
Abstract
Background
Allogeneic stem cells are the most potent sources for replacing cell, tissue, and organ malfunctions. The clinical use of these stem cells has been limited due to the risk of immune system rejection due to the incompatibility of human leukocyte (HLA) antigens between donors and recipients. To overcome this limitation, we used the CRISPR/Cas9 system to eliminate the β2 microglobulin (B2M) gene, which plays a vital role in the expression of HLA class I.
Results
Non-viral transfer of two gRNAs targeting the first exon and intron in the B2M gene results in large deletions in the target region. In addition, the results of this study showed that 11.11% and 22.22% of cells received genomic changes as homozygous and heterozygous, respectively.
Conclusion
In conclusion, we have shown that the dual guide RNA strategy is a simple and efficient method for modifying genes. As a result, these cells can be proposed as universal cells that are not detectable in the cell therapy system and transplantation by the receptor immune system.
Similar content being viewed by others

Background
Organ transplantation (OTP) is a primary and high-risk medical procedure for the treatment of patients with treatable diseases, including organ failure [1, 2]. OTP has major limitations, including a lack of appropriate donors in terms of safety, which reduces its use. Autologous and allogeneic stem cells were initially thought to be effective in reducing these limitations [3]. In principle, allogeneic cell products are more beneficial compared with autologous cell products by providing safe and consistent products [4, 5]. However, rejection of the transplant due to a mismatch of human leukocyte antigen (HLA) between donor and recipient is a significant challenge in using these cells. Accordingly, host T cells can elicit immune responses against donor major/minor histocompatibility antigens and lead to a quick rejection. In addition, the likelihood of graft progression versus host disease (GVHD) must be considered [6].
Many studies have shown that preventing HLA mismatch can reduce graft rejection by downregulating donor HLA class expression [7, 8]. Disrupting the expression of HLA type I by targeting the β2-microglobulin (B2M) locus, which encoding invariable and pivotal component of HLA I, would result in the incomplete formation of protein and thus lead to address immune rejection [9]. So far, various methods have been used for this purpose, including Cre-LoxP system [10], small interfering RNA (siRNA) [11], transcription activator-like effector nucleases (TALEN) [12, 13], and zinc-finger nuclease (ZFN) [14]. Despite many advances, these approaches have limitations, including unintended recombination through the Cre-LoxP system [10, 15], incomplete deletion, and low efficacy in targeting target genes using the RNAi method [16]. In addition, the design of TALEN and ZFN cores is challenging, so these methods are less used for research or therapeutic purposes. To overcome these limitations, clustered regularly interspaced short palindromic repeats (CRISPR) system has been introduced as a revolutionary technology. Due to its simplicity and programmability, this system has become a versatile gene-editing tool with promising results. The CRISPR system is constituted of a ribonucleoprotein complex, which cleaves double-stranded DNA molecules harboring sequences complementary to the guide RNA (crRNA) within the ribonuclease. So, in this system, DNA recognition is only dependent on the presence of an engineered 20-nucleotide sequence, leading to vast application in therapeutic trials. Utilizing CRISPR/Cas9 technology by designing two gRNAs is an easy way to target two sites simultaneously, followed by complete and large deletion [17]. The CRISPR/Cas9 system, with its single or dual gRNA approach, can create double-strand breaks (DSBs) and cause non-homologous end joining (NHEJ) through the repair path. As this pathway is prone to error; as a result, there is a possibility of indel mutations and gene deletion [7, 18, 19].
In this study, we used the CRISPR/Cas9 system to target the B2M gene in the HEK293T (human embryonic kidney cell line) cell line via two gRNAs and paved the way toward generating a universal cell model using the CRISPR/Cas9-mediated non-homologous end-joining repair system. One of the reasons for using the HEK293T cell line is the high rate of transfection efficiency of this cell by different methods. In addition, this cell line is easily cultured and propagated. Overall, the characteristics of the HEK293T cell line are very suitable for gene editing by CRISPR/Cas9 system [20].
Methods
gRNA design and CRISPR/Cas9 construction
The online CRISPR tool (http://crispor.tefor.net/) was used to design a pair of guide RNAs for exon 1 of B2M gene (BME). These tools rank all gRNAs and evaluate the potential off-target sites through a bioinformatics BLAST search with the whole genome DNA sequences. For this purpose, we perform a genome-scale screen to measure gRNA activity. Accordingly, we screened six gRNAs targeting B2M using online CRISPR tools and identified specific gRNA sequences highest editing efficiency for target deletion without carrying off-target effects. Then, selected gRNAs were tested in a single experiment, and the gRNAs with the most efficiency in targeted deletion were opted (Table 1). The designed gRNAs were cloned into the pSpCas9 (BB)-2A-GFP (PX458) (plasmid #48138; Addgene, Watertown, MA, USA) using standard cloning protocol with single-step digestion–ligation. In this method, oligonucleotides encoding the gRNA were annealed and ligated into the BbsI (# ER1011; Thermo Scientific, Waltham, MA, USA) restriction sites in the PX458 plasmid. Proper vector construction was confirmed by PCR using primers described in Table 2. Finally, Sanger sequencing was carried out to validate the cloning results.
Cell culture and transfection
HEK293T cells were cultured in high glucose Dulbecco's modified Eagle's medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco), 100 units penicillin/ml, 100 μg streptomycin/ml (Sigma-Aldrich, Arklow, Ireland) and incubated at 37 °C with 5% CO2. Twenty-four hours prior to transfection, a six-welled plate was seeded with 5 × 105 cells in each well and cultivated in 2 mL growth media devoid of the Penicillin–Streptomycin antibiotics. Then, transfection of cells was performed, using 1 μg of plasmid and Lipofectamine.
Isolation of transfected cells via fluorescence-activated cell sorting (FACS) and single-cell preparation
After 48 h, the approximate transfection efficiency was estimated using a fluorescence microscope. Subsequently, cells exhibiting enhanced green fluorescent protein (eGFP) expression were isolated, using a FACSAria III flow cytometer, and subsequently were cultivated in complete growth media for 3 to 4 days to expand into 80% of confluency. Then, the cell suspension was subjected to serial dilution to determine the zygosity of single cells.
Genotyping of clonal cell lines by PCR and Sanger sequencing
The genomic DNA of the 31 expanded single cells was extracted using a DNAeasy kit (Qiagen, Manchester, UK). The extracted DNAs were amplified by PCR using primers described in (Table 2). The PCR product was visualized using gel electrophoresis. According to the manufacturer’s instruction, the gel-purified PCR products were cloned into a TA vector using the TOPO TA cloning kit (Thermo Fisher Scientific). The results demonstrated the zygosity state of each clone, including homozygous, heterozygous, and wild-type states, each signifying single allele mutation, double allele mutation, and no mutation, respectively (Fig. 1).

A summary of B2M gene knockout as covered in this study indicates the process, which would ultimately lead to gene knockout by CRISPR/Cas9 system. Analysis of the gene deletion via PCR and Sanger sequencing confirms approximately 2.2 bp segment deletion separating the two guide RNAs. Indels in targeted segment is slightly variable between different single clones due the incomplete Cas9 cleavage efficiency
Results
Preparation of the CRISPR/Cas9 plasmid for targeting B2M gene
The 20-bp oligonucleotides encoding gRNA that targets exon 1 of B2M gene were cloned into the PX458 plasmid. Based on the results of Sanger sequencing, this designed gRNA can direct the Cas9 enzyme to generate the DSB at target sites.
NHEJ-mediated B2M gene editing in HEK293T cells
Transfection efficiency was measured by counting eGFP expressing cells, which revealed the efficiency to be approximately 30% (48 h after transfection) (Fig. 2). We used serial dilution to separate single cell clones from heterogeneous cell populations to assess the function of the CRISPR/Cas9 system. Upon introducing the Cas9-gRNAs system into cells, we expected to see four potential cell lines: cell lines without mutation, with heterozygous mutation, biallelic mutation, and with homozygous mutation. Dual DSB induction in both alleles within the intended target resulted in B2M gene expression ablation.

