
Abstract
Background
We had earlier described the growth-promoting and -depressive effects of replacing soybean meal (SBM) with low (12.5% and 25%) and high (50% and 100%) inclusion levels of black soldier fly larvae meal (BSFLM), respectively, in Ross x Ross 708 broiler chicken diets. Herein, using 16S rRNA gene amplicon sequencing, we investigated the effects of replacing SBM with increasing inclusion levels (0-100%) of BSFLM in broiler diets on the cecal bacterial community composition at each growth phase compared to broilers fed a basal corn-SBM diet with or without the in-feed antibiotic, bacitracin methylene disalicylate (BMD). We also evaluated the impact of low (12.5% and 25%) inclusion levels of BSFLM (LIL-BSFLM) on the prevalence of selected antimicrobial resistance genes (ARGs) in litter and cecal samples from 35-day-old birds.
Results
Compared to a conventional SBM-based broiler chicken diet, high (50 to100%) inclusion levels of BSFLM (HIL-BSFLM) significantly altered the cecal bacterial composition and structure, whereas LIL-BSFLM had a minimal effect. Differential abundance analysis further revealed that the ceca of birds fed 100% BSFLM consistently harbored a ~ 3 log-fold higher abundance of Romboutsia and a ~ 2 log-fold lower abundance of Shuttleworthia relative to those fed a BMD-supplemented control diet at all growth phases. Transient changes in the abundance of several potentially significant bacterial genera, primarily belonging to the class Clostridia, were also observed for birds fed HIL-BSFLM. At the finisher phase, Enterococci bacteria were enriched in the ceca of chickens raised without antibiotic, regardless of the level of dietary BSFLM. Additionally, bacitracin (bcrR) and macrolide (ermB) resistance genes were found to be less abundant in the ceca of chickens fed antibiotic-free diets, including either a corn-SBM or LIL-BSFLM diet.
Conclusions
Chickens fed a HIL-BSFLM presented with an imbalanced gut bacterial microbiota profile, which may be linked to the previously reported growth-depressing effects of a BSFLM diet. In contrast, LIL-BSFLM had a minimal effect on the composition of the cecal bacterial microbiota and did not enrich for selected ARGs. Thus, substitution of SBM with low levels of BSFLM in broiler diets could be a promising alternative to the antibiotic growth promoter, BMD, with the added-value of not enriching for bacitracin- and macrolide-associated ARGs.
Similar content being viewed by others


Introduction
Global consumption of meat protein is expected to increase over the next decade, mainly due to increasing population growth and dietary changes associated with growing affluence [1]. Currently, poultry meat comprises 59% of total global meat production and is expected to rise to 62% by the year 2032, largely driven by growing consumer demand[1, 2]. This demand will undoubtedly be met with an increase in production of poultry feed, which primarily consists of corn and soybean meal (SBM) [1]. SBM is routinely added to poultry feed as the main source of crude protein and amino acids [3]. However, soybean production and its supply chain are associated with negative environmental consequences, including deforestation, biodiversity loss, pesticide use, soil depletion, water usage, and greenhouse gas emissions [4,5,6,7,8]. In addition to these environmental costs, there is also a growing global health concern regarding in-feed antibiotic use in poultry diets for growth promotion and prophylactic purposes, as the inappropriate use of antibiotics has been linked to promoting the emergence and dissemination of antibiotic-resistant bacteria and antibiotic resistance genes (ARGs) that threaten modern medicine [9,10,11]. Faced with the double burden of finding sustainable protein feed and the growing restrictions and bans on antibiotic use in poultry production, it is crucial to identify and evaluate alternative feedstuffs with functional properties to produce poultry meat sustainably and profitably.
An emerging and novel sustainable protein alternative to SBM in poultry feed is meal prepared from the larvae of black soldier flies [Hermetia illucens (Diptera: Stratiomyidae)]. Black soldier fly larvae are capable of efficiently upcycling food wastes and organic wastes such as fruits, vegetables, and manure into a high-quality proteinaceous feed [12,13,14]. Apart from this, black soldier fly larvae are also a source of antimicrobial and immunomodulatory compounds such as chitin [15], lauric acid [16], and antimicrobial peptides [17]. Focusing on its potential as a substitute ingredient of conventional poultry feeds, we have recently shown that replacement of SBM with low inclusion levels of black soldier fly larvae meal (LIL-BSFLM; defined here as 12.5% and 25%) can provide for a positive growth response in broiler chickens during the starter period of growth, comparable to those fed a conventional corn-SBM-based diet supplemented with the antibiotic growth promoter, bacitracin methylene disalicylate (BMD) [18]. Likewise, Facey et al. demonstrated that substituting SBM in broiler chicken diets with 12.5% and 25% BSFLM fortified with BSFLM oil also had performance-enhancing effects similar to a conventional SBM-based diet supplemented with in-feed BMD and narasin during the starter phase [19]. Conversely, we observed that high inclusion levels of BSFLM (HIL-BSFLM, defined here as 50% and 100% BSFLM) in broiler chicken diets had a detrimental impact on the overall growth performance of broiler chickens, a finding that had also been reported by several other studies [18,19,20,21].
In recent years, the intestinal microbiota of poultry has received considerable attention as it plays an important role in various processes that affect the overall health, growth, and performance of poultry. These processes include nutrient digestion and absorption [22], nitrogen recycling by metabolising urea [23], supplying B vitamins to their hosts [24], production of essential amino acids [24], modulation of the immune system [25], and colonization resistance to enteric pathogens [26]. Within the intestinal tract, the twin ceca (located where the small and large intestines meet) contains the largest number and diversity of bacteria (1011-1012 colony forming units/g) [27]. It has been suggested that a well-functioning cecum is responsible for providing 10% of the energy needs for chickens, as it is an important region for microbial fermentation of non-starch polysaccharides and the subsequent production of short chain fatty acids (SCFAs) [28, 29]. Additionally, the chicken cecal microbiota also acts as a major reservoir of chicken and zoonotic pathogens, as well as antimicrobial resistance determinants [30]. Thus, the cecal microbiota and their associated metabolites have a profound influence on health and disease and is increasingly regarded as a potential modifiable risk factor and therapeutic target to promote broiler health and performance [31]. Herein, to gain a deeper understanding of the chicken intestinal microbiome and its dynamics in response to dietary BSFLM, we analyzed the bacterial composition of the cecal microbiota of broilers fed increasing inclusion levels of BSFLM (0-100%) in search of any potential links between gut bacteria and the BSFLM-mediated growth promotion and depression. In the context of antimicrobial resistance mitigation, we also examined the impact of performance-enhancing amounts of dietary BSFLM on selected antimicrobial resistance gene (ARG) abundance in chicken cecal and litter samples with the purpose of providing insight into a potential value-added benefit of using BSFLM as an alternative feed ingredient.
