
Abstract
Background
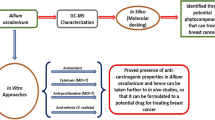
Breast cancer ranks top among newly reported cancer cases and most of the women suffers from breast cancer. Development of target therapy using phytochemicals with minimal side effects is trending in health care research. Phytochemicals targets complex multiple signalling events in cancer and are pleiotropic in nature. Thus, the present study was conducted to check the effectivity of curcumin analogues (Capsaicin, Chlorogenic acid, Ferulic acid, Zingerone, Gingerol) against the receptors that are expressed in breast cancer cells and prove its ethno-medicinal value by using bioinformatic tools and softwares like PDB, Patch Dock, PubChem, Chimera and My Presto.
Result
Out of the various curcumin analogues studied, Ferulic acid showed best binding affinity with all the breast cancer cell specific receptors (FGF, MMP9, RNRM1, TGF-beta, DHFR, VEGF and aromatase) which was confirmed through the docking studies.
Conclusion
The current work was a preliminary step towards screening suitable drug candidate against breast cancer using in silico methods. This information can be used further to carry out in vivo studies using selected natural analogues of curcumin as a suitable drug candidate against breast cancer saving time and cost.
Similar content being viewed by others


Background
Cancer is the world's second leading cause of death. In the last 5 years, breast cancer had been diagnosed in 7.8 million women out of which Indian women are having higher breast cancer malignancy and less survival rate at the stage 3 or 4 of breast cancer [25]. Variety of therapies such as surgery, radiation, or chemo are used to treat cancer which will not always result in tumour reduction but can cause undesirable side effects such as nausea, drug resistance, cytotoxicity, relapse and lowering life quality, etc.
Target therapy is nowadays a promising method for developing anti-cancer drug where major proteins and receptors of cancer cells are promising targets and have been approved by the US Food and Drug Administration (FDA) [23]. The receptors such as Fibroblast Growth Factor (FGF), Matrix metallo proteinase 9 (MMP9), Ribonucleotide reductase subunit M1 (RNRM1), Transforming Growth Factor beta (TGF-β), Dihydrofolate Reductase (DHFR), Vascular Endothelial Growth Factor (VEGF) and aromatase are selected for the present study because all these receptors are getting expressed during breast cancer cell formation FGF plays roles in inflammation, Single nucleotide polymorphism (SNP), angiogenesis, proliferation etc. [33]. VEGF is involved in angiogenesis of cancer cells, while the epithelial mesenchymal transition is aided by TGF-β [13, 20]. Presences of aromatase have an unavoidable role in breast cancer development mainly in post-menopausal women [12]. DHFR and RNRm1 are essential for nucleotide synthesis in cancerous cells [15]. In addition to Matrix remodelling, and angiogenesis, cancer invasion and progression are aided by MMP9 [26]
Phytochemicals or their analogues are the potential source of medicinal compounds with less side effects [8, 28]. Phytochemicals from plants that are used as drug either directly or with slight modifications are known as “green drug” (E.g.: Curcumin, resveratrol, paclitaxel, and vincristine) [38]. Most of the G8 countries are involved in the green drugs research for cancer by spending ~ 1–3% of their Gross Domestic Product (GDP) [4].
Curcumin is a well-researched anti-cancer substance having a broad range of pharmacological and biological effects found in the rhizome of Curcuma longa L., a member of the zingiberaceae family [31]. Breast carcinoma, colon carcinoma, renal & basal cell carcinoma, T cell & acute myelogenous leukaemia, B cell lymphoma, melanoma, and prostate carcinoma are among the cancers that curcumin inhibits [19]. Bioavailability of curcumin is a major concern coming to clinical application. In order to overcome this, curcumin analogues can be used in cancer studies [27]. Curcumin analogues with higher bioavailability and wide spectrum pharmacological activity such as Capsaicin, Chlorogenic acid, Ferulic acid, Zingerone and Gingerol are selected here. Hence, in the present study, using bioinformatics tools, curcumin analogues were founded out and its effectiveness against breast cancer receptors and their ethno-medicinal usefulness were studied.
Unlike the studious and expensive wet laboratory experiments, the structure-based analysis is now easy with in silico technologies [10].
Methods
Selection and retrieval of receptors
Literature works were searched for potential anti-cancer targets [24]. Three-dimensional structure (in PDB format) of each of the seven selected receptors was retrieved from the protein data bank (PDB) using respective PDB ID (Aromatase: 3EQM, DHFR: 1DRF, FGF: 1QQL, MMP9: 1L6J, RNRM1: 4X3V, TGF-beta: 1TGK, VEGF: 1VPF).
Retrieval of ligands and screening
Pharmacologically active natural analogues of curcumin and established anti-cancer drugs which are used as controls were identified from previous literature works [14]. Structure of all the analogues was retrieved from PubChem compound database using PubChem ID (Capsaicin: 1,548,943, Chlorogenic acid: 1,794,427, Ferulic acid: 445,858, Gingerol: 442,793 and Zingerone: 31,211). 3D structure of control drugs used for the study was downloaded from Drug bank database using respective Drug bank ID (Pentosan polysulfate: DB00686, Proguanil: DB01131, Axitinib: DB06626, Galunisertib: DB11911, Marimastat: DB00786, Triapine: DB11940 and Testolactone: DB00894).
Molecular docking
PatchDock [30] was used which is based on geometry-based molecular docking algorithm. The potential complexes are sorted by shape complementarity criteria. Number of hydrogen bonds formed between the ligand and the receptor were determined by using chimera tool and the delta G values calculated using my Presto5 software [11]: [21]. Clustering RMSD—a positive number that specifies the radius of the RMSD clustering in angstroms. This value is used in the final clustering stage of the algorithm. It ensures that the distance between any two output solutions will be at least the specified clustering RMSD value. The default value for this parameter is 4 A°. Each receptor was docked against respective control drugs and all the five curcumin analogues used for the study.
Interactions of the receptor with ligand were analysed after molecular docking using chimera and my Presto5 software. Position of hydrogen bond formed between the ligand and receptor was identified using chimera software and marked using default options. Delta G value of the docked protein–ligand complex was analysed using my Presto Portal. Maximum negative delta G indicates higher binding affinity.
Determination of binding site
Active site or catalytic sites of each receptor were identified from the previous literature works [35]: [18]. This was done to find out the magnitude of interaction of receptor and ligand (by comparing the position of binding). Best were selected based on the position of H bond and free energy value of docked complex. In our study, during docking, the cofactors for the receptors DHFR, i.e. folate and for Aromatase, i.e. androstenedione were included. All other receptors do not have any cofactors.
Result
The work was done to screen important curcumin analogues depending on their hydrogen bond formation and delta G values. The best analogue will be used further for ADMET analysis and wet laboratory studies. The results obtained after patch dock analysis were studied with respect to following parameters: GSC score, AI area. The best were analysed using chimera to understand the hydrogen bond, and using my Presto 5 software, binding energy was predicted.
The following results were obtained for each receptor interaction with curcumin analogues and the control drug against each receptor.
Hydrogen bond formation and Delta G value obtained from the interaction of control with the receptor was taken as a standard to understand the interaction of other curcumin analogues (Fig. 1).

Docked pictures of curcumin analogues and pentosan polysulfate (control) against FGF receptor. (Ligand represents the blue sticks, hydrogen bond interaction is represented in blue line, and receptor is represented in solid white ribbon form)
Control drug (pentosan polyslfate) formed 4 hydrogen bonds with FGF receptor. Out of the five analogues, chlorogenic acid formed three hydrogen bonds and ferulic acid formed two hydrogen bonds and Zingerone formed one hydrogen bond with the FGF receptor. Gingerol didn’t form any H bond with the FGF receptor. Position of H bonds; LYS 194.A of ferulic acid and SER 113.A of chlorogenic acid are same as that of control. During the interaction with FGF receptor, the control liberates − 2.15 kcal/mol energy (Table.1). Among analogues, the highest energy is liberated by chlorogenic acid (− 1.74 kcal/mol), followed by Zingerone (− 1.08 kcal/mol) and ferulic acid (− 0.99 kcal/mol).
Marimastat (control) is capable of forming 6 H bonds with receptor MMP-9 (with A chain) (Fig. 2).

Docked pictures of curcumin analogues and marimastat (control) against MMP-9 receptor. (Ligand represents the blue sticks, hydrogen bond interaction is represented in blue line, and receptor is represented in solid white ribbon form)
H bond formed by ferulic acid is at GLU 274.A; the nearest amino acid of SER.A 273 (formed by control). Chlorogenic acid formed three bonds with the receptor at Thr 245.A, SER 242.A. Gingerol formed one hydrogen bond at position Thr 426.A. On comparing the delta G values, control drug liberated − 3.57 kcal/mol, which is slightly lesser compared to the energy liberated by Ferulic acid (− 3.68 kcal/mol) (Table.1). Chlorogenic acid liberated − 3.02 kcal/mol energy. Also, gingerol and zingerone showed almost equal amount of delta G (− 1.9 kcal/mol). Least value was shown by capsaicin (Fig. 3).

Docked pictures of curcumin analogues and Triapine (control) against RNRM1 receptor. (Ligand represents the blue sticks, hydrogen bond interaction is represented in blue line, and receptor is represented in solid white ribbon form)
Triapine (control) formed 4 H bonds at positions at ASP 287.A, GLN 288.A,TYR 285.A and SER 269.B with the receptor RNRM1. Ferulic acid formed two H bonds at positions SER 269.B and TYR 285.A same as the position of control. Single H bonds are formed by both gingerol and zingerone with the RNRM1 receptor. Chlorogenic acid formed 3 H bonds at different position ASN 207.B, THR 210.B, ASN 291.B. Ferulic acid liberated − 2.81 kcal/mol energy which is higher than the delta G of all other ligands studied except the control (− 3.27 kcal/mol) on interaction with the RNRM1 receptor (Table.1).
Galunisertib (control) formed a single H bond with the TGF- β receptor at position ASN 42.A and 1 H bond of ferulic acid also formed a single H bond at the same position of the receptor (Fig. 4). Chlorogenic acid formed a single H bond with the receptor at TYR 40.A. The remaining 3 ligands gingerol, capsaicin and zingerone didn’t form any bond with the receptor. The curcumin analogues liberated less energy in a range of − 1.19–0.09 kcal/mol (Table.1) when docked with TGF-β receptor. Capsaicin has a positive value of delta G, i.e. 0.48 kcal/mol. Control had − 4.5 kcal/mol. In this case, ferulic acid showed better delta G values of − 1.19 kcal/mol.

Docked pictures of curcumin analogues and galunisertib (control) against TGF-β receptor. (Ligand represents the blue sticks, hydrogen bond interaction is represented in blue line, and receptor is represented in solid white ribbon form)
Interacting with the VEGF receptor, control (Axitinib), ferulic acid, as well as chlorogenic acid, is forming bonds at same position that is CYS 61.A (Fig. 5). Control forms 2 H bond with the receptor, chlorogenic acid formed 3 H bonds and the remaining analogues formed single H bond with the receptor. Here, the control Axitinib and Chlorogenic acid have almost similar range of delta G value, i.e. − 2.69 kcal/mol and − 2.01 kcal/mol, respectively (Table.1). Ferulic acid has a delta G value of − 1.54 kcal/mol. All the remaining analogues liberated a delta G greater than − 1 kcal/mol.

Docked pictures of curcumin analogues and axitinib (control) against VEGF receptor. (Ligand represents the blue sticks, hydrogen bond interaction is represented in blue line, and receptor is represented in solid white ribbon form)
Two H bonds at ASP 22.A were formed by the control with the receptor DHFR(Fig. 6). Ferulic acid has also formed single H bond with the receptor at the same position (ASP 22.A) same as control. Zingerone and capsaicin are not forming H bonds with the receptor. Chlorogenic acid formed four hydrogen bonds with the receptor at SER 4.A, LYS 133.A, MET 58.A and ARG 37.A. Comparing the delta G values, Ferulic acid has more negative delta G (− 1.68 kcal/mol) followed by chlorogenic acid (− 1.54 kcal/mol) (Table.1). The Control liberated the maximum delta G (− 2.73 kcal/mol) among the studied compounds.

Docked pictures of curcumin analogues and proguanil (control) against human DHFR receptor. (Ligand represents the blue sticks, hydrogen bond interaction is represented in blue line, and receptor is represented in solid white ribbon form)
Control (testolactone) was found to form two H bonds with aromatase receptor at ASSN 136.A and SER 90.A. Similar bonds are formed by ferulic acid at SER 90.A and ASN 136.A with the receptor (Fig. 7). Capsaicin and zingerone didn’t form any bonds. Chlorogenic acid formed single bond at TYR 361.A with the receptor. Gingerol formed bond with the receptor at position VAL 422.A. Control liberated − 4.06 kcal/mol energy which was the highest compared to other analogues (Table.1). Among the five analogues studied, a significant value of delta G was liberated by ferulic acid (2.46 kcal/mol) followed by chlorogenic acid (− 2.12 kcal/mol), respectively.

Docked pictures of curcumin analogues and testolactone (control) against human aromatase receptor. (Ligand represents the blue sticks, hydrogen bond interaction is represented in blue line, receptor is represented in solid white ribbon form)
Binding affinity of ferulic acid only in case of MMP9 receptor is more compared to the control but in all other cases, the binding affinity of ferulic acid is less than the control. And on comparing with other curcumin analogues, the ferulic acid showed good binding affinity to all the receptors.
Discussion
Being the most prominent cancer among women, with high metastasis and less rate of survival, it is the need of the hour to develop potential drugs against breast cancer [37, 42]. From many reports, it has proven that curcumin analogues have anti-cancer activity and inhibit tumour growth by inducing apoptosis, preventing/reducing recurrence and also found to regulate intracellular components/pathways that control tumour growth [1, 7, 29].
From the five analogues of the curcumin studied, ferulic acid and chlorogenic acid showed promising results, based on the number of hydrogen bond formation and delta G values.
Hydrogen bonding is an exchange reaction whereby the hydrogen bond donors and acceptors of the free protein and ligand break their hydrogen bonds with water and form new ones in the protein–ligand complex [16].
The hydrogen bonds formed by the ferulic acid with all the receptor have screened ferulic acid as the best curcumin analogue compared with the other analysed analogues. In case of the docking study between ferulic acid and FGF receptor, the amino acid residues involved in hydrogen bond formation were Lysine, Serine, Glutamate, Tyrosine, Asparagine, Cysteine and Asparatic acid. Except lysine, all other amino acids are non-essential. All these amino acids are capable of forming hydrogen bonds. Other types of interaction also occur like ionic bond formation due to the presence of asparatic acid and disulphide bond formed by cysteine residues. These residues do not match the amino acid residues which are present in the active site of all the seven receptors [17, 32, 34, 36, 40, 41], 2.
The reason for the non-matching of residues could be due to non-availability of X-ray structure of this receptors with natural substrate or other inhibitors. The presence of water molecules can also be a reason for the difference in the amino acid residues involved in the bond formation.
The water molecules were considered during the docking studies. Water has an important role in ligand binding thermodynamics [6], even in the environment of a lipophilic binding cavity [3]. Moreover, water related H-bonding networks have a significant influence in the structure–activity relationship [5] and optimizing the ligand taking into account the surrounding water network may result in enhanced binding affinity and prolonged residence time [22].
Compound that liberates highest negative delta G (or highest amount of free energy) is ideal for drug designing [9]. Binding free energy gives us the idea of strength and affinity of the interaction between the ligand and the receptor. The greater the binding free energy is, the weaker the interaction is and vice versa. Ligand which displayed least binding energy showed best affinity among the test molecules. Binding affinity determines that a high concentration of weakly interacting partners cannot replace the effect of a low concentration of the specific partner interacting with high affinity [39].
All these seven receptors get expressed during breast cancer cell development. Hence, ferulic acid can be a suitable drug candidate against breast cancer studies.
Conclusion
From the current study, it can be concluded that as Ferulic acid has better delta G value and position of binding with the active site of receptors, it is a better inhibitor for all the seven receptors studied. Further ADMET analysis is essential to study the pharmacokinetic properties of these compounds. As this is a preliminary stage of screening, further in vitro and in vivo studies are recommended to reinforce the therapeutic value.
Availability of data and materials
The data can be shared.
Abbreviations
- FGF:
-
Fibroblast growth factor
- MMP9:
-
Matrix metallo proteinase 9
- RNRM1:
-
Ribonucleotide reductase subunit M1
- TGF-β:
-
Transforming growth factor beta
- DHFR:
-
Dihydrofolate reductase
- VEGF:
-
Vascular endothelial growth factor
- ADMET:
-
Absorption, distribution, metabolism, elimination, toxicity
- H bond:
-
Hydrogen bond
References
Allegra A, Innao V, Russo S, Gerace D, Alonci A, Musolino C et al (2017) Anticancer activity of curcumin and its analogues: preclinical and clinical studies. Cancer Invest 35(1):1–22
Baroi S, Saha A, Bachar R, Bachar SC et al (2020) Cannabinoid as potential aromatase inhibitor through molecular modeling and screening for anti-cancer activity. Dhaka Univ J Pharm Sci 19(1):47–58
Baron R, Setny P (2010) McCammon A (2010) Water in cavity—ligand recognition. J Am Chem Soc 132:12091–12097
Basu T, Mallik A, Mandal N et al (2017) Evolving importance of anticancer research using herbal medicine: a scientometric analysis. Scientometrics 110(3):1375–1396
Biela A, Nasief NN, Betz M, Heine A, Hangauer D (2013) Klebe G (2013) Dissecting the hydrophobic effect on the molecular level: The role of water, enthalpy, and entropy in ligand binding to thermolysin. Angew Chem Int Ed Engl 52:1822–1828
Breiten B, Lockett MR, Sherman W, Fujit S, Al-Sayah M, Lange H, Bowers CM, Heroux A, Krilov G (2013) Whitesides GM (2013) water networks contribute to enthalpy/entropy compensation in protein—ligand binding. J Am Chem Soc 135:15579–15584
Chandru H, Sharada AC, Bettadaiah BK, Kumar CA, Rangappa KS, Jayashree K et al (2007) In vivo growth inhibitory and anti-angiogenic effects of synthetic novel dienone cyclopropoxy curcumin analogues on mouse Ehrlich ascites tumor. Bioorg Med Chem 15(24):7696–7703
Chaudhuri D, Ghate NB, Panja S, Das A, Mandal N et al (2015) Wild edible fruit of Prunus nepalensis Ser. (Steud), a potential source of antioxidants, ameliorates iron overload-induced hepatotoxicity and liver fibrosis in mice. PLoS ONE 10(12):e0144280
Cournia Z, Allen B, Sherman W (2017) Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J Chem Inf Model 57(12):2911–2937
de Souza Neto LR, Moreira-Filho JT, Neves BJ, Maidana RLBR, Guimarães ACR, Furnham N, Silva FP Jr et al (2020) In silico strategies to support fragment-to-lead optimization in drug discovery. Front Chem 8:93
Eric P, Thomas DG, Conred CH, Greg CD et al (2004) UCSI chimera- visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612
Gerard C, Brown KA (2018) Obesity and breast cancer–Role of estrogens and the molecular underpinnings of aromatase regulation in breast adipose tissue. Mol Cell Endocrinol 466:15–30
Gulei D, Mehterov N, Ling H, Stanta G, Braicu C, Berindan-Neagoe I et al (1861) (2017) The “good-cop bad-cop” TGF-beta role in breast cancer modulated by non-coding RNAs. Biochim et Biophys Acta (BBA)-Gen Subj 7:1661–1675
Gupta AP, Khan S, Manzoor MM, Yadav AK, Sharma G, Anand R, Gupta S (2017) Anticancer curcumin: natural analogues and structureactivity relationship. Stud Nat Prod Chem. https://doi.org/10.1016/B978-0-444-63929-5.00010-3
He L, Shi X, Liu Z, Ren X, Zhang C, Yang Z, Li Z et al (2019) Roles of EAAT1, DHFR, and Fetuin-A in the pathogenesis, progression and prognosis of chondrosarcoma. Onco Targets Ther 12:8411
Zhao H, Huang D (2011) Hydrogen bonding penalty upon ligand binding. PLoS ONE. https://doi.org/10.1371/journal.pone.0019923
Iheagwam FN, Ogunlana OO, Ogunlana OE, Isewon I, Oyelade J et al (2019) Potential anti-cancer flavonoids isolated from Caesalpinia bonduc young twigs and leaves: molecular docking and in silico studies. Bioinform Biol Insights 13:1177932218821371
Jian L, Wen Yu, Gao L, Gao L, He F, Zhou J, Wang J, Dai R, Chen X, Kang Di, Lihong Hu (2020) Design, synthesis and biological evaluation of novel 1H–1, 2, 4-triazole, benzothiazole and indazole-based derivatives as potent FGFR1 inhibitors via fragment-based virtual screening. J Eenzyme Inhib Med Chem 35(1):72–84
Jiang JH, Deng P (2019) Discovery of new inhibitors of transforming growth factor-beta type 1 receptor by utilizing docking and structure-activity relationship analysis. Int J Mol Sci 20(17):4090
Kawczyk-Krupka A, Kwiatek B, Czuba ZP, Mertas A, Latos W, Verwanger T, Sieroń A et al (2018) Secretion of the angiogenic factor VEGF after photodynamic therapy with ALA under hypoxia-like conditions in colon cancer cells. Photodiagn Photodyn Ther 21:16–18
Kota K, Benson M, Kota G, Bhaskar D, Junichi H, Ikuo F, Tadaaki M et al (2016) myPresto/omegagene: a GPU-accelerated molecular dynamics simulator tailored for enhanced conformational sampling methods with a non-Ewald electrostatic scheme. Biophys Physicobiol 13:209–216
Krimmer SG, Cramer J, Betz M, Fridh V, Karlsson R, Heine A, Klebe G (2016) Rational design of thermodynamic and kinetic binding profiles by optimizing surface water networks coating protein-bound ligands. J Med Chem 59:10530–10548
Lee YT, Tan YJ, Oon CE (2018) Molecular targeted therapy: treating cancer with specificity. Eur J Pharmacol 834:188–196
Ballas MS, Chachoua A (2011) Rationale for targeting VEGF, FGF, and PDGF for the treatment of NSCLC. Onco Targets Ther. https://doi.org/10.2147/OTT.S18155
Mathur P, Sathishkumar K, Chaturvedi M, Das P, Sudarshan KL, Santhappan S, Roselind FS et al (2020) ICMR-NCDIR-NCRP Investigator group: cancer statistics, 2020: report from national cancer registry programme, India. JCO Glob Oncol 6:1063–1075
Nazir SU, Kumar R, Singh A, Khan A, Tanwar P, Tripathi R, Hussain S et al (2019) Breast cancer invasion and progression by MMP-9 through Ets-1 transcription factor. Gene. https://doi.org/10.1016/j.gene.2019.143952
Padhye S, Banerjee S, Chavan D, Pandye S, Swamy KV, Ali S, Sarkar FH et al (2009) Fluorocurcumins as cyclooxygenase-2 inhibitor: molecular docking, pharmacokinetics and tissue distribution in mice. Pharm Res 26(11):2438–2445
Panja S, Ghate NB, Mandal N et al (2016) A microalga, Euglena tuba induces apoptosis and suppresses metastasis in human lung and breast carcinoma cells through ROS-mediated regulation of MAPKs. Cancer Cell Int 16(1):1–13
Ramasamy TS, Ayob AZ, Myint HHL, Thiagarajah S, Amini F et al (2015) Targeting colorectal cancer stem cells using curcumin and curcumin analogues: insights into the mechanism of the therapeutic efficacy. Cancer Cell Int 15(1):1–15
Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) PatchDock and SymmDock servers for rigid and symmetric docking. Nucleic Acids Res 33:W363–W367. https://doi.org/10.1093/nar/gki481
Soheil ZM, Habsah AK, Pouya H, Hassan T, Sazaly A, Keivan Z (2014) A review on antibacterial, antiviral, and antifungal activity of curcumin. Biologic activity and biotechnological development of natural products 2014. Review Article. https://doi.org/10.1155/2014/186864
Srivastava V, Kumar A, Mishra BN, Siddiqi MI et al (2008) Molecular docking studies on DMDP derivatives as human DHFR inhibitors. Bioinformation 3(4):180
Sun Y, Fan X, Zhang Q, Shi X, Xu G, Zou C et al (2017) Cancer-associated fibroblasts secrete FGF-1 to promote ovarian proliferation, migration, and invasion through the activation of FGF-1/FGFR4 signaling. Tumor Biol 39(7). https://doi.org/10.1177/1010428317712592
Tandon A, Sinha S (2011) Structural insights into the binding of MMP9 inhibitors. Bioinformation 5(8):310–314
Ulviye AC, Betül KC, Begüm NS, Derya OS, Sinem I, Yusuf O, Zafer AK (2020) Synthesis, docking studies and biological activity of new benzimidazole-triazolothiadiazine derivatives as aromatase inhibitor. Molecules 25:1642. https://doi.org/10.3390/molecules25071642
Vadija R, Mustyala KK, Nambigari N, Dulapalli R, Dumpati RK, Ramatenki V, Vuruputuri U et al (2016) Homology modeling and virtual screening studies of FGF-7 protein-a structure-based approach to design new molecules against tumor angiogenesis. J Chem Biol 9(3):69–78
Veronesi U, Zucali R, Luini A (1986) Local control and survival inearly breast cancer: the Milan trial. Int J Radiat Oncol* Biol* Phys 12(5):717–720
Wang T, Gong X, Jiang R, Li H, Du W, Kuang G et al (2016) Ferulic acid inhibits proliferation and promotes apoptosis via blockage of PI3K/Akt pathway in osteosarcoma cell. Am J Transl Res 8(2):968
Xing Du, Li Yi, Xia Y-L, Ai S-M, Liang J, Sang P, Ji X-L, Liu S-Q (2016) Insights into protein-ligand interactions: mechanisms models, and methods. Int J Mol Sci 17(2):144
Yamini L, Vijjulatha M (2008) Inhibitors of human dihydrofolate reductase: a computational design and docking studies using glide. E-J Chem 5(2):263–270
Yan W, Li SX, Wei M, Gao H et al (2018) Identification of MMP9 as a novel key gene in mantle cell lymphoma based on bioinformatic analysis and design of cyclic peptides as MMP9 inhibitors based on molecular docking. Oncol Rep 40(5):2515–2524
Yin L, Duan JJ, Bian XW, Yu SC et al (2020) Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res 22(1):1–13
Acknowledgements
We would like to thank Dr. Sethulekshmy Nair, Dr. Harish Madhav and Dr. Deepthi DC for their support to carry out this work
Funding
No funds, grants, or other support was received.
Author information
Authors and Affiliations
Contributions
SN and DUK contributed to the study design, methodology, investigation, data analysis and wrote the first draft of the manuscript. PNG contributed to methodology, data analysis, validation and resources. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Praseetha, N.G., Divya, U.K. & Nair, S. Identifying the potential role of curcumin analogues as anti-breast cancer agents; an in silico approach. Egypt J Med Hum Genet 23, 100 (2022). https://doi.org/10.1186/s43042-022-00312-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00312-x