The GFP protein expression of HEK293 cells following transfection. The transfection was carried out using Lipofectamine reagents. This figure illustrates a small proportion of the total cells, absorbing the vectors containing the gRNAs. A Untransfected HEK293T cells with UV visible. B Transfected HEK293T cells with UV visible. C Untransfected HEK293T cells with GFP−. D Transfected HEK293T cells with GFP+
B2M Zygosity determination in HEK293T cell line
The genomic DNAs from all single-cell derived expanded clones were analyzed by PCR and Sanger sequencing. According to PCR results, cells with homozygote and heterozygote deletions were visualized as a single band (473 bp) and double bands (473 bp and 2765 bp), respectively, on agarose gel electrophoresis. In addition, wild-type cells were visible as a single band (2765 bp). Finally, the sequencing of surveyed clones indicated that among a total of thirty-six clones, four clones (11.11%) received homozygote, and eight clones (22.22%) received heterozygote genome modifications. The rest of the cells had intact genomes (wild type). In addition, due to the off-target effects of the Cas9 enzyme, different types of small indel were observed in several clones.
Discussion
This study aimed to evaluate the efficiency of CRISPR/Cas9-mediated genome editing with dual guide RNA as a simple method for genetic manipulation to produce null HLA class 1 donor cells that could potentially address graft-related problems. Due to the B2M subunit's structural importance, the reduced B2M expression has led to the incorrect formation and inactivation of HLA type 1 [21,22,23]. As a result of the eradication of cell surface expression of polymorphic HLA class I molecules (HLA-A,-B,-C), allograft cells have the chance to evade the host immune system and are not recognized by the CD8 + T cells [24, 25]. The CRISPR/Cas9 system allows us to delete the B2M gene and cut a relatively large portion of DNA by simply inserting two guide RNAs into the cell (Fig. 3). In this study, a third portion—12 out of 36 clones—of the cells was mutated by dual Cas9 cleavage. Notably, the creation of relatively large deletion within the target region would enable us to employ a fast and straightforward method such as PCR for genotyping the modified genes. Our data and findings from other studies clearly revealed the benefits of utilizing the dual gRNAs approach in gene knockout. It was already shown that this efficient strategy could be employed to study repetitive sequences in which genetic manipulations via single-guide RNA results in increased off-target effects. In addition, the successful applicability of dual gRNA CRISPR/Cas9 technology in perturbing non-coding regions, including silencers, enhancers, and long non-coding RNAs, has been reported [7, 26]. According to previous studies, small indel mutations are more likely to lead to the loss of function in non-coding regions using a gRNA-based CRISPR/Cas9 system [7, 27]. In addition, the use of single gRNAs in CRISPR/Cas9 systems can lead to the production of various small indels in target sequences that require laborious T7 endonuclease 1 (T7E1) assay to detect Cas9 activity [28]. Furthermore, employing a single gRNA method cannot efficiently abate b2m in specific cell types such as T cells [7].

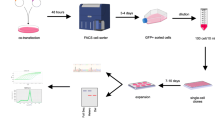
A Schematic flowchart of the experimental procedure for knocking out B2M gene out by CRISPR/Cas9 genome editing system. A Generating the desired gRNA through designed guide oligos under temperature condition. B Inserting the gRNA into backbone of PX458 vector (red region) followed by ejection of two-cutting selective sites. C Transforming the resulting vectors into E. coli DH5α and isolating vectors from colonies. D Co-transfecting the collected vectors into HEK293T cell line using commercially transfection reagent, Lipofectamine. E Directing the gRNA into the specific genomic site and editing the targeted DNA through Cas9 protein. F Rejoining the fragment sites by error-prone NHEJ pathway and knocking out B2M gene
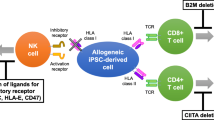
Hong et al. simultaneously produced HLA class I cells by targeting HLA-A/B/C genes using six gRNAs. The efficiency of the method used in this study was less than the method used in our study. According to the results of their study, the simultaneous transfer of six gRNAs to the cell reduced the survival of target cells. Also, selecting cells simultaneously under the influence of six gRNAs would be technically demanding [29]. In other studies conducted by Xu et al. and Jang et al., the HLA-A and/or HLA-B was successfully ablated in inducible pluripotent stem cells (iPSC) instead of complete disruption of HLA class I [25, 30]. Although they could attain cells with less immunogenicity, there will still be HLA disparity for alleles retaining between donor and recipient, which should be assessed.
Another alternative way to provide an optimal source of donor cells is to remove HLA class II under certain conditions. Although targeting the CIITA gene can lead to an expansion of the immune system, elimination of HLA class II due to impaired maturation of CD4 + T cells can lead to lymphopenia [25, 31].
