
Abstract
Objective
This study was conducted to accomplish a better insight into the impact of single nucleotide polymorphisms (SNPs) of nicotinic acetylcholine receptors (nAChR) at the risk of Alzheimer’s disease (AD) and their possible pathogenicity.
Methods
We carried out a systemic review of accessible studies. The case–control studies were assessed by an electronic search of international and local databases to identify relevant studies on SNPs relating to nAChR genes in AD. Two reviewers evaluated the inclusion/exclusion criteria, summarized, and analyzed the extracted data. We used odds ratios (ORs) with 95% confidence intervals (CIs) for reporting our data. Online databases were checked for possible pathogenicity of statistically significant SNPs. Also, online databases, including NCBI, NIH, ClinVar, RegulomeDB, and Ensemble, were used to analyze and identify structure and function, DNA features, and flank sequencing in SNPs.
Results
Among all collected SNPs, rs4779978 and rs1827294 on CHRNA7, rs1044394 on CHRNA4, and rs1127314 on CHRNB2 showed statistically significant between AD cases and controls.
Conclusions
Some SNPs from the reviewed reports show evidence supporting their possible involvement in AD pathology. However, more comprehensive studies are necessary to identify the exact correlation and their role on the pathogenicity of disease.
Similar content being viewed by others


Introduction
The most common form of dementia is Alzheimer’s disease (AD), which is pathologically characterized by the loss of extensive neuronal cells, especially in the neocortex and hippocampus, and the accumulation of senile plaques, synaptic loss, oxidative stress, and inflammation [1,2,3]. A selective reduction of cholinergic neurotransmission resulting from the loss of cholinergic neurons and nicotinic acetylcholine receptors (nAChR) represents the critical neurochemical changes during AD [4].
The neuronal nAChR is a family of ligand-gated cationic channels found throughout the central nervous system (CNS), involved in many diseases, including AD, schizophrenia, and Parkinson’s disease [5, 6]. These receptors are pentameric structures formed from a combination of 12 potential subunits (α2–α10 and β2–β4) to produce many ranges of subtypes, each of them fulfilling different functions [7,8,9]. There are only 11 nAChR subunits in the human CNS (α8 nAChR is absent from mammalian genomes) [10]. α7 nAChRs are made of five α7 subunits and encoded by the CHRNA7 gene [11]. They are expressed throughout the CNS with high levels in the regions related to memory functions and involved in the processing of sensory information, such as the hippocampus [5].
It has been displayed that beta-amyloid (Aβ) and α7 nAChR interaction change Ca2+ homeostasis and acetylcholine release, thereby modulating neuronal physiological functions implicated in memory processes. Chronic inhibition of cholinergic signaling by Aβ contributes to the cognitive deficits associated with AD [4]. Reports have described decreases in α7 nAChR expression in the hippocampus and temporal cortex of the AD brain [12]. Decreased levels of α4 and β2 nAChR subunits have also been observed in the aged and AD brains [6].
Single nucleotide polymorphisms (SNPs) usually occur within a wild-type DNA sequence [1] and may be associated with essential signals of the presence and severity of an illness. Specific SNPs in genes can detect various diseases, including metabolic disorders, childhood leukemia, and neurodegenerative diseases [13,14,15]. Moreover, several online bioinformatics databases evaluate the role of SNPs related to selective genes in AD disease. In the same vein, studies have used a series of bioinformatics methods to identify key genes and potential mechanisms related to AD [16, 17].
In the CNS, α4β2 and α7 receptors are the most abundant nAChRs, directly correlated with AD pathophysiology. To identify the possible associations of polymorphisms of CHRNA7, CHRNA4, and CHRNB2 genes with AD and recognize their possible pathogenicity, we performed a systematic review on available literature and analysis in the related databases.
Methods
Search strategy
The electronic databases, including PubMed, Scopus, and Google Scholar, were searched. These keywords were used to identify all case–control studies on mutations related to nAChR genes in AD: (Alzheimer’s disease OR Alzheimer OR AD) AND (nicotinic acetylcholine receptor OR nAChR) AND (SNP OR single nucleotide polymorphism) AND (CHRNA7 OR CHRNB2 OR CHRNA4). Also, we searched www.alzgene.org to confirm and double-check our findings. The search was restricted to English language but not a year.