Results
Impact of diet on the development of the core cecal bacterial microbiota of broiler chickens
A total of 288 cecal samples were collected (1 ceca x 2 birds per replicate x 8 replicates x 6 diets x 3 sampling times). Of these, twenty samples were excluded from analyses due to a lack of digesta present in the cecum, and four samples were excluded after 16S gene amplicon sequencing and quality filtering. Therefore, a total of 264 cecal samples were included in the 16S rRNA gene amplicon analysis. The detailed distribution of cecal samples analyzed for each diet, including the respective sample sizes (n) for days 14, 28 and 35, were as follows: BMD (n = 14, 15, 15), 0% BSFLM (n = 13, 16,15), 12.5% BSFLM (n = 12, 15, 16), 25% BSFLM (n = 14, 16, 16), 50% BSFLM (n = 14, 16, 15), and 100% BSFLM (n = 14, 16, 16).
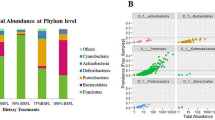
By profiling the bacterial composition of over 260 cecal samples collected from broiler chickens subjected to conventional corn-SBM-based diets and the BSFLM-substitution diets throughout a three-stage production cycle (see Additional file 1, Table S1, for 16S rRNA gene sequencing metrics), it was consistently observed that Firmicutes (72.33% ± 0.21%, mean ± SEM) and Bacteroidetes (22.24% ± 0.22%) remained to be the predominant phyla in ceca spanning all three growth phases (Fig. 1A). At the genus level, a shift in the chicken cecal bacterial composition that was driven by age was revealed. For instance, at the end of the starter phase (day 14), Lactobacillus had the highest overall abundance and comprised an estimated 13.20-19.08% of the total bacterial content of the ceca, whereas Barnesiella was the most prevalent genus detected in birds sampled at later stages of growth (i.e., grower and finisher phases) (Fig. 1B). There was also a noticeable expansion of Alistipes bacteria in the ceca of 35-day-old chickens compared to those of the younger cohorts (Fig. 1B). The partial/complete replacement of SBM with BSFLM in chicken diets appeared to have little influence on the age-dependent dynamics of the core bacterial constituents of the boiler’s cecal bacterial microbiota, illustrating the stability and resilience of the chicken gut microbiome to dietary changes.

Taxonomic composition of the broiler chicken cecal bacterial communities at the phylum (A) and genus (B) level. Data are grouped by phase of growth (Day 14, 28 and 35) and diet (BMD, 0%, 12.5%, 25%, 50% and 100% BSFLM), with the results presented as averaged values obtained from up to sixteen individual biological replicates per group
Impact of diet on the bacterial diversity of the broiler chicken cecum
To better explore and compare the cecal bacterial community structure of broiler chickens raised on different diets, alpha-diversity indices including taxon richness, Shannon and Simpson indices, and Pielou’s evenness were estimated using genus-level taxonomic data. On day 14, no significant difference between dietary groups were detected for any of the computed diversity metrics, with the exception that a significant increase in bacterial richness observed for chickens fed 100% BSFLM relative to those in the 0% BSFLM group (p < 0.05) (Fig. 2A). On day 28 and 35, both the 50% and 100% BSFLM groups displayed a significantly higher level of bacterial richness relative to those receiving a SBM-based diet with (i.e., BMD group) and without (i.e., 0% BSFLM group) supplementation with the antibiotic growth promoter, BMD (p < 0.05) (Fig. 2B and C), although that did not coincide with any statistically meaningful differences in both evenness and Simpson’s diversity index. Shannon’s index, on the contrary, was sensitive to the BSFLM content of the chicken diets on day 28 and 35, with significant diversity variance being detected mainly between high and low BSFLM-content diet groups (Fig. 2B and C).

Alpha diversity metrics of the cecal bacterial community recovered from 14- (A), 28- (B), and 35-day-old (C) broiler chickens fed varied amounts of BSFLM and a conventional soybean-based diet with or without BMD. The box-and-whisker plots show the first and third quartile (bottom and top lines of the box) and the median (horizontal middle line) values of the indicated diversity metrics. Each broiler chicken cecal sample is represented by a solid black dot. Statistical significance of between-group differences were detected using the non-parametric Kruskal-Wallis test followed by post-hoc pairwise Dunn test with Bonferroni correction and are indicated with an asterisk as follows: * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. BMD, bacitracin methylene disalicylate; BSFLM, black soldier fly larvae meal; %, percentage of soybean meal replaced with black soldier fly larvae meal (BSFLM)
While there was a general lack of difference in all four alpha-diversity indicators between the cecal bacterial communities derived from the BMD and 0% BSFLM diet groups throughout the duration of a 5-week sampling period (Fig. 2), bacterial richness was observed to significantly increase overtime only for the broiler chicken populations without dietary exposure to BMD (i.e., the 0% BSFLM group)(see Additional File 2, Figure S1A and S1B). This age-dependent increase in bacterial richness was also conserved among the other four antibiotic-free BSFLM diet groups (see Additional file 2, Figure S1C-F). Together these data suggest that BMD abrogates the developmental expansion of bacterial community richness in the broiler’s cecum.
Based on the principal coordinate analysis (PCoA) performed using Bray-Curtis’s distance matrices and the corresponding PERMANOVA statistics (see Additional file 2, Figures S2 and S3), the observed variance in the chicken gut microbial community between individual groups can be explained by differences in both age (Figure S2) and diet (Figure S3). No significant interaction of these two explanatory variables was detected (see Additional file 1, Table S2), implying the age- and diet-factors likely contribute independently to the microbiome dissimilarities. As indicated by the error-corrected multiple pairwise comparisons (Table 1), the bacterial composition of the ceca of 14-day-old broiler chickens fed 100% BSFLM differed significantly from the BMD (p < 0.015), 0% BSFLM (p < 0.015) and 12.5% BSFLM (p < 0.030) groups. On day 28, the bacterial composition significantly differed between the majority of diets, with the exceptions being among the BMD, 12.5%, and 25% BSFLM groups and between the 50% and 100% BSFLM groups (Table 1). Finally, at the finisher-phase, broiler chickens fed 0%, 12.5%, and 25% BSFLM diets and the conventional antibiotic-supplemented SBM-based diet (BMD group), exhibited a more similar cecal bacterial composition than broiler chickens fed HIL-BSFLM (i.e., 50–100% BSFLM) (Table 1). Taken together, these data demonstrate that HIL-BSFLM, particularly at 100% BSFLM, results in significant changes in the composition of the cecal microbiota, whereas LIL-BSFLM have only a minimal effect.