Although many in vivo studies on animal models are solely concentrated on T cells, some studies recognized B cells in the transplanted organ for their ability to develop into long-lived plasma cells that produce high-affinity alloantibodies. Also, in addition to T cells, B cells are recognized to have a role in transplant rejection by donor-specific antibody production and otherwise may lead to tolerance when acting as an antigen-presenting cell [32].
The generation of universal cells using the dual gRNAs approach can resolve the issues such as the shortage of suitable donors and graft rejections. Moreover, these HLA class I null-cells can serve as a source of artificial antigen-presenting cells to produce cytotoxic T cells. However, this area has many limitations, including the identification and destruction of cells by natural killer cells (NK cells). To solve this problem, overexpression of non-classical HLA type 1 protein has been suggested to reduce the emerging status of NK cells [31]. Tumor formation due to HLA class I deficiency after transplantation is another problem in this field. The artificial introduction of suicide or apoptosis genes has been proposed to prevent tumor formation. In addition, it has been suggested that artificially engineered Caspase 9-induced artificial apoptosis has shown promising results because it is more effective, safer, and more immunogenic than the previous approach [10, 33].
Conclusion
In conclusion, in this study, we targeted specific loci in the B2M gene through two gRNAs that eradicate the expression of HLA class I molecules (HLA-A, -B, -C) on the cell surface. In the future, this cell model can be used to create a universal cell for therapeutic purposes in different patients, regardless of the nature of HLA. However, further studies in this area need to be conducted to explore other aspects of functional and immune assays.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- B2M:
-
Beta-2 microglobulin
- Cas9:
-
CRISPR-associated protein 9 nuclease
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- crRNA:
-
CRISPR RNA
- DNA:
-
Deoxyribonucleic acid
- DSB:
-
Double-strand break
- FACS:
-
Fluorescence-activated cell sorting
- GFP:
-
Green fluorescent protein
- gRNA:
-
Single-guide RNA
- GVHD:
-
Graft-versus-host disease
- HEK293T:
-
Human embryonic kidney
- HLA:
-
Human leukocyte antigen
- iPSC:
-
Inducible pluripotent stem cells
- MHC:
-
Major histocompatibility complex
- NHEJ:
-
Non-homologous end joining
- NK cell:
-
Natural killer cell
- OTP:
-
Organ transplantation
- PCR:
-
Polymerase chain reaction
- PAM:
-
Protospacer adjacent motif
- RNA:
-
Ribonucleic acid
- siRNA:
-
Small interfering RNA
- T7E1:
-
T7 endonuclease 1
- TALEN:
-
Transcription activator-like effector nucleases
- ZFN:
-
Zinc-finger nuclease
References
Black CK et al (2018) Solid organ transplantation in the 21(st) century. Ann Transl Med 6(20):409–409
Bezinover D, Saner F (2019) Organ transplantation in the modern era. BMC Anesthesiol 19(1):32
Girlanda R (2016) Deceased organ donation for transplantation: challenges and opportunities. World J Transplant 6(3):451–459
Tullis GE, Spears K, Kirk MD (2014) Immunological barriers to stem cell therapy in the central nervous system. Stem Cells Int 2014:507905
Li C et al (2021) Allogeneic vs. autologous mesenchymal stem/stromal cells in their medication practice. Cell Biosci 11(1):187
Kot M et al (2019) The importance of HLA assessment in “off-the-shelf” allogeneic mesenchymal stem cells based-therapies. Int J Mol Sci 20(22):5680
Mandal PK et al (2014) Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 15(5):643–652
Torikai H et al (2016) Genetic editing of HLA expression in hematopoietic stem cells to broaden their human application. Sci Rep 6(1):1–11
Quach DH et al (2019) A strategy to protect off-the-shelf cell therapy products using virus-specific T-cells engineered to eliminate alloreactive T-cells. J Transl Med 17(1):240
Riolobos L et al (2013) HLA engineering of human pluripotent stem cells. Mol Ther 21(6):1232–1241
Haga K et al (2006) Permanent, lowered HLA class I expression using lentivirus vectors with shRNA constructs: averting cytotoxicity by alloreactive T lymphocytes. Transpl Proc 38(10):3184–3188
Cui D et al (2016) Generating hESCs with reduced immunogenicity by disrupting TAP1 or TAPBP. Biosci Biotechnol Biochem 80(8):1484–1491
Wang D et al (2015) Targeted disruption of the β2-microglobulin gene minimizes the immunogenicity of human embryonic stem cells. Stem Cells Transl Med 4(10):1234–1245
Torikai H et al (2013) ZFN-driven gene editing prevents HLA-A expression on hematopoietic stem cells -improving the chance of finding an HLA-matched donor. Blood 122(21):1655–1655
McLellan MA, Rosenthal NA, Pinto AR (2017) Cre-loxP-mediated recombination: general principles and experimental considerations. Curr Protoc Mouse Biol 7(1):1–12
Zimmer AM et al (2019) Loss-of-function approaches in comparative physiology: is there a future for knockdown experiments in the era of genome editing? J Exp Biol 222(7):175737
Amiri F et al (2021) HLA-A gene knockout using CRISPR/Cas9 system toward overcoming transplantation concerns. Egypt J Med Human Genet 22(1):37
Ran FA et al (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11):2281–2308
Qiao H et al (2021) The advance of CRISPR-Cas9-based and NIR/CRISPR-Cas9-based imaging system. Front Chem 9:1069
Ooi A et al (2016) A guide to transient expression of membrane proteins in HEK-293 cells for functional characterization. Front Physiol 7:300
Torikai H et al (2013) Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 122(8):1341–1349
Qian X, Villa-Diaz LG, Krebsbach PH (2013) Advances in culture and manipulation of human pluripotent stem cells. J Dent Res 92(11):956–962
Koga K, Wang B, Kaneko S (2020) Current status and future perspectives of HLA-edited induced pluripotent stem cells. Inflam Regen 40(1):23
Chen X et al (2014) Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in caenorhabditis elegans. Sci Rep 4:7581
Xu H et al (2019) Targeted disruption of HLA genes via CRISPR-Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell 24(4):566-578.e7
Beerman I et al (2014) Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 15(1):37–50
Bauer DE et al (2013) An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 342(6155):253–257
Sentmanat MF et al (2018) A survey of validation strategies for CRISPR-Cas9 editing. Sci Rep 8(1):888
Hong CH et al (2017) Antigen presentation by individually transferred HLA class I genes in HLA-A, HLA-B, HLA-C null human cell line generated using the multiplex CRISPR-Cas9 system. J Immunother 40(6):201–210
Jang Y et al (2019) Development of immunocompatible pluripotent stem cells via CRISPR-based human leukocyte antigen engineering. Exp Mol Med 51(1):1–11
Mattapally S et al (2018) Human leukocyte antigen class I and II knockout human induced pluripotent stem cell-derived cells: universal donor for cell therapy. J Am Heart Assoc 7(23):e010239–e010239
Karahan GE, Claas FHJ, Heidt S (2017) B cell immunity in solid organ transplantation. Front Immunol 7:686
Hicklin DJ et al (1998) beta2-microglobulin mutations, HLA class I antigen loss, and tumor progression in melanoma. J Clin Investig 101(12):2720–2729
Acknowledgements
We would like to express our very great appreciation to Dr. Mohsen Mazloom Rezaei for his valuable guidance and support and Mr. Saeed Ataei for his help in writing the article. We would also like to thank all staff of the Department of Immunology and Student Research Committee in Shiraz University of Medical Sciences for their help in the research work and financial support.
Funding
This manuscript was extracted from the MSc thesis of Maryam Ranjbar and was supported by a grant (95-01-01-13944) from the Vice-Chancellor for Research Affairs of Shiraz University of Medical Sciences, Shiraz, Iran.
Author information
Authors and Affiliations
Contributions
MD and MR conceived and designed the experiments. MR, FA and MTD carried out the experiments. MR, FA and MN wrote the manuscript with input from all authors. FT designed the graphical abstract as well as edited the final version of article. MD supervised the project. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ranjbar, M., Amiri, F., Nourigorji, M. et al. B2M gene knockout in HEK293T cells by non-viral delivery of CRISPR-Cas9 system for the generation of universal cells. Egypt J Med Hum Genet 23, 62 (2022). https://doi.org/10.1186/s43042-022-00267-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00267-z