Inclusion and exclusion criteria
We included all case–control studies which evaluated CHRNA7, CHRNB2, and CHRNA4-related SNPs in AD patients. All acquired papers from the mentioned databases were imported into Endnote X8 software, and duplicates were removed. Two investigators independently screened the title and abstract to delete irrelevant articles to our objective. Disputes among selected studies between reviewers were resolved by discussion or by a senior researcher's comments. The primary outcome for this study was the identification of SNPs, and the secondary outcome was the differences of identified SNPs between case and control groups. The exclusion criteria for studies were defined as follows: (I) duplicate, review, conference, and irrelevant papers; (II) studies of familial AD, early-onset AD, and mild cognitive impairment; (III) animal or cell line studies, and (IV) irrelevant studies to nAChR genes.
Data extraction
The raw data were obtained from each publication. Two independent authors recorded the extracted data in specific forms. The following information was extracted: author, publication date, number of cases or controls, sex, SNP, genotype, or allele mutation.
From the National Center for Biotechnology Information (NCBI) dbSNP database, we extracted the complementary information of SNPs, including ID, position, and clinical significance.
Methodological quality of studies
The JBI criteria appraisal checklist assessed selected articles for case–control studies, and their results were reported as a percentage based on evaluating the questions according to JBI criteria.
Statistics
The strength of association between identified SNPs and AD was measured by odds ratios (ORs) with 95% confidence intervals (CIs). Results for reported SNPs were not subjected to the meta-analysis due to the lack of sufficient data. However, OR, CI, and p-value for each study were calculated with MedCalc software (Version 19.1, Belgium).
Database analysis
We also used online databases and webservers to determine the function and structure of all selected SNPs on the selective genes. The SNPs collected from the literature were examined in more details through the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov), National library of medicine (NIH) (https://pubmed.ncbi.nlm.nih.gov), and RegulomeDB (https://regulomedb.org). Furthermore, ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) helps to analyze human genetic variations along with annotation of variant disease associations. We extracted all information about SNPs, including mutated alleles, chromosomes, genes, functional consequences, mutation locations, and frequencies related to the pathogenic and non-pathogenic mutant alleles. Also, in the Ensemble site (http://www.ensembl.org/index.html), we evaluated the sequences of the selected genes.
Results
General study characteristics
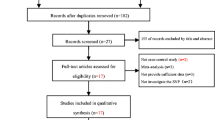
We collected 2090 articles from the above databases, and 6 articles were identified by reviewing the references. Of these, 1881 articles (77%) were deleted based on title and abstract, and 215 (50%) remained. In the next step, 187 articles were eliminated due to irrelevant full text. The full text of the remaining 28 (30%) articles were screened accurately, and 16 articles were removed because the SNPs on the selective gene were not reported. Finally, 12 articles (15%) matched our inclusion criteria, and related SNP to nAChR genes remained for future reviews (Fig. 1). In the screened papers, 60, 30, and 10% of studies were focused on α7, α4, and β2, respectively.

PRISMA flowchart and algorithm for included studies
Study quality
The JBI criteria appraisal checklist for case–control studies was used to evaluate the quality of the studies. This checklist includes ten questions and evaluates the articles through the following items: criteria for data collection, inclusion criteria, matching between case–control, criteria for case–control selection, how to evaluate studies, how to deal with confounding factors, exclusion criteria, output evaluation time, and statistical analysis methods. The options used in this checklist were Yes, No, Unclear, and not applicable. The quality assessment showed that the JBI criteria appraisal checklist for the case–control score of each study was not less than 65%. Among them, there were 8 papers categorized above 90%. The basic specifications and methodological quality of the included studies are shown in Fig. 2.

Evaluation of the selected articles via the JBI criteria appraisal checklist. All studies with a high quality of 65–100% were reported. JBI: Joanna Briggs Institute
Significant SNPs
According to the results, frequency of rs4779978 and rs1827294 SNPs on CHRNA7 gene were statistically different between cases and controls, [OR] = 0.59, 95% CI = (0.39–0.90), p = 0.016 and [OR] = 1.70, 95% CI = (1.15–2.53), p = 0.008, respectively. We also studied CHRNA4 SNPs connection with the risk of AD. rs1044394 showed statistically significant [OR] = 4.35, 95% CI = (2.07–9.14), p = 0.000 for genotype C/C and [OR] = 0.28, 95% CI = (0.12–0.65), p = 0.003 for genotype C/T. On CHRNB2 gene, the rs1127314 SNP showed the significant difference in frequency between cases and controls, [OR] = 0.70, 95% CI = (0.50–0.98), p = 0.043. Besides, no approved clinical evidence was reported to identify whether these SNPs have drastic roles in the prevalence or the pathogenicity of AD. Table 1 illustrates the detailed features of the included studies.
Database analysis outcomes
The study consisted of 2 intronic SNPs regions on the CHRNA7 gene, one SNP within missense non-coding transcript variant synonymous reign on the CHRNA4 gene and one SNP within the 3 untranslated reigns on the CHRNB2 gene.
In the Ensemble site, we evaluated all the sequences of selected genes, CHRNA7, CHRNB2, and CHRNA4, under the record numbers of NM_000746.6, NM_000744.7, and NM_000748.3, respectively, and finding flanking sequences of SNPs (Table 2).
We used the RegulomeDB to determine regulatory DNA elements, including regions of DNase hypersensitivity and promoter regions. Three SNPs are categorized in rank 4 (TF binding + DNase peak) and 1 SNPs in rank 5 (TF binding or DNase peak). The results are shown in Table 3.
Discussion
The impaired function of several proteins in CNS seems to serve a molecular basis of AD [18]. We reviewed the genetic polymorphisms of the CHRNA4, 7, and CHRNB2 genes, frequently reported in AD pathogenesis. Analysis of the allelic differences of rs477978 and rs1827294 SNPs of the CHRNA7 gene has shown a significant difference between the AD and controls [6]. Carson et al. have reported that rs1827294 tend to have an association with the risk of AD [1]. According to the study of Weng et al., statistical analysis of the allelic differences of rs1044394 and rs1127314 SNPs of the CHRNA4 and CHRNB2 genes has a significant difference between AD and control groups [19].
The decreased levels of α4 nAChRs in the neocortex of the AD patients may result from the reduction of neuron numbers in the area [2]. Moreover, previous studies have shown that genetic polymorphisms of the CHRNA4 gene are linked to the pathogenesis of AD [6, 20]. Gelfman et al. employed Transcript-inferred Pathogenicity (TraP) score to assess pathogenic non-coding variants in genic areas. This approach can evaluate synonymous and intronic variants when searching for disease risk. The existence of high TraP scores in cases is probably correlated with disease risk [21]. Another recent study by Spielmann et al. identified a typical Parkinson’s disease-associated risk variant in a non-coding distal enhancer element, regulating the expression of a-synuclein [22]. Recently, Jacob et al., for the first time, reported a deleterious synonymous variant in the final nucleotide of an exon, which results in exon skipping [23]. Therefore, it seems that synonymous and intronic variants may be novel risk factors for neurodegenerative diseases.
The SNPs are normally situated either synonymous or with intronic or untranslated regions, lacking any obvious direct functional effect. However, they may affect protein production at the transcriptional and or translational levels. Yang et al. reported variants that cause amino-acid changes, among which rs56210055 (p. A312T) and rs55633891 (p. A315V) may affect the permeability of the ion channel or nAChR stability, which can additionally alternate the nAChR function and manipulate downstream signaling [24]. Li et al. evaluated whether retained introns could be translated into proteins or not. They found that the expression levels of intron-retained transcripts of 12 AD-associated genes were increased due to the deficiency of splicing machinery in AD cases. Their findings present an initial attempt to exploit the association of intron-retained with AD [25].
We also used the NCBI site to examine the clinical significance of the selected SNPs. We found that two SNPs regions of the CHRNA7 were an intronic SNP, which may affect the expression of α7 nAChR via a pre-mRNA splicing mechanism [1, 26], indicating that the rs477978 and rs1827294 may modulate genetic risk by changing RNA splicing or stability as well as the subsequent protein production [3]. According to synthesized results, there was one SNP within missense non-coding transcript variant synonymous reign of the CHRNA4 gene and one SNP within the 3 untranslated reigns of the CHRNB2 gene. These SNPs might affect the promoter and hence the transcription mechanism, cause mismatched folding of protein, or cause the production of shorter proteins. Though these SNPs are clinically benign, further extensive in vitro, in vivo, and clinical studies are necessary to find their exact functions.
Bioinformatics studies have utilized NCBI and NIH to obtain related information such as genomes, protein sequences, and structures [27]. Chowdhury et al. have collected two different gene expression microarray datasets of AD from the Gene Expression Omnibus of NCBI [28]. Mohammed et al. have utilized the RegulomeDB database to know and predict regulatory DNA elements, including binding sites of the transcription factors and promoter regions [29]. In addition to NCBI and NIH, we used ClinVar to collect the necessary information to recognize binding sites of transcription factors, promoter regions, folding protein, and splicing process.
Conclusion
In general, SNPs are crucial for the pathophysiology of disorders. Our findings revealed a weak correlation between polymorphisms in dominant neuronal nAChR genes and the pathogenesis of AD. However, further studies with complementary and detailed evaluations should be performed to determine their exact clinical effects.
Availability of data and materials
This item is not applicable to this study.
References
Carson R, Craig D, McGuinness B, Johnston JA, O’Neill FA, Passmore AP et al (2008) Alpha7 nicotinic acetylcholine receptor gene and reduced risk of Alzheimer’s disease. J Med Genet 45(4):244–248
Cook LJ, Ho LW, Taylor AE, Brayne C, Evans JG, Xuereb J et al (2004) Candidate gene association studies of the alpha 4 (CHRNA4) and beta 2 (CHRNB2) neuronal nicotinic acetylcholine receptor subunit genes in Alzheimer’s disease. Neurosci Lett 358(2):142–146
Sumirtanurdin R, Thalib AY, Cantona K, Abdulah R (2019) Effect of genetic polymorphisms on Alzheimer’s disease treatment outcomes: an update. Clin Interv Aging 14:631–642
Guan ZZ, Zhang X, Ravid R, Nordberg A (2000) Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer’s disease. J Neurochem 74(1):237–243
Fehér A, Juhász A, Rimanóczy A, Csibri E, Kálmán J, Janka Z (2009) Association between a genetic variant of the alpha-7 nicotinic acetylcholine receptor subunit and four types of dementia. Dement Geriatr Cogn Disord 28(1):56–62
Kawamata J, Shimohama S (2002) Association of novel and established polymorphisms in neuronal nicotinic acetylcholine receptors with sporadic Alzheimer’s disease. J Alzheimer’s Dis JAD 4(2):71–76
Mihailescu S, Drucker-Colı́n R (2000) Nicotine, brain nicotinic receptors, and neuropsychiatric disorders. Arch Med Res 31(2):131–144
Schaaf CP (2014) Nicotinic acetylcholine receptors in human genetic disease. Genet Med 16(9):649–656
Weiland S, Bertrand D, Leonard S (2000) Neuronal nicotinic acetylcholine receptors: from the gene to the disease. Behav Brain Res 113(1):43–56
Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89:73
Carson R, Craig D, Hart D, Todd S, McGuinness B, Johnston JA et al (2008) Genetic variation in the α7 nicotinic acetylcholine receptor is associated with delusional symptoms in Alzheimer’s disease. NeuroMol Med 10(4):377–384
Hellström-Lindahl E, Mousavi M, Zhang X, Ravid R, Nordberg A (1999) Regional distribution of nicotinic receptor subunit mRNAs in human brain: comparison between Alzheimer and normal brain. Brain Res Mol Brain Res 66(1–2):94–103
Amber S, Zahid S (2018) Data integration for functional annotation of regulatory single nucleotide polymorphisms associated with Alzheimer’s disease susceptibility. Gene 672:115–125
Deng N, Zhou H, Fan H, Yuan Y (2017) Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 8(66):110635
Yang Q, Chen K, Zhang H, Zhang W, Gong C, Zhang Q et al (2019) Correlations between single nucleotide polymorphisms, cognitive dysfunction, and postmortem brain pathology in Alzheimer’s disease among Han Chinese. Neurosci Bull 35(2):193–204
Jiang Z, Tan G, Wang Z (2020) Comprehensive analysis reveals a six-gene signature and associated drugs in alzheimer disease
Quan X, Liang H, Chen Y, Qin Q, Wei Y, Liang Z (2020) Related network and differential expression analyses identify nuclear genes and pathways in the hippocampus of Alzheimer disease. Med Sci Monit Int Med J Exp Clin Res 26:e919311–e919321
Dorszewska J, Florczak J, Różycka A, Jaroszewska-Kolecka J, Trzeciak WH, Kozubski W (2005) Polymorphisms of the CHRNA4 gene encoding the α4 subunit of nicotinic acetylcholine receptor as related to the oxidative DNA damage and the level of apoptotic proteins in lymphocytes of the patients with Alzheimer’s disease. DNA Cell Biol 24(12):786–794
Weng PH, Chen JH, Chen TF, Sun Y, Wen LL, Yip PK et al (2016) CHRNA7 polymorphisms and dementia risk: interactions with apolipoprotein ϵ4 and cigarette smoking. Sci Rep 6:1
Dorszewska J, Florczak J, Rózycka A, Jaroszewska-Kolecka J, Trzeciak WH, Kozubski W (2005) Polymorphisms of the CHRNA4 gene encoding the α4 subunit of nicotinic acetylcholine receptor as related to the oxidative DNA damage and the level of apoptotic proteins in lymphocytes of the patients with Alzheimer’ s disease. DNA Cell Biol 24(12):786–794
Gelfman S, Wang Q, McSweeney KM, Ren Z, La Carpia F, Halvorsen M et al (2017) Annotating pathogenic non-coding variants in genic regions. Nat Commun 8(1):1–11
Spielmann M, Mundlos S (2016) Looking beyond the genes: the role of non-coding variants in human disease. Hum Mol Genet 25(R2):R157–R165
Jacob A, Pasquier J, Carapito R, Auradé F, Molitor A, Froguel P et al (2020) A de novo synonymous variant in EFTUD2 disrupts normal splicing and causes mandibulofacial dysostosis with microcephaly: case report. BMC Med Genet 21(1):1–8
Yang J, Wang S, Yang Z, Hodgkinson CA, Iarikova P, Ma JZ et al (2015) The contribution of rare and common variants in 30 genes to risk nicotine dependence. Mol Psychiatry 20(11):1467–1478
Li HD, Funk CC, McFarland K, Dammer EB, Allen M, Carrasquillo MM et al (2021) Integrative functional genomic analysis of intron retention in human and mouse brain with Alzheimer’s disease. Alzheimer’s Dement 17:984
Parvizpour S, Jomah AF, Razmara J (2020) Structural and functional analysis of mutated human Pyrin B30. 2 domain. Curr Proteomics 17(1):78–85
Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I (2019) GenBank. Nucleic Acids Res 47(D1):D94–D99
Chowdhury UN, Islam MB, Ahmad S, Moni MA (2020) Systems biology and bioinformatics approach to identify gene signatures, pathways and therapeutic targets of Alzheimer’s disease. Inform Med Unlocked 21:100439
Mohammed MY, Khaier MAM (2021) Comprehensive data analysis of single nucleotide polymorphism (Snps) in human FOXP3 gene. Saudi J Biomed Res 6(5):73–84
Acknowledgements
The authors would like thanks to NSRC, TUOMS, for supporting of this research.
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
SSE and SM contributed to study conception and design; JM and MA were involved in data collection; SSE, FF, PP, and SFK contributed to analysis and interpretation of results; and SM and SP prepared the draft and manuscript. All authors reviewed the results and approved the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This item is not applicable to this study.
Consent for publication
The authors provide consent to publish the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohammadi, S., Mahmoudi, J., Farajdokht, F. et al. Polymorphisms of nicotinic acetylcholine receptors in Alzheimer’s disease: a systematic review and data analysis. Egypt J Med Hum Genet 23, 144 (2022). https://doi.org/10.1186/s43042-022-00357-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00357-y