
Abstract
Background
Glioblastoma (GBM) is the commonest primary malignant cerebral tumor in adults. Detection of genetic mutations in liquid biopsy is endorsed rapidly throughout several solid neoplasms but still limited in GBM. Our study provides insight for the genetic alterations in liquid biopsy of the newly diagnosed GBM patients using next generation sequencing technology together with identification of the microsatellite instability (MSI) status in those patients.
Results
Eighteen variants detected in 15 genes which were (4, 12 and 2) missense, coding silent and intronic mutations, respectively. The 4 substitution–missense mutations were as follows: Drug responsive TP53 (p.Pro72Arg) variant was detected in 6 patients (85.7%). KDR (p.Gln472His) variant was noted in 4 patients (57.1%) as a result of substitution at c.1416A > T. Two patients revealed KIT (p.Met541Leu) variant which result from substitution at c.1621A > C. Only one patient showed mutation in JAK3 gene which was (p.Val718Leu) variant resulting from c.2152G > C substitution. Regarding MSI status, four cases (57.1%) were MSI-Low and three cases (42.9%) were MSI-High.
Conclusions
This study identifies the molecular landscape and microsatellite instability alternations in Egyptian brain tumor patients, which may have an important role in improving the outcome, survival and may help in evolving a characteristic individual therapy.
Similar content being viewed by others

Background
Glioblastoma (GBM) is very aggressive and has a median of 12- to 15-month survival with less than 5% of 5 year survival [1]. Patients had a highly different therapeutic response and rates of survival which could be due to tumor heterogeneity [2]. Clinical, pathological examination and imaging techniques are the standard techniques for GBM diagnosis. Invasive tissue biopsy procedure has many risks to those patients, as affecting neurological functions, hemorrhage, etc. [3], with some tumors may be inaccessible due to their location or close to risk organ [4]. Also, imaging techniques cannot discriminate pseudo- and true progression after treatment to prevent unnecessary operations and further useless treatment [5]. Therefore, the need appears for more simple techniques to assess biomarkers from non-surgical samples. Isolation of circulating tumor DNA (ctDNA) from blood has a number of benefits such as decreasing invasive damages and obstacles of getting sufficient tumor tissues. Also, blood sampling is attainable and easy to reiterate when needed which provides a persuasive and achievable method to estimate the characteristics of cerebral tumors [6]. However, few presence of ctDNA in blood is a significant challenge for tumor biomarker testing availability and its translation clinically [7]. Incorporation of histological and genetic evaluation is recommended in GBM, with many genes involved such as isocitrate dehydrogenase 1 and 2 mutations, 1p/19q chromosomal codeletion and point mutations in tumor protein 53, etc. [8]. Using next generation sequencing (NGS) in estimation of genetic alternations in liquid biopsy has been grown rapidly across several solid tumors [9] but still limited in GBM especially in Egypt. Many studies revealed mutations in blood of GBM patients such as Piccioni et al. [10] who has used NGS in ctDNA analysis of advanced glioblastoma patients and has observed mutations in TP53, PDGFRA and NF1 genes, etc. Another study had 33 GBM patients showed mutations in TP53, EGFR and MET genes, etc. [11].
Study objectives
Our study provides insight for the genetic alterations in liquid biopsy of the newly diagnosed GBM patients using targeted next generation sequencing technology together with identification of the microsatellite instability (MSI) status in those patients.
Methods
Participants and sample preparation
This pilot prospective study included 7 newly diagnosed brain tumor patients and was performed from Dec. 2019 to Jun. 2020. DNA was isolated from blood samples by QIAamp® DNA Mini kit—Catalogue Number ID: 51304 as stated by the manufacturer guidance. Concentration, quality and amplifiability of the isolated DNA samples have been tested before further processing [12].
Sequencing and data analysis
Preparation of the libraries was done by Illumina AmpliSeq™ Cancer Hotspot Panel v2, Catalogue Number: 20019161 detecting 50 genetic mutations. Libraries were examined by 2100 Bioanalyzer instrument using DNA 1000 kit—Catalogue Code: 5067-1504 with the anticipated PCR yield 186–277 bp. Patients’ libraries were combined to reach a final sequencing library which ran using MiSeqDx system with read length of 2 × 150 bp and approximately 17 h to finalize the run [9]. Checking each run quality was done by determining specifications depending on PhiX libraries that provide a cluster density of 865–965 k/mm2 clusters passing filter for v2 technology, as well as, run’s quality score is evaluated. The percentage of bases more than the Q30 is averaged over the whole run with a quality score for v2 technology more than 80% bases higher than the Q30 on 2 × 150 bp. Sequence reads was aligned to the Genome Reference Consortium Human Build 37 (GRCh37).
Detection of microsatellite instability (MSI) in studied patients
Mononucleotide markers were recommended in the detection of MSI. Thus, we identified 3 mononucleotide markers: BAT25, BAT26 and NR27, according to manufacturer protocol and data were analyzed using Agilent 2100 Bioanalyzer system [12].
Results
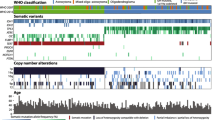
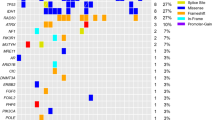
Seven patients were included with 6/7 (85.7%) patients were males. The median patient age was 50 years (range 23–58). Right-sided tumor site was common among our patients 5/7 (71.4%). By MRI brain scan, the median size of the tumor was 5 cm (4–6 cm). The clinicopathological features of our patients are described in Table 1. Variant allele frequency (VAF) of each variant (Table 2) and primary analysis revealed 28 mutations (Table 3). Four variants out of 28 were not found in Catalogue of Somatic Mutations in Cancer (COSMIC) database with 5 non-coding variants were noted in the intron of a transcript and only 1 variant was a SNP in COSMIC database. Across 15 genes, there were (4, 12 and 2) missense, coding silent and intronic mutations, respectively (Fig. 1). Searching in ClinVar database, 13/18 was benign mutations, 1 variant has conflicting interpretations of pathogenicity, 3 mutations were not recorded and only 1 variant was drug responsive one. Regarding missense variants, Tumor protein TP53 (TP53 p.Pro72Arg) was detected in 6 patients (85.7%) which was a drug response mutation resulted from c.215C > G. Mutation in Kinase Insert Domain Receptor (KDR) gene was found in 4 patients (57.1%); 3 patients were glioblastoma multiforme grade IV and one patient was astrocytoma grade II. This mutation was p.(Gln472His) resulting from c.1416A > T and was not recorded in ClinVar. Two patients had mutation in KIT Proto-Oncogene, Receptor Tyrosine Kinase (KIT) gene. It was p.(Met541Leu) variant which results due to c.1621A > C and was known as a benign/likely benign mutation in ClinVar. Only one patient revealed mutation in Janus Kinase 3 (JAK3) gene which was p.(Val718Leu) variant resulting from c.2152G > C and recorded in ClinVar as conflicting interpretations of pathogenicity, likely benign or uncertain significance variant. Benign coding silent variants were noticed in the following genes: Isocitrate dehydrogenase (NADP(+)) 1 (IDH1), APC Regulator of WNT Signaling Pathway (APC), Epidermal Growth Factor Receptor (EGFR), Cyclin Dependent Kinase Inhibitor 2A (CDKN2A), HRas Proto-Oncogene, GTPase (HRAS). These mutations were p.Gly105=, p.Thr1493=, p.Gln787=, p.Arg58=, and p.His27=, respectively, which result from c.315C > T, c.4479G > A, c.2361G > A, c.174A > C and c.81T > C, respectively. MET Proto-Oncogene, Receptor Tyrosine Kinase (MET) gene mutation showed 2 benign variants p.(Ile377=) and p.(Ser178=) due to c.1131C > T and c.534C > T, respectively. Two benign variants were detected in platelet-derived growth factor receptor A (PDGFRA) gene, p.Val824= and p.Pro567= which result from c.2472C > T, and c.1701A > G, respectively. Ret Proto-Oncogene (RET) gene revealed 2 benign variants, p.Ser904 = due to c.2712C > G and p.Leu769 = due to c.2307G > T. Fibroblast growth factor receptor 3 (FGFR3) gene revealed p.(Thr653=) due to c.1959G > A which is not recorded in ClinVar. Two intronic mutations were noticed in Fms-Related Receptor Tyrosine Kinase 3 (FLT3) gene in 5 patients due to c.1310-3T > C and SWI/SNF-Related, Matrix-Associated, Actin-Dependent Regulator of Chromatin, Subfamily B, Member 1 (SMARCB1) gene in 2 cases due to c.1119-41G > A. As regards MSI status, 4/7 (57.1%) cases had MSI-Low and 3/7 (42.9%) cases had MSI-high (Fig. 1).

Different studied activating mutations and MSI status in GBM patients
Discussion
Recently, our information about the genetic features of cerebral tumors has raised dramatically by using next generation sequencing platforms [13]. Liquid biopsy has been widely used in solid tumors to identify driver mutations, but is still limited in glioblastoma multiforme (GBM) patients [14]. Our pilot study aimed to assess the activating variants in blood samples of the newly diagnosed GBM using targeted next generation sequencing together with identification of the microsatellite instability (MSI) status. We found the drug responsive variant p.(Pro72Arg) of the tumor suppressor TP53 gene in 6/7 (85.7%) patients. Previous reports showed that TP53 is mutated in 29% of the GBM samples and another study showed TP53 mutation in 38% of gliomas, including 23% of primary glioblastomas and 80% of secondary glioblastomas [15, 16]. Other studies showed that p53 is the commonest mutation noticed in the blood derived ctDNA samples of gliomas [17]. The drug responsive variant p.(Pro72Arg) of the TP53 gene was found in 47.94% ependymoma grade III and also detected in a young medulloblastoma patient [18]. Kinase Insert Domain Receptor (KDR) gene which is a Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) gene has a role in tumor initiation and neovascularization [19]. We found KDR gene mutation in 4 patients (57.1%) which is p.(Gln472His) as a result of c.1416A > T. KDR p.(Gln472His) is a germline variant observed in fifty percent of GBM and forty-seven percent of grade 2–3 astrocytomas [20]. A correlation between KDR p.(Gln472His) and risk of glioma has been observed, as unusual angiogenesis may be implicated in primary tumorigenesis [21] with the more angiogenic activity, the worse the survival rate. Thus, GBM patients with the p.(Gln472His) substitution have poor prognosis and this may be related to increases in micro vessel density [22]. Other reports found better survival in positive KDR p.(Gln472His) head and neck squamous cell carcinomas [23]. KIT mutations have been described in tumor cell proliferation, such as cancer stem cell proliferation, and proliferation of endothelium in gliomas and assisting tumor-related angiogenesis [24]. Here, we observed KIT p.(Met541Leu) variant in 2 (28.6%) patients which result from substitution at c.1621A > C. This variant has been described to enhance the receptor affinity to its ligand, stem cell factor (SCF) [25]. Zaman et al. [20] observed KIT M514L in 43.75% of both patients of GBM and glioma grade 2–3 and this variant may be used as a marker of aggressiveness which result from mechanisms that do not include regulation of angiogenesis. In our study, only one patient revealed JAK3 p.(Val718Leu) variant resulting from c.2152G > C substitution. JAK3 is a gene encodes a protein-tyrosine kinase which functions in cytokine receptor-mediated signal transduction and altered in 1.90% of all tumors [26]. As regards MSI status in our GBM patients, 4/7 (57.1%) had MSI-Low and 3/7 (42.9%) had MSI-High. Viana-Pereira et al. [27] found that 13.5% of high-grade glioma samples presented instability, with (< 1%, 12.5% and 86.8%) are MSI-H, MSI-L stable tumors, respectively. Previous study noticed about 27% MSI in 45 pediatric high-grade gliomas using mononucleotide (BAT25 and BAT26) markers [28], another study did not note MSI in 41 cases using (CAT25, BAT25 and BAT26) [29]. Further studies are needed to explore whether liquid biopsy in brain tumor patients could potentially defeat the natural difficulty developed accompanied by the standard tissue biopsy. Larger sample size and longer follow-up period are recommended to compare genetic mutations and MSI status in liquid based versus tissue-based biopsy by targeted next generation sequencing.
Conclusions
Development of noninvasive or minimally invasive approaches to discover and monitor tumors is a major challenge and still limited in our brain tumor patients. This study identifies the molecular landscape and microsatellite instability status in a sample of Egyptian brain tumor patients, which may have an important role in improving the outcome, survival rate and to develop new personalized treatments.
Availability of data and materials
Available upon reasonable request.
Abbreviations
- APC:
-
APC Regulator of WNT Signaling Pathway
- CNS:
-
Central nervous system
- COSMIC:
-
Catalogue of Somatic Mutations in Cancer
- ctDNA:
-
Circulating tumor DNA
- EGFR:
-
Epidermal Growth Factor Receptor
- FLT3:
-
Fms-Related Receptor Tyrosine Kinase 3
- GBM:
-
Glioblastoma multiforme
- GRCh37:
-
Genome Reference Consortium Human Build 37
- HNSCC:
-
Head and neck squamous cell carcinoma
- HRAS:
-
HRas Proto-Oncogene, GTPase
- IDH1/2:
-
Isocitrate dehydrogenase (NADP(+)) 1/2
- IRB:
-
Institutional Review Board
- KDR:
-
Kinase Insert Domain Receptor
- MET:
-
MET Proto-Oncogene, Receptor Tyrosine Kinase
- MGMT:
-
O6-methylguanine methyltransferase
- MSI:
-
Microsatellite instability
- NCI:
-
National Cancer Institute
- NGS:
-
Next generation sequencing
- PDGFRA:
-
Platelet-derived growth factor receptor alpha
- RET:
-
Ret Proto-Oncogene
- SCF:
-
Stem cell factor
- SMARCB1:
-
SWI/SNF-Related, Matrix-Associated, Actin-Dependent Regulator of Chromatin, Subfamily B, Member 1
- TP53:
-
Tumor protein TP53
- VCF:
-
Variant Call Format
- VEGFR2:
-
Vascular Endothelial Growth Factor Receptor 2
References
Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA, Turner MC, Walsh KM et al (2014) The epidemiology of glioma in adults: a “state of the science” review. NeuroOncol 16:896–913
Donato V, Papaleo A, Castrichino A, Banelli E, Giangaspero F, Salvati M, Delfini R (2007) Prognosticimplication of clinical and pathologic features in patients with glioblastomamultiforme treated withconcomitant radiation plus temozolomide. Tumori J 93:248–256
Shankar GM, Balaj L, Stott SL, Nahed B, Carter BS (2017) Liquid biopsy forbrain tumors. Expert Rev Mol Diagn 17:943–947
Nieder C, Grosu AL, Astner S, Molls M (2005) Treatment of unresectableglioblastomamultiforme. Anticancer Res 25:4605–4610
Delgado-Lopez PD, Rinones-Mena E, Corrales-Garcia EM (2018) Treatment related changes in glioblastoma: a review on the controversies in responseassessment criteria and the concepts of true progression, pseudoprogression, pseudoresponse and radionecrosis. Clin Transl Oncol 20:939–953
Gorgannezhad L, Umer M, Islam MN, Nguyen NT, Shiddiky MJA (2018) Circulating tumor DNA and liquid biopsy: opportunities, challenges, and recent advances in detection technologies. Lab Chip 18:1174–1196
Gyanchandani R, Kvam E, Heller R et al (2018) Whole genome amplification of cell-free DNA enables detection of circulating tumor DNA mutations from fingerstick capillary blood. Sci Rep 8:17313
Weller M, Stupp R, Hegi ME, van den Bent M, Tonn JC, Sanson M et al (2012) Personalized care in neuro-oncology coming of age: why we needMGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neuro Oncol 14(4):100–108
Kassem N, Kassem H, Kassem L et al (2021) Detection of activating mutations in liquid biopsy of Egyptian breast cancer patients using targeted next-generation sequencing: a pilot study. J Egypt Natl Cancer Inst 33:10
Piccioni DE, Achrol AS, Kiedrowski LA, Banks KC, Boucher N, Barkhoudarian G, Kelly DF, Juarez T, Lanman RB, Raymond VM et al (2019) Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol 8(2):34
Schwaederle M, Husain H, Fanta PT, Piccioni DE, Kesari S, Schwab RB, Banks KC, Lanman RB, Talasaz A, Parker BA et al (2016) Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget 7:9707–9717
Kassem NM, Emera G, Kassem HA et al (2019) Clinicopathological features of Egyptian colorectal cancer patients regarding somatic genetic mutations especially in KRAS gene and microsatellite instability status: a pilot study. Egypt J Med Hum Genet 20:20
Nikiforova MN, Wald AI, Melan MA, Roy S, Zhong S, Hamilton RL, Lieberman FS, Drappatz J, Amankulor NM, Pollack IF, Nikiforov YE, Horbinski C (2016) Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol 18(3):379–387
Wan JCM, Massie C, Garcia-Corbacho J et al (2017) Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 17:223–238
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404
Zacher A, Kaulich K, Stepanow S, Wolter M, Kohrer K, Felsberg J, Malzkorn B, Reifenberger G (2017) Molecular diagnostics of gliomas using next generation sequencing of a glioma-tailored gene panel. Brain Pathol 27:146–159
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM et al (2014) Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6:224ra24
Butt E, Alyami S, Nageeti T, Saeed M, AlQuthami K et al (2020) Mutation profiling of anaplastic ependymoma grade III by Ion Proton next generation DNA sequencing [version 2; peer review: 2 approved]. F1000Research 8:613
Yao X, Ping Y, Liu Y et al (2013) Vascular endothelial growth factor receptor 2 (VEGFR-2) plays a key role in vasculogenic mimicry formation, neovascularization and tumor initiation by Glioma stem-like cells. PLoS ONE 8(3):e57188
Zaman N, Dass SS, Du Parcq P et al (2020) The KDR (VEGFR-2) genetic polymorphism Q472H and c-KIT polymorphism M541L are associated with more aggressive behaviour in astrocytic gliomas. Cancer Genom Proteom 17(6):715–727
Oliveira Rosario DE, DA Rosa BG, Goncalves TL, Matias DIL, Freitas C, Ferrer VP (2020) Glioblastoma factors increase the migration of human brain endothelial cells in vitro by increasing MMP-9/CXCR4 levels. Anticancer Res 40(5):2725–2737
Leon SP, Folkerth RD, Black PM (1996) Microvessel density is a prognostic indicator for patients with astroglial brain tumors. Cancer 77(2):362–372
Plate K, Scholz A, Dumont D (2012) Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol 124(6):763–775
Gomes AL, Reis-Filho JS, Lopes JM, Martinho O, Lambros MB, Martins A, Pardal F, Reis RM (2007) Molecular alterations of KIT oncogene in gliomas. Cell Oncol 29(5):399–408
Chatterjee A, Ghosh J, Kapur R (2015) Mastocytosis- a mutated KIT receptor induced myeloproliferative disorder. Oncotarget 6(21):18250–18264
The AACR Project GENIE Consortium (2017) AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov 7(8):818–831
Viana-Pereira M, Lee A, Popov S et al (2011) Microsatellite instability in pediatric high grade glioma is associated with genomic profile and differential target gene inactivation. PLoS ONE 6(5):e20588
Alonso M, Hamelin R, Kim M, Porwancher K, Sung T et al (2001) Microsatellite instability occurs in distinct subtypes of pediatric but not adult central nervous system tumors. Cancer Res 61:2124–2128
Eckert A, Kloor M, Giersch A, Ahmadi R, Herold-Mende C et al (2007) Microsatellite instability in pediatric and adult high-grade gliomas. Brain Pathol 17:146–150
Acknowledgements
We are grateful for our molecular laboratory & IT teams.
Funding
No funding obtained.
Author information
Authors and Affiliations
Contributions
NK and MH elucidated the patient data. HS picked up the clinical data. HK analyzed the genetic data and was the main writer of the manuscript. All authors read and accepted the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was accepted by Kasr Al Ainy Clinical Oncology department Institutional Review Board (IRB)-11-2019. Informed consent in a written form was taken from all patients involved in this work.
Consent for publication
This consent was obtained from all patients involved in this work.
Competing interests
No conflict of interest has been declared.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kassem, N.M., Kassem, H.A., Selim, H. et al. Targeted next generation sequencing provides insight for the genetic alterations in liquid biopsy of Egyptian brain tumor patients. Egypt J Med Hum Genet 23, 23 (2022). https://doi.org/10.1186/s43042-022-00214-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00214-y