad range of HOXA9 mRNA expression (range = 0.22–921.1; median = 14.6) and MEIS1 mRNA expression (range = 0–1716; median = 33.9) were observed in AML patients (Table 2, Figs. 2 and 3).

Expression of HOXA9 mRNA among adult AML cases and controls

Expression of MEIS1 mRNA among adult AML cases and controls
Correlation between the expression of HOXA9 and MEIS1 genes
As shown in Fig. 4, we have found a significant positive intermediate correlation between HOXA9 and MEIS1 gene expression.

Correlation between HOXA9 and MEIS1 genes expression
Correlation between elevated HOXA9 and MEIS1 gene expression and clinicopathological characteristics of AML patients
The AML patients were divided based on the median expression level of the HOXA9 gene into two groups: low gene expression (< 14.6) and high gene expression (> 14.6).
There were no significant differences in age, total leukocytic count (TLC), hemoglobin (Hb) level, platelet count, and blast percentage in the peripheral blood or the bone marrow between the low and high HOXA9 gene expression groups (p > 0.05) (Table 3). Additionally, there were no significant differences regarding the distribution of FAB subtypes (M1, M2, M4, M5, M7), sex, organomegaly (hepatomegaly, splenomegaly, lymphadenopathy), BM cellularity, and IPT characteristics (p > 0.05). Furthermore, cytogenetic risk (favorable, intermediate, and adverse) and FLT3-ITD gene mutation did not significantly differ (p > 0.05) (Table 3).
Patients were classified based on the median expression level of the MEIS1 gene into low gene expression (< 33.9) and high gene expression (> 33.9) groups.
The results revealed no significant differences in age, TLC, Hb level, platelet count, and blast percentage in peripheral blood and BM between the low and high expression groups (p > 0.05) (Table 4). Furthermore, no significant differences were observed regarding the distribution of FAB subtypes (M1, M2, M4, M5, M7), sex, organomegaly (hepatomegaly, splenomegaly, lymphadenopathy), bone marrow cellularity, and IPT characteristics (p > 0.05). However, patients with low gene expression had aberrant markers with a percentage of (43.8%), while patients with high gene expression had aberrant markers with a percentage of (14.7%), (p = 0.014). Aberrant IPT markers were significantly associated with low MEIS1 gene expression. No significant differences were reported regarding cytogenetic risk (favorable, intermediate, and adverse) and FLT3-ITD gene mutation (p > 0.05) (Table 4).
Impact of HOXA9 and MEIS1 gene expression on response to initial treatment
Regarding the response to initial treatment, 68 patients (74.7%) showed CR, 23 patients (25.3%) showed no CR, and 20 patients relapsed from those who achieved CR. No significant differences were detected regarding response to treatment (CR and no CR) between the high and low HOXA9 and MEIS1 gene expression groups (Tables 3 and 4).
Impact of HOXA9 and MEIS1 gene expression on the prognosis of AML patients
Follow-up was performed for 40 months; there were 19 patients died during the induction: 15 patients died from infection and 4 died from bleeding. The median follow-up period was 6.7 months (range, 0.3–40 months).
Kaplan–Meier analysis and log-rank test showed that there was no significant association between low expression and over-expression of the HOXA9 gene and OS (p = 0.799). The median OS was 11.070 and 11.4 months, respectively, while the cumulative OS after 3 years was 15.9% and 30.4%, respectively. In addition, there was no association between low expression and over-expression of the HOXA9 gene and DFS (p = 0.264). The median DFS was 9.230 and 16.8 months, respectively, while the cumulative DFS after 3 years was 0.0% and 44.1%, respectively, in AML patients (Fig. 5A,B and Table 3, Additional file 1: Table S2).

Impact of HOXA9 gene expression on A DFS and B OS. Impact of MEIS1 gene expression on C DFS and D OS
Regarding the MEIS1 gene, there was no association between low expression and over-expression of the gene and OS (p = 0.695). The median OS was 9.0 and 12.47 months, respectively, while the cumulative OS after 3 years was 21.1% and 31.8%, respectively. Also, there was no association between low expression and over-expression of the MEIS1 gene and the DFS (p = 0.351). The median survival was 14.070 and 25.630, respectively, while the cumulative DFS after 3 years was 25.4% and 44.7%, respectively, in AML patients (Fig. 5C,D and Table 4, Additional file 1: table S2).
Discussion
AML therapeutic options are still limited due to its heterogeneity and complex biologic conduct. Despite the continuous efforts to develop novel specified therapy, the rates of AML-associated morbidity and mortality remain high [18].
The current advancement in molecular techniques is essential to provide an in-depth comprehension of leukemia pathogenesis and to offer novel molecular markers for diagnosis and prognosis [18].
Considering their documented role in hematopoiesis, aberrations of HOXA9 and its cofactor MEIS1 have been assessed in AML, focusing on their clinical intimation [19, 20]. Their synergetic expression leads to aggressive leukemia in mice [21].
HOXA9 gene expression pattern has been investigated in AML [22]. Yet, there is still a lack of evidence concerning its association with MEIS1 gene expression [23].
The present study has shown significantly higher expression of HOXA9 mRNA and MEIS1 mRNA levels in de novo AML patients compared to the control group, suggesting its pathogenic role in AML. These data are consistent with Gao et al. [24], who demonstrated higher HOXA9 and MEIS1 expression in AML patients than in healthy BM donors. Additionally, Kandel et al. [25] examined HOXA9 in sixty samples of AML patients and reported higher expression than in healthy controls.
Ward et al. [18] observed the highest differential expression of HOXA9 and MEIS1 genes in AML patients. This finding has insightful repercussions since these genes are fundamentals in preserving and progressing variable myeloid disorders, and their combination results in AML occurrence [26].
Considerable data suggest that HOXA9 is an oncogene that is associated with hematological malignancies, including chronic myeloid leukemia (CML) [27], acute lymphoblastic leukemia (ALL) [28], and AML [10, 11, 18]. Ismail et al. [27] indicated that HOXA9 expression is encountered in 44.6% of de novo CML cases. Rozovskaia et al. [28] reported that HOXA9 and MEIS1 genes are expressed in ALL and AML. Loftus et al. [29] reported high expression of HOXA9 and MEIS1 genes in samples with [lysine (K)-specific methyltransferase 2A-mixed lineage leukemia, translocated to 3 (KMT2A—MLLT3) and KMT2A-MLLT1 fusions]. However, the non-fusion samples showed normal HOXA9 and MEIS1 gene expression levels.
An interesting finding in the present study was the significant positive intermediate correlation between HOXA9 and MEIS1 gene expression in AML cases. Similarly, Chen et al. [30] stated that the HOXA9 gene expression positively correlates with the MEIS1 gene expression.
In the current study, no significant differences were observed between the high and low HOXA9 gene expression groups regarding age, sex, organomegaly, complete blood picture, bone marrow examination, immunophenotypic characteristics, cytogenetic risk, and FLT3-ITD gene mutation. Similar results were reported between the high and low MEIS1 gene expression groups. However, the low-expression group demonstrated a significantly higher percentage of aberrant IPT markers than the high-expression group.
In agreement with our results, Gao et al. [24] concluded no significant associations of the baseline demographic, clinical, and molecular patterns with HOXA9 and MEIS1 gene mutations. However, lower transcript levels are shown in patients with M3 than in those with other FAB subtypes. On the other hand, Kandel et al. [25] demonstrated that HOXA9 gene expression is associated with hepatosplenomegaly in AML patients. According to Ismail et al. [27], HOXA9 gene expression is significantly associated with a larger size of the spleen, a higher breakpoint cluster region-Abelson proto-oncogene (BCR-ABL) expression, and a higher Sokal score.
The current study reported no significant differences between subjects with high and low HOXA9 and MEIS1 gene expression regarding response to initial treatment (CR and no CR). In contrast, Gao et al. [24] demonstrated a significant difference between patients with high and low HOXA9 gene expression levels regarding complete remission after one cycle of chemotherapy and 1-year relapse. This contradiction may be due to different treatment protocols or may be related to different geographical distributions.
Regarding the association of HOXA9 and MEIS1 gene expression levels and AML outcome, there was no association between the expression levels of HOXA9 and MEIS1 genes and DFS and OS. In contrast, Gao et al. [24] stated that the high expression of HOXA9 gene in AML is related to shorter relapse-free survival and OS compared to its low expression and acts as a powerful prognostic marker for response to therapy and survival. Similar results from previous studies demonstrated that high HOXA9 gene expression has a shorter OS in AML [31, 32].
This discrepancy may be caused by the small size of our sample. Additionally, the MEIS1 gene expression behavior in malignant cells is influenced by many factors, including the immune status of the patients and the tumor microenvironment [33].
Conclusion
HOXA9 and MEIS1 genes are highly expressed in AML cases than in controls. However, their expression level was not associated with treatment response or survival rate of AML patients. Therefore, based on the results of the current study, HOXA9 and MEIS1 could be used as diagnostic biological markers for AML, but could not be used for assessment of the disease prognosis. Analyzing HOXA9 and MEIS1 gene mutations in further studies, including larger sample sizes, is recommended to assess the mechanism of aberrant HOXA9 and MEIS1 gene expression and to explore the molecular pathway linking these genes to leukemia progression. This will offer novel treatment modalities targeting the affected genes. Finally, longer follow-up may reveal other associations not depicted in the present study.
Availability of data and materials
Data analyzed in the current work are available from the corresponding author upon reasonable request.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- AUC:
-
Area under the curve
- BCR-ABL :
-
Breakpoint cluster region-Abelson proto-oncogene
- BM:
-
Bone marrow
- BMT:
-
Bone marrow transplantation
- CD:
-
Cluster of differentiation
- cDNA:
-
Complementary DNA
- CML:
-
Chronic myeloid leukemia
- CR:
-
Complete remission
- Ct:
-
Cycle threshold
- DFS:
-
Disease-free survival
- ELN:
-
European Leukemia Net
- FAB:
-
French–American–British
- FISH:
-
Fluorescence in situ hybridization
- Hb:
-
Hemoglobin
- HOXA9 :
-
Homeobox A 9
- IPT:
-
Immunophenotyping
- K-EDTA:
-
Potassium ethylenediaminetetraacetic acid
- KMT2A :
-
Lysine (K)-specific methyltransferase 2A
- MEIS1 :
-
Myeloid ecotropic integration site 1
- MLLT3 :
-
Mixed lineage leukemia, translocated to, 3
- mRNA:
-
Messenger ribonucleic acid
- NA:
-
Not applicable
- NPV:
-
Negative predictive value
- OS:
-
Overall survival
- PPV:
-
Positive predictive value
- RNA:
-
Ribonucleic acid
- ROC:
-
Receiver operating characteristic
- RT-PCR:
-
Real-time polymerase chain reaction
- RQ:
-
Relative quantitation
- TLC:
-
Total leukocytic count
- WHO:
-
World Health Organization
References
Newell LF, Cook RJ (2021) Advances in acute myeloid leukemia. BMJ 375:n2026
Surveillance, epidemiology, and end results (SEER) program cancer stat facts: Leukemia - acute myeloid leukemia (AML) https://seer.cancer.gov/statfacts/html/amyl.html; 2018, Accessed 18 Apr 2022
Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM (2019) Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev 6:70–87
Fu W, Cheng G, Ding Y, Deng Y, Guo P (2020) Identification of hub genes and its correlation with the prognosis of acute myeloid leukemia based on high-throughput data analysis. Prec Radiat Oncol 4:49–56
Argiropoulos B, Yung E, Humphries RK (2007) Unraveling the crucial roles of Meis1 in leukemogenesis and normal hematopoiesis. Genes Dev 21(22):2845–2849
Abramovich C, Humphries RK (2005) Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol 12:210–216
Bijl J, Thompson A, Ramirez-Solis R, Krosl J, Grier DG, Lawrence HJ et al (2006) Analysis of HSC activity and compensatory Hox gene expression profile in Hoxb cluster mutant fetal liver cells. Blood 108(1):116–122
Wang GG, Pasillas MP, Kamps MP (2005) Meis1 programs transcription of FLT3 and cancer stem cell character, using a mechanism that requires interaction with Pbx and a novel function of the Meis1 C-terminus. Blood 106(1):254–264
Sitwala KV, Dandekar MN, Hess JL (2008) HOX proteins and leukemia. Int J Clin Exp Pathol 1(6):461–474
Fujino T, Yamazaki Y, Largaespada DA, Jenkins NA, Copeland NG, Hirokawa K et al (2001) Inhibition of myeloid differentiation by Hoxa9, Hoxb8, and Meis homeobox genes. Exp Hematol 29(7):856–863
Wang GG, Pasillas MP, Kamps MP (2006) Persistent transactivation by meis1 replaces hox function in myeloid leukemogenesis models: evidence for co-occupancy of meis1-pbx and hox-pbx complexes on promoters of leukemia-associated genes. Mol Cell Biol 26:3902–3916
Daniel AA, Attilio O, Robert H, Jürgen T, Borowitz MJ, et al (2016) Revision to the World Health Organization classification of myeloid neoplasms and acute Leukemia. Blood 127(20):2391–2405
Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH et al (2010) Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116:354–365
Cheson BD, Bennett JM, Kopecky KJ, Büchner T, Willman CL, Estey EH et al (2003) Revised recommendations of the international working Group for Diagnosis, standardization of Response Criteria, Treatment outcomes, and Reporting standards for therapeutic Trials in Acute Myeloid leukemia. J Clin Oncol 21(24):4642–4649
Döhner H, Estey E, Amadori S, Appelbaum FR, Büchner T, Burnett AK et al (2010) Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel: on behalf of the European leukemia Net. Bood 115(3):453–474
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T et al (2017) Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129(4):424–447
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−∆∆CT) method. Methods 25:402–408
Ward C, Cauchy P, Garcia P, Frampton J, Esteban M, Volpe G (2020) High WBP5 expression correlates with elevation of HOX genes levels and is associated with inferior survival in patients with acute myeloid leukaemia. Sci Rep 10:3505
Hassawi M, Shestakova EA, Fournier M, Lebert-Ghali CÉ, Vaisson G, Frison H et al (2014) Hoxa9 collaborates with E2A-PBX1 in mouse B cell leukemia in association with Flt3 activation and decrease of B cell gene expression. Dev Dyn 243(1):145–158
Thorne RM, Milne TA (2015) Dangerous liaisons: cooperation between Pbx3, Meis1, and Hoxa9 in leukemia. Haematologica 100(7):850–853
Garcia-Cuellar MP, Steger J, Füller E, Hetzner K, Slany RK (2015) Pbx3 and Meis1 cooperate through multiple mechanisms to support Hox-induced murine leukemia. Haematologica 100(7):905–913
Spencer DH, Young MA, Lamprecht TL, Helton NM, Fulton R, O’Laughlin M et al (2015) Epigenomic analysis of the HOX gene loci reveals mechanisms that may control canonical expression patterns in AML and normal hematopoietic cells. Leukemia 29(6):1279–1289
Schneider E, Staffas A, Röhner L, Malmberg ED, Ashouri A, Krowiorz K et al (2018) Micro-ribonucleic acid-155 is a direct target of Meis1, but not a driver in acute myeloid leukemia. Haematologica 103(2):246–255
Gao L, Sun J, Liu F, Zhang H, Ma Y (2016) Higher expression levels of the HOXA9 gene, closely associated with MLL-PTD and EZH2 mutations, predict inferior outcome in acute myeloid leukemia. Onco Targets Ther 9:711–722
Kandel S, Ahmedy I, Tayel S, Mohamed A (2021) Role of homeobox-A9 gene expression in patients with de novo acute myeloid leukemia. Menoufia Med J 34:291–296
Arbor A (2017) Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Curr Opin Hematol 23:354–361
Ismail M, Moneer M (2013) Does HOXA9 gene expression in egyptian chronic myelogenous leukemia patients affect disease progression? A retrospective cohort study. Turk J Haem 30(4):359–365
Rozovskaia T, Feinstein E, Mor O, Foa R, Blechman J, Nakamura T et al (2001) Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4:11) abnormality. Oncogene 20(7):874–878
Loftus JP, Yahiaoui A, Brown PA, Niswander LM, Bagashev A, Wang M et al (2021) Combinatorial efficacy of entospletinib and chemotherapy in patient-derived xenograft models of infant acute lymphoblastic leukemia. Haematologica 106(4):1067–1078
Chen SL, Qin ZY, Hu F, Wang Y, Dai YJ, Liang Y (2019) The role of the HOXA gene family in acute myeloid leukemia. Genes (Basel) 10(8):621
Li Z, Huang H, Li Y, Jiang X, Chen P, Arnovitz S et al (2012) Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood 119(10):2314–2324
Ahmedy I, Tayel S (2021) Prognostic impact of homeobox and PR domain-containing protein 16 genes expressions in patients with acute myeloid leukemia. Gene Rep 25:101425
Meng L, Tian Z, Wang J, Liu X, Zhang W, Hu M et al (2021) Effect of myeloid ecotropic viral integration site (MEIS) family genes on tumor microenvironment remodeling and its potential therapeutic effect. Transl Androl Urol 10(2):594–608
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
A.M.A., F.M.T., H.M.K., and N.M.H. put the idea, designed the study, and supervised the work. M.S.A. shared the molecular work and performed the statistical analysis. H.A.A. collected the data, performed the molecular work, and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Benha University research ethics committee, and the included patients gave the consent to participate.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary table 1. ROC curve analysis. Supplementary table 2. Cumulative OS and DFS.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdelrahman, A.M.N., Tolba, F.M., Kamal, H.M. et al. Evaluation of the HOXA9 and MEIS1 genes as a potential biomarker in adult acute myeloid leukemia. Egypt J Med Hum Genet 24, 11 (2023). https://doi.org/10.1186/s43042-023-00391-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-023-00391-4