Impact of BSFLM diet on the abundance of individual bacterial taxa in the broiler chicken cecum
To further characterize the changes in the bacterial community structure associated with dietary exposure to various levels of BSFLM at different ages, differential abundance analysis was conducted using ANCOM-BC [32] to identify key bacterial taxa whose absolute abundance was impacted by the BSFLM-based diets relative to the BMD control diet. On day 14, no significant differences in the differential abundance of taxa at the genus level were detected for any of the 050% BSFLM groups compared to the BMD control (Fig. 3A). In contrast, birds fed 100% BSFLM exhibited a significantly lower abundance of bacteria belonging to the genera Shuttleworthia, [Eubacterium] coprostanoligenes group, Lachnospiraceae UCG-010, Tyzzerella 3, Anaerostipes, Butyricicoccus, [Ruminococcus] gauvreauii group and Ruminiclostridium 5 (Fig. 3A). In addition, there was a greater than 2 log-fold increase in the absolute abundance of Romboutsia bacteria in birds fed 100% BSFLM relative to the BMD-fed birds.

Cecal bacterial genera significantly affected by the dietary exposure to different levels of BSFLM on day 14 (A), day 28 (B), and day 35 (C). Horizontal bars represent log fold change ± standard error (shown as error bars) of absolute abundance associated with 0%, 12.5%, 25%, 50%, or 100% BSFLM diets relative to the BMD diet (reference) as determined by differential abundance analysis performed using ANCOM-BC2. Only taxa with statistically significant changes in their abundance relative to the reference group were displayed
On day 28, a higher level of Bifidobacterium and lower abundance of uncultured Clostridia bacterium and Intestinimonas were associated with birds fed 0% BSFLM (i.e., absence of both BMD and BSFLM) (Fig. 3B). The abundance of only a few bacterial genera were altered for chickens fed 12.5%, 25%, and 50% BSFLM diets relative to the BMD control (Fig. 3B). For example, Erysipelatoclostridium was found to be more abundant in birds fed 12.5% BSFLM, while both Christensenellaceae R-7 group, and Ruminococcaceae UCG-009 bacteria had greater abundance in the 50% BSFLM group (Fig. 3B). For birds receiving a 100% BSFLM diet, a total of 14 genera, including Romboutsia (also identified on day 14), and Ruminococcaceae UCG-010, which was also commonly observed in the 25% and 50% BSFLM groups, displayed significant increases in their absolute abundance relative to BMD-fed birds (Fig. 3B). Like day 14, Shuttleworthia was less abundant in the 100% BSFLM group compared to BMD, on day 28.
On day 35, Enterococcus bacteria were more abundant in the ceca of chickens fed a non-BMD diet, regardless of the BSFLM percentage content (Fig. 3C). Intriguingly, birds that received 50% and 100% BSFLM displayed several similar changes in the abundance of specific bacterial genera when compared to those fed the BMD control diet. Excluding the genera Enterococcus and Ruminiclostridium, whose elevated abundance appeared to be a common response to a non-BMD diet rather than a BSFLM-dependent one, Candidatus Soleaferrea, Anaerofilum, Ruminococcaceae UCG-005, Intestinimonas, Romboutsia, Bilophila, together with two uncultured genera, were all found to be more abundant in absolute-terms among both the 50% and 100% BSFLM diet groups (Fig. 3C). Several additional genera were observed to increase in abundance in the 100% BSFLM group and included Streptococcus, Ruminococcaceae UCG-010, Angelakisella, Ruminococcaceae UCG-009, and Christensenellaceae R-7 group (Fig. 3C). On the contrary, in addition to the Shuttleworthia population that had remained diminished since day 14, the abundance of bacteria belonging to the genera Anaerostipes, Lactobacillus, and Ruminicoccaceae UCG-013 were also reduced in the 100% BSFLM group (Fig. 3C).
Effect of diet on antibiotic resistance gene abundance in broiler chicken ceca and litter
Previously, we had demonstrated that replacement of SBM in broiler chicken diets with LIL-BSFLM had some growth performance-enhancing properties comparable to chickens raised on a conventional SBM-based diet supplemented with BMD [33]. To further evaluate whether substitution of SBM with BSFLM could influence the levels of antimicrobial resistance (AMR) determinants within the poultry production system, quantitative real-time PCR assays were performed on both cecal and litter DNA samples taken from 35-day-old chickens and the abundance of fourteen AMR-associated gene targets were measured. According to the normalized gene quantification data derived from the litter samples collected, no significant differences in the relative abundance of all gene targets evaluated were observed between chickens that received BMD and 0, 12.5, or 25% BSFLM diets (Table 2). However, the bacitracin resistance gene, bcrR, and the macrolide resistance gene, ermB, were found to be less abundant in the ceca of chickens following the 0%, 12.5% and 25% BSFLM dietary regime relative to those fed the BMD supplemented diet (Table 2). This observation suggests that elimination of BMD from conventional poultry feed can indeed diminish the abundance of bacitracin- and macrolide-associated resistance genes and that partial replacement of SBM with BSFLM has no significant effect on influencing the levels of selected gene targets within the chicken gut.
Discussion
Earlier work had shown that replacement of SBM with LIL-BSFLM in broiler chicken diets resulted in growth-promoting effects similar to chickens fed a conventional corn-SBM-based diet supplemented with BMD [18, 19], whereas HIL-BSFLM substitution negatively impacted growth performance [19, 20]. In the present study, we demonstrated that the cecal bacterial communities of chickens fed low amounts of BSFLM remained comparable to those that received the BMD control diet throughout a three-phase growth cycle. This suggests that the growth-enhancing properties of BSFLM and those of the antibiotic growth promoter, BMD, can not be sufficiently distinguished based on genus-level variations in the cecal bacterial community composition. In stark contrast, replacement of SBM with high amounts of BSFLM in chicken diets induced a significant modification of the cecal bacterial structure, including increased community richness and Shannon’s diversity (Fig. 1) and long-term enrichment/depletion of specific bacterial genera (Fig. 3). Together these observations indicate that high amounts of BSFLM results in an imbalanced cecal bacterial microbiota (i.e., dysbiosis) that could be potentially disruptive to the optimal growth of broiler chickens.
In agreement with our observations associated with diets containing high amounts of BSFLM on the cecal bacterial diversity of male broiler chickens, similar studies examining the effect of complete replacement of SBM or fish meal with BSFLM in Lohmann Brown Classic laying hen [21] and ISA brown layer chick [34] diets, have also reported increased in bacterial richness and Shannon’s diversity index, together with significant changes in beta diversity of gut bacterial communities [34]. In contrast, for diets with low BSFLM content, Biasato et al. showed that replacement of SBM with 15% BSFLM (but not 5% and 10%) was associated with a reduction in Shannon’s diversity in the cecal microbiota of 35-day-old broiler chickens [35]. However, we observed no significant differences in any of the diversity metrics between the 12.5% BSFLM diet group and both BSFLM-free control groups (i.e., BMD and 0% BSFLM groups). Other related studies investigating a different approach of supplementing (instead of substituting) corn-SBM-based diets with lower (1–5%) inclusion levels of BSFLM in poultry diets have also provided some evidence of BSFLM-dependent changes in the cecal bacterial community diversity [36, 37]. Nonetheless, due to variations in experimental conditions such as bird species, BSFLM inclusion levels and source, sampling frequency, and diversity indices evaluated, comparing studies regarding the effect of BSFLM towards shaping the chicken microbiome remains challenging. Furthermore, despite the findings reported here and elsewhere, it is unclear if these changes in the cecal bacterial diversity of broiler chickens is driven directly by the introduction of BSFLM into the diet, and/or indirectly due to factors such as lower feed intake (i.e., related to changes in appetite) and other host-specific responses to a modified diet.
Notably, the two genera Shuttleworthia and Romboutsia were consistently suppressed or enriched, respectively, in birds receiving 100% BSFLM at all phases of growth (Fig. 3). Previous studies have shown that the abundance of Shuttleworthia in the cecum of chickens positively correlates with body weight gain [38], body weight [39], average daily feed intake, average daily gain, and negatively correlates with the feed-to-gain ratio [40]. It is tempting to speculate, then, that depletion of this genus in birds receiving 100% BSFLM contributes to the poor performance observed in this group. In addition to depletion of Shuttleworthia, a temporary reduction in the genera Eubacterium coprostanoligenes group, Lachnospiraceae UCG-010, Tyzerella 3, Anaerostipes, Butyricicoccus, Ruminococcus gauvreauii group, and Ruminiclostridium 5 of birds fed HIL-BSFLM was observed. These genera have also previously been positively correlated with poultry performance [41,42,43,44], and collectively, their short-term depletion in birds receiving high amounts of BSFLM may contribute, in part, to the poor performance exhibited for these birds. In fact, many species belonging to these genera are significant producers of SCFAs, which play a significant role in the intestinal health and growth performance of poultry. Yet, in the earlier study [18], we failed to detect any significant differences in the cecal SCFA levels between birds fed 100% BSFLM and all other control diets, suggesting that the growth-depressive effects of high amounts of BSFLM is independent of the reduced metabolic capacity of the cecal microbiota towards SCFA production.
Romboutsia is a genus that is commonly detected in the poultry gut [45]; however, the functional role of this genus in the intestinal tract and bird performance is currently unclear. Intriguingly, the intestinal abundance of this genus has been shown to increase in certain intestinal diseases such as dextran sulphate sodium-induced colitis in mice [46] and gastric cancer in humans [47]. Moreover, the species Romboutsia timonensis was first identified and isolated from the colon of patient suffering from severe anemia with melaena (gastrointestinal bleeding) [48]. To this end, a prolonged increase in the cecal abundance of Romboutsia in broiler chicken may serve as a bio-indicator of a compromised digestive system and, ultimately, poor growth performance.
Apart from the genus Romboutsia, during the grower and finisher phases, HIL-BSFLM was also associated with an increased cecal abundance of several genera including, Bilophila, Intestinimonas, Ruminococcaceae UCG-005/UCG-009/UCG-010, Anaerofilum, Candidatus Soleaferrea, and Christensenellaceae R-7 group. Bacterial species belonging to several of these genera have previously been functionally linked to the health and disease status of their host including Bilophila, Christensenellaceae R-7 group, Ruminococcaceae and Intestinimonas and these are discussed below. To the best of our knowledge, the genera Anaerofilum and Candidatus soleaferra are poorly described in the literature. Therefore, it is difficult to ascertain the possible impact that the enrichment of these two genera might have on poultry health and performance.
Among the Bilophila genus, high levels of the pathobiont Bilophila wadsworthia have been observed in fecal samples taken from humans suffering from Kwashiorkor, a severe form of malnutrition caused by a lack of protein [49]. Intriguingly, transplantation of the fecal flora high in B. wadsworthia from an individual suffering from Kwashiorkor to germ-free mice resulted in marked weight loss in recipient mice —an indication that the flora itself can directly and negatively impact body weight [49]. Additionally, transplantation of this fecal was also accompanied by perturbations in amino acid and carbohydrate metabolism in mice [49]. More recently, B. wadsworthia was shown to synergize with a high fat diet to promote inflammation, intestinal barrier dysfunction, bile acid and glucose dysmetabolism and hepatic steatosis in mice [50]. In 35-day-old broiler chickens, complete replacement of SBM with BSFLM was previously shown to affect amino acid metabolism [18, 19] and increase liver weight (i.e. hepatic hypertrophy) [19]. These findings are somewhat consistent with the negative effects that B. wadsworthia has on mouse physiology [49, 50]. Therefore, it is tempting to speculate that HIL-BSFLM-mediated expansion of Bilophila bacteria may interfere with broiler chicken body weight gain.
While the role of the genus Christensenellaceae R-7 group in poultry health is currently unclear, an inverse relationship between the relative abundance of gut Christensenellaceae and the body mass index of humans has previously been established [51]. Interestingly, it has been shown that amendment of an obese-associated microbiome with the species Christensenellaceae minuta in mice, can reduce weight gain when subsequently transferred into germ-free mice [52]. As well, a higher prevalence of Christensenellaceae was found in goats with lower growth rates [53]. Taken together, the Christensenellaceae R-7 group bacteria may have significant implications modulating body weight of broiler chicken, which may, in turn, explain the reduced performance associated with a high BSFLM content diet.
Of significance, three members of the taxonomically related members of the family Ruminococcaceae including UCG-005/UCG-009/UCG-010 were enriched in birds fed HIL-BSFLM. Ruminococcaceae family are capable of degrading and fermenting fibre containing complex polysaccharides [54] and members of this family have been reported to positively associated with poultry performance [55, 56]. Recently, Yan et al. demonstrated an increase in abundance of cecal Ruminococcaceae in hens fed diets supplemented with 2% and 3% BSFLM [37]. The authors of this study speculated that Ruminococcaceae may potentially degrade chitin (a linear polysaccharide) in BSFLM, thus explaining the increased abundance of members in this family [37]. Consistent with this, Ruminococcaceae were shown to increase in abundance in the human gut in response to chitin isolated from cricket powder [57]. Similarly, when frogs were switched from an herbivorous to an insect-enriched diet, Ruminococcaceae, which were shown to contain genes coding for putative chitin-digestion enzymes, increased in abundance within the gut [58]. These findings collectively point towards the possibility that BSFLM-derived chitin may be enriching for chitin-degrading Ruminococcaceae as an adaptive response to an insectivorous diet in broiler chickens.
The Intestinimonas genus has been shown to negatively correlate with average daily gain from days 1–48 in yellow-feathered broiler chickens [59]. Among this genus, Intestinimonas strain AF211 was shown to convert the Amadori product, Nε-fructosylysine, into butyrate [60]. Nε-fructosylysine is a sugar-amino acid product abundantly formed in heated foods via the non-enzymatic Maillard reaction, whereby the amino group of amino acids react with the carbonyl group of reduced sugar [60]. The Maillard reaction can cause degradation of nutritional protein quality as a result of destroying essential amino acids or reducing their availability [61], reduce protein digestibility [62], or inhibit the activity of digestive enzymes [62,63,64]. Importantly, commercial BSFLM undergoes common processing procedures, including heat treatment [65], which may have the potential to generate Amadori products (e.g., Nε-fructoselysine). Given that Intestinomonas was enriched in birds fed high amounts of BSFLM and that complete replacement of SBM with BSFLM in broiler chicken diets have been shown to interfere with protein digestibility [18, 20, 66], one possibility is that Maillard reaction products present in BSFLM impeded protein digestibility and this manifested in a decrease in performance and enrichment of Nε-fructoselysine-degrading Intestinimonas. Thus, future research efforts assessing the impact of heating BSFLM on the levels of Maillard reaction products warrants further investigation.
In the present study, dietary supplementation of the antibiotic BMD did not affect the overall diversity of the cecal microbiota for each phase of growth when compared to the otherwise identical diet of 0% BSFLM. In agreement with this, Proctor et al. demonstrated that the overall microbial diversity of the cecum remained unchanged in young broiler chickens fed therapeutic levels (200 g/tonne) of BMD compared to “raised without antibiotic” production [67]. Likewise, several other studies have found that the overall intestinal microbial diversity is not significantly altered by in-feed bacitracin [68,69,70,71]. Notably, Enterococci were found to be more abundant in chickens raised without antibiotic, regardless of the dietary exposure to BSFLM, compared to chickens fed BMD at the finisher phase (Fig. 3). This is consistent with the antibacterial activity of BMD against Enterococci spp., several of which are known to cause serious diseases in chickens [72]. In fact, bacitracin has previously been shown to eliminate E. faecalis from the intestine of broiler chickens, but not E. faecium, owing to an increased sensitivity of the former species to bacitracin [73]. However, the taxonomic resolution of the methods used in this study cannot discriminate between different Enterococci species.
In the present study, LIL-BSFLM did not impact the relative abundance of selected ARGs, indicating that inclusion of BSFLM, unlike BMD, in chicken diets has the added benefit of not enriching for ARGs important to human and animal health. This should be viewed as a positive outcome. In the ceca of birds fed BMD, the bacitracin resistance gene, bcrR, and the macrolide-lincosamide-streptogramin B resistance gene, ermB, were more abundant. In E. faecalis, the bcrAB genes encode for an ABC-type transporter (BcrAB) that is responsible for extruding bacitracin from the bacterial cell [74]. The gene bcrD encodes for an undecaprenol kinase, whose expression confers low levels of bacitracin resistance [75]. Expression of the bcrABD operon is positively regulated by the membrane-bound one-component BcrR regulator, whose gene is located upstream of this operon [76]. The gene ermB encodes for a ribosomal methyltransferase that dimethylates a single adenine in 23S rRNA, resulting in reduced affinity between macrolide-lincosamide-streptogramin (MLS) antibiotics and the ribosome [77]. The bcrABDR locus has previously been found on transferable plasmids in E. faecalis and Clostridium perfringens strains isolated from chicken samples [78, 79], as well as on the chromosome of bacitracin-resistant C. perfringens strains isolated from turkeys and broiler chickens [80]. Intriguingly, a multidrug resistance plasmid, pXD5, carrying both the bcrABDR and ermB genes has also been reported in E. faecium and E. facecalis of both human and swine origin [81]. This clearly indicates that bacitracin resistance and macrolide resistance can be genetically linked [81]. Given our observation that both bcrR and ermB were elevated in birds fed bacitracin, this could be evidence of BMD-mediated co-selection for bacitracin and macrolide resistance determinants.
The present study has several limitations. PCR amplification and sequencing of the V3-4 variable region of the 16S rRNA gene can introduce biases in taxonomic identification and does not provide bacterial species level resolution [82, 83]. To overcome these limitations, full-length 16S rRNA gene sequencing by PacBio and Nanopore long-read technology would have simultaneously improved the taxonomic resolution (i.e., species-level identification) of this study, while eliminating biases commonly associated with 16S rRNA gene amplicon-sequencing data [83,84,85]. Alternatively, a shotgun metagenomics approach would have also provided species-level resolution of the cecal microbiota [86] and would have provided insight into the potential functional metabolic capacity of the microbiota in response to dietary BSFLM. Another limitation of this study is that the bacterial microbiota of other regions along the gastrointestinal tract including the main nutrient absorption sites (duodenum, jejunum, and ileum) were not examined. Finally, while the poultry gastrointestinal tract is dominated by bacteria, other microorganisms such as fungi [87], viruses [88] and archaea [89, 90] participate in the poultry gut microbiome. Although little is known about the role of these other microorganisms in the chicken gut, elucidating the impact of dietary BSFLM as well as in-feed BMD on these microbial communities may provide insight into their performance-modulating properties.
Conclusions
In summary, our data indicate that 12.5% or 25% BSFLM can be incorporated into broiler chicken diets with minimal impact on the cecal bacterial community and without enriching for specific antimicrobial resistance determinants. In contrast, HIL-BSFLM impacted the cecal bacterial microbiota, leading to the formation of an imbalanced microbiota that, in turn, may compromise the overall health and performance of broiler chickens. While it is unclear how the dynamic changes in the cecal microbiome contribute to BSFLM-mediated effects on broiler performance, the genera that were differentially abundant in the cecal of broiler chickens fed HIL-BSFLM may serve as biomarkers for poor growth performance. Future studies aimed at deciphering causal relationships between these candidate microbial biomarkers is crucial for a better understanding of the cecal bacterial microbiota and its function in poultry health and performance.
Materials and methods
Study design and sampling
Experimental details outlining bird housing, composition of experimental diets, and broiler chicken performance data have been described previously [18]. Briefly, a total of 480, day-old male Ross x Ross 708 broiler chicks obtained from a commercial hatchery (Maple Leaf Foods, New Hamburg, On, Canada) were placed in forty-eight identical metabolic cages (10 birds per cage, 8 cages per diet) and divided into 6 experimental diets in a completely randomized design. The chicks were vaccinated with Bronchitis and Marek’s vaccines at the hatchery. Mash diets were formulated for a three-phase feeding program: Starter: d 0–14, Grower: d 15–28, and Finisher: d 29–35 that met or exceeded nutrient specification for Ross x Ross 708 [91]. The diets consisted of a corn-SBM based diet with BMD (BMD group) or without BMD (0% BSFLM), and four diets where the SBM was replaced with, 12.5%, 25%, 50% and 100% commercial BSFLM (Enterra Feed Inc., Vancouver, B.C., Canada., Lot #: L191203M-1). Black soldier fly larvae were reared on a 100% plant-based diet consisting of pre-consumer food wastes.
Sample collection
On days 14, 28, and 35, corresponding to the end of the starter, grower, and finisher phases, respectively, two birds from each cage (8 replicate cages per diet) were selected and weighed individually before being sacrificed by cervical dislocation. From each bird, ceca were aseptically collected and transferred into a sterile Whirl-Pak plastic bag (Nasco, Fort Atkinson, WI, USA), immediately placed on dry ice for transportation, and subsequently stored frozen at -80°C until further analysis. Day 35 broiler chicken excreta was also collected aseptically from slide-in trays that were placed underneath each of the 48 wire-mesh-floor cages and stored at -20°C for later analysis.
Microbial DNA isolation
Total microbial genomic DNA was extracted from 250 mg of cecal digesta and litter samples using the QIAmp Power Fecal Kit Pro DNA kit (Qiagen, Toronto, ON, Canada) and the DNeasy PowerSoil Pro Kit (Qiagen, Canada), respectively, according to the manufacturer’s instructions and using a final elution volume of 100 µl. In several instances, when the cecal digesta available was less than 250 mg, the entire digesta sample (40–190 mg) was used for extracting microbial DNA with the elution volume adjusted accordingly. Unless otherwise specified, the quality of the extracted nucleic acid samples were individually assessed using a Nanodrop™ One/OneC Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific, Toronto, ON, Canada) (A260/280) and by agarose gel-electrophoresis. The DNA concentrations of samples were determined using the Qubit double-stranded DNA high-sensitivity assay kit with a Qubit 4.0 Fluorometer (Thermo Fisher Scientific, Canada).
Microbial profiling 16S rRNA gene sequencing
To determine the composition of the cecal microbial communities, an Illumina 16S metagenomics sequencing workflow (Illumina Canada, Vancouver, BC, Canada) targeting the V3 and V4 variable region of the 16S rRNA gene was employed [92]. Briefly, the V3 and V4 region of the 16S rRNA gene (~ 460 bp) was amplified from the cecum-derived microbial genomic DNA in a 25 µl PCR mixture containing 12.5 ng of template DNA, 200 nM of each of the 16S amplicon PCR forward and reverse primers, and 1 x KAPA HiFi Hot start Ready Mix (Roche, Millipore-Sigma, Mississauga, ON). The PCR mixtures were heated for 3 min at 95oC, followed by 25 cycles of 30 s at 95°C, 30 s at 55°C, 30 s at 72°C, before finishing with 5 min at 72°C. The 16S rRNA gene amplicons were purified using AMPure XP magnetic beads (Beckman Coulter, Mississauga, ON, Canada), and adaptor-incorporated unique dual-indices were added to the amplicons using the Nextera XT index Kit v2 (Illumina, Canada) in accordance with the Illumina protocol. Indexed amplicon libraries were then further purified and normalized using the NGS normalization 96-well kit (Norgen Biotek, Thorold, ON, Canada) before pooling to a final concentration of 4 nM. High-throughput sequencing was performed on a MiSeq sequencer (Illumina, Canada) using the MiSeq v3 kit (600-cycle) and a loading concentration of 8 pM with 10% PhiX spike-in, to generate 2 × 300 bp paired-end sequences, targeting an output of 100,000 raw reads/sample.
Bioinformatics analyses
QIIME2, an end-to-end bioinformatics pipeline, with custom settings and the DADA2 option chosen for denoising, was used for downstream processing of the 16S rRNA gene sequencing output [93]. To assign taxonomy to the amplicon sequence variants, SILVA SSU reference database (release 132) was used for training classifiers. To remove primer sequences and any low-quality bases at the 5’ end, the first 21 bases from the forward read and the first 21 bases from the reverse-read were discarded. For quality trimming at the 3’ end, the forward and reverse reads were truncated at position 280 and 219, respectively, based on manual quality inspection.
Detection and quantification of antimicrobial resistance-associated gene targets
The abundance of 14 selected gene targets associated with antibiotic resistance (sul1, strA, strB, blaOXA-20, ermB, ermF, aadA, blaCMY-2, tetA, sul2, mphE, msrE and bcrR) or horizontal gene transfer (int1), were determined by quantitative real-time PCR (qPCR) using a Bio-Rad CFX96 real-time PCR instrument (Bio-Rad Laboratories, Mississauga, ON, Canada) with Bio-Rad CFX Maestro software version 3.0, as previously described [94]. The relative abundance of each gene was determined as the ratio of targeted gene copy number per total 16S rRNA gene, rrnS, copy numbers in the reaction. The primers and hydrolysis probes used in the present study are listed in Additional file 1, Table S3. All primers and hydrolysis probes were synthesized by Sigma (Sigma–Aldrich, Toronto, ON, Canada). The qPCR assays were performed using the Brilliant II QPCR Master Mix (Agilent) for TaqMan PCR and the Brilliant II SYBR Green® Low ROX qPCR Master Mix (Agilent, Toronto, ON, Canada) for SYBR Green qPCR. Two microlitres of template DNA (corresponding to 0.1–10 ng DNA) were added to each qPCR reaction mixture to reach a final volume of 25 µl. Each sample reaction, including the template-free control reaction, was run in triplicate with the following cycle conditions: 1 cycle at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and the established annealing temperature and extension time as specified for each primer set in Additional file 1. For the SYBR Green assay, melt curve analyses was performed as previously described [94]. When using TaqMan chemistry, the identity of the quantified gene targets were ensured on the basis of hybridization. The expected PCR product for each target gene was synthesized and subcloned into the vector, pBlueScript II SK (+) (Bio Basic Inc., Markham, Ontario, Canada). Plasmid copy numbers were calculated using the measured DNA concentration from a NanoDrop N-1000 spectrophotometer. Both amplification efficiencies (i.e., acceptable amplification efficiency between 95 and 105%) and gene target copy numbers were determined using standard curves consisting of a 10-fold serial dilution of a known target plasmid (from 107 to 100 copies/µl). Gene targets that were detected at copy numbers below 1–4 copies per reaction were considered to be below the limit of quantification [94].
Statistical analysis and data visualization
Unless otherwise specified, microbial compositional analysis was conducted using the open-source program R and RStudio (ver. 1.4.1106) [95]. The QIIME2-derived taxonomic assignment outputs were processed using R-based microbiome analytical packages qiime2R ver. 0.99.4 [96] and phyloseq ver. 1.34.0 [97], before graphical visualization by ggplot2 ver. 3.3.3 [98]. Contaminating sequences from negative control samples were identified and removed using the “ prevalence method” of the R decontam package [99]. To estimate alpha diversity, diversity indices were computed using R package vegan ver. 2.5-7 [100] and the statistical tests of non-parametric Kruskal-Wallis test and pairwise Dunn’s post-hoc test, with bonferroni correction to adjust p values for multiple pairwise comparisons, performed using the functions “kruskal_test” and “dunn-test” from R package rstatix ver. 0.7.0 [101]. Beta-diversity was estimated by performing principal coordinate analysis (PCoA) based on the Bray-Curtis dissimilarity using the “vegdist” and “pcoa” functions of R packages vegan and ape ver. 5.4-1 [102]. Significance of dissimilarity between groups were examined by permutational multivariate analysis of variance (PERMANOVA) using vegan function “adonis2” with 999 permutation, while the statistical significance of difference detected between any two groups was determined using the function “pairwise adonis” of R package pairwiseAdonis ver. 0.4 [103] with the Bonferroni method chosen for error correction and 999 permutations. The differential abundances of individual taxa between groups associated with different diets were determined using the Analysis of Compositions of Microbiomes with Bias Correction (ANCOM-BC2) methodology [32], conducted by running the “ancombc2” function of R package ANCOMBC ver. 2.02 with default options to take into account the independent variables of diet and cage separation as fixed- and random-effect, respectively. Any differential abundance with statistical significance at the bacterial taxonomic ranks of genus based on the mixed-directional false discovery rate-controlled Dunnett’s type of test output of ANCOM-BC2 were reported as log fold changes (with standard errors) relative to the reference BMD diet group. Non-parametric Mann Whitney U tests were used to compare antibiotic resistance-associated gene abundance between BSFLM-fed birds and BMD-fed birds, using GraphPad Prism v9.2 (La Jolla, CA, United States).
Data availability
All biological sequence data are accessible on the NCBI server under BioProject identifier (ID)PRJNA998860. Nucleotide sequences for 16S rRNA amplicon sequence data were submitted to the Sequence Read Archive (SRA) under SAMN36715391–SAMN36715686. All other data generated or analyzed during this study are included in this published article and its supplementary information files.
References
OECD/FAO, OECD-FAO Agricultural. Outlook 2023–2032. Paris: OECD Publishing; 2023. https://doi.org/10.1787/08801ab7-en.
Mottet A, Tempio G. Global poultry production: current state and future outlook and challenges. Worlds Poult Sci J. 2017;73(2):245–56.
Aguirre L, Cámara L, Smith A, Arroyo JJ, de Juan AF, Fondevila G, et al. Chemical composition, protein quality indicators and in vitro protein digestibility of commercial soybean meals from different origins for use in poultry feeding. Anim Feed Sci Technol. 2022;293:115473.
Prudêncio da Silva V, van der Werf HMG, Spies A, Soares SR. Variability in environmental impacts of Brazilian soybean according to crop production and transport scenarios. J Environ Manage. 2010;91(9):1831–9.
da Silva RFB, Viña A, Moran EF, Dou Y, Batistella M, Liu J. Socioeconomic and environmental effects of soybean production in metacoupled systems. Sci Rep. 2021;11(1):18662.
Mostert PF, Bos AP, van Harn J, de Jong IC. The impact of changing toward higher welfare broiler production systems on greenhouse gas emissions: a Dutch case study using life cycle assessment. Poult Sci. 2022;101(12):102151.
Singh B, Kaur G, Quintana-Ashwell NE, Singh G, Lo TH, Nelson KA. Row spacing and irrigation management affect soybean yield, water use efficiency and economics. Agric Water Manag. 2023;277:108087.
Zhang Q, Hong J, Zhang T, Tian X, Geng Y, Chen W, et al. Environmental footprints of soybean production in China. Envir Dev Sustain. 2022. https://doi.org/10.1007/s10668-022-02424-1.
Wu C, Wang Y, Shi X, Wang S, Ren H, Shen Z, et al. Rapid rise of the ESBL and mcr-1 genes in Escherichia coli of chicken origin in China, 2008–2014. Emerg Microbes Infect. 2018;7(1):1–10.
Dutil L, Irwin R, Finley R, Ng LK, Avery B, Boerlin P, et al. Ceftiofur resistance in Salmonella enterica Serovar Heidelberg from chicken meat and humans, Canada. Emerg Infect Dis. 2010;16(1):48–54.
Levy SB, Fitzgerald GB, Macone AB. Spread of antibiotic-resistant plasmids from chicken to chicken and from chicken to man. Nature. 1976;260(5546):40–2.
Van Looveren N, Verbaet L, Frooninckx L, Van Miert S, Van Campenhout L, Van Der Borght M, et al. Effect of heat treatment on microbiological safety of supermarket food waste as substrate for black soldier fly larvae (Hermetia illucens). Waste Manag. 2023;164:209–18.
Parodi A, Gerrits WJJ, Van Loon JJA, De Boer IJM, Aarnink AJA, Van Zanten HHE. Black soldier fly reared on pig manure: Bioconversion efficiencies, nutrients in the residual material, greenhouse gas and ammonia emissions. Waste Manag. 2021;126:674–83.
Parra Paz AS, Carrejo NS, Gómez Rodríguez CH. Effects of larval density and feeding rates on the bioconversion of vegetable waste using black soldier fly larvae Hermetia illucens (L.), (Diptera: Stratiomyidae). Waste Biomass Valori. 2015;6(6):1059–65.
Kemboi VJ, Kipkoech C, Njire M, Were S, Lagat MK, Ndwiga F, et al. Biocontrol potential of chitin and Chitosan extracted from black soldier fly pupal exuviae against bacterial wilt of tomato. Microorganisms. 2022;10(1):165.
Borrelli L, Varriale L, Dipineto L, Pace A, Menna LF, Fioretti A. Insect derived lauric acid as promising alternative strategy to antibiotics in the antimicrobial resistance scenario. Front Microbiol. 2021;12.
Moretta A, Salvia R, Scieuzo C, Di Somma A, Vogel H, Pucci P, et al. A bioinformatic study of antimicrobial peptides identified in the black soldier fly (BSF) Hermetia illucens (Diptera: Stratiomyidae). Sci Rep. 2020;10(1):16875.
Fruci M, Kithama M, Kiarie EG, Shao S, Liu H, Topp E, et al. Effects of partial or complete replacement of soybean meal with commercial black soldier fly larvae (Hermetia illucens) meal on growth performance, cecal short chain fatty acids, and excreta metabolome of broiler chickens. Poul Sci. 2023;102(4):102463.
Facey H, Kithama M, Mohammadigheisar M, Huber L-A, Shoveller AK, Kiarie EG. Complete replacement of soybean meal with black soldier fly larvae meal in feeding program for broiler chickens from placement through to 49 days of age reduced growth performance and altered organs morphology. Poult Sci. 2023;102(1):102293.
Velten S, Neumann C, Bleyer M, Gruber-Dujardin E, Hanuszewska M, Przybylska-Gornowicz B, Liebert F. Effects of 50% substitution of soybean meal by alternative proteins from Hermetia illucens or Spirulina platensis in meat-type chicken diets with graded amino acid supply. Open J Anim Sci. 2018;8:119–36.
Borrelli L, Coretti L, Dipineto L, Bovera F, Menna F, Chiariotti L et al. Insect-based diet, a promising nutritional source, modulates gut microbiota composition and SCFAs production in laying hens. 2017;7(1):1–11.
Yin Z, Ji S, Yang J, Guo W, Li Y, Ren Z, et al. Cecal microbial succession and its apparent association with nutrient metabolism in broiler chickens. mSphere. 2023;8(3):e00614–22.
Karasawa Y. Significant role of the nitrogen recycling system through the ceca occurs in protein-depleted chickens. J Exp Zool. 1999;283(4–5):418–25.
Clench MH, Mathias JR. The avian cecum: a review. Wilson Bull. 1995;107(1):93–121.
Pan D, Yu Z. Intestinal microbiome of poultry and its interaction with host and diet. Gut Microbes. 2014;5(1):108–19.
Rubinelli PM, Kim SA, Park SH, Roto SM, Nealon NJ, Ryan EP, et al. Differential effects of rice bran cultivars to limit Salmonella Typhimurium in chicken cecal in vitro incubations and impact on the cecal microbiome and metabolome. PLoS ONE. 2017;12(9):e0185002.
Yadav S, Jha R. Strategies to modulate the intestinal microbiota and their effects on nutrient utilization, performance, and health of poultry. J Anim Sci Biotech. 2019;10(1):2.
Józefiak D, Rutkowski A, Martin SA. Carbohydrate fermentation in the avian ceca: a review. Anim Feed Sci Techn. 2004;113(1):1–15.
Hegde SN, Rolls BA, Coates ME. The effects of the gut microflora and dietary fibre on energy utilization by the chick. Br J Nutr. 1982;48(1):73–80.
Diarra MS, Rempel H, Champagne J, Masson L, Pritchard J, Topp E. Distribution of antimicrobial resistance and virulence genes in Enterococcus spp. and characterization of isolates from broiler chickens. Appl Environ Microbiol. 2010;76(24):8033–43.
Rubio LA. Possibilities of early life programming in broiler chickens via intestinal microbiota modulation. Poult Sci. 2019;98(2):695–706.
Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11(1):3514.
Tiihonen K, Kettunen H, Bento MHL, Saarinen M, Lahtinen S, Ouwehand AC, et al. The effect of feeding essential oils on broiler performance and gut microbiota. Br Poult Sci. 2010;51(3):381–92.
Ndotono EW, Khamis FM, Bargul JL, Tanga CM. Gut microbiota shift in layer pullets fed on black soldier fly larvae-based feeds towards enhancing healthy gut microbial community. Sci Rep. 2022;12(1):16714.
Biasato I, Ferrocino I, Dabbou S, Evangelista R, Gai F, Gasco L, et al. Black soldier fly and gut health in broiler chickens: insights into the relationship between cecal microbiota and intestinal mucin composition. J Anim Sci Biotech. 2020;11(1):11.
He C, Lei J, Yao Y, Qu X, Chen J, Xie K et al. Black soldier fly (Hermetia illucens) larvae meal modulates intestinal morphology and microbiota in Xuefeng black-bone chickens. 2021;12.
Yan Y, Zhang J, Chen X, Wang Z. Effects of black soldier fly larvae (Hermetia illucens larvae) meal on the production performance and cecal microbiota of hens. Vet Sci. 2023;10(5):364.
Liu YS, Li S, Wang XF, Xing T, Li JL, Zhu XD, et al. Microbiota populations and short-chain fatty acids production in cecum of immunosuppressed broilers consuming diets containing γ-irradiated Astragalus polysaccharides. Poult Sci. 2021;100(1):273–82.
Lee K-C, Kil DY, Sul WJ. Cecal microbiome divergence of broiler chickens by sex and body weight. J Microbiol. 2017;55(12):939–45.
Zhu C, Huang K, Bai Y, Feng X, Gong L, Wei C, et al. Dietary supplementation with berberine improves growth performance and modulates the composition and function of cecal microbiota in yellow-feathered broilers. Poult Sci. 2021;100(2):1034–48.
Yan J, Zhou B, Xi Y, Huan H, Li M, Yu J, et al. Fermented feed regulates growth performance and the cecal microbiota community in geese. Poul Sci. 2019;98(10):4673–84.
Han H, Zhou Y, Liu Q, Wang G, Feng J, Zhang M. Effects of ammonia on gut microbiota and growth performance of broiler chickens. 2021;11(6):1716.
Madigan-Stretton J, Mikkelsen D, Soumeh EA. Multienzyme super-dosing in broiler chicken diets: the implications for gut morphology, microbial profile, nutrient digestibility, and bone mineralization. Animal. 2020. https://doi.org/10.3390/ani11010001.
Xiang H, Gan J, Zeng D, Li J, Yu H, Zhao H, et al. Specific microbial taxa and functional capacity contribute to chicken abdominal fat deposition. Front Microbiol. 2021. https://doi.org/10.3389/fmicb.2021.643025.
Wiersema ML, Koester LR, Schmitz-Esser S, Koltes DA. Comparison of intestinal permeability, morphology, and ileal microbial communities of commercial hens housed in conventional cages and cage-free housing systems. Poult Sci. 2021;100(2):1178–91.
Wang H-G, Zhang M-N, Wen X, He L, Zhang M-H, Zhang J-L, et al. Cepharanthine ameliorates dextran sulphate sodium-induced colitis through modulating gut microbiota. Microb Biotechn. 2022;15(8):2208–22.
Zhang X, Li C, Cao W, Zhang Z. Alterations of gastric microbiota in gastric cancer and precancerous stages. Front Cell Infect Microbiol. 2021. https://doi.org/10.3389/fcimb.2021.559148.
Ricaboni D, Mailhe M, Khelaifia S, Raoult D, Million M. Romboutsia Timonensis, a new species isolated from human gut. New Microbes New Infect. 2016;12:6–7.
Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, et al. Gut microbiomes of Malawian twin pairs discordant for Kwashiorkor. Science. 2013;339(6119):548–54.
Natividad JM, Lamas B, Pham HP, Michel M-L, Rainteau D, Bridonneau C, et al. Bilophila wadsworthia aggravates high fat diet induced metabolic dysfunctions in mice. Nat Commun. 2018;9(1):2802.
Waters JL, Ley RE. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019;17(1):83.
Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–99.
Wang D, Tang G, Zhao L, Wang M, Chen L, Zhao C, et al. Potential roles of the rectum keystone microbiota in modulating the microbial community and growth performance in goat model. J Anim Sci Biotechnol. 2023;14(1):55.
Biddle A, Stewart L, Blanchard J, Leschine S. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity. 2013;5(3):627–40.
Jacquier V, Nelson A, Jlali M, Rhayat L, Brinch KS, Devillard E. Bacillus subtilis 29784 induces a shift in broiler gut microbiome toward butyrate-producing bacteria and improves intestinal histomorphology and animal performance. Poult Sci. 2019;98(6):2548–54.
Yang J, Huang K, Wang J, Wu D, Liu Z, Yu P, et al. Combined use of Bacillus subtilis yb-114,246 and Bacillus licheniformis yb-214,245 improves body growth performance of Chinese Huainan partridge shank chickens by enhancing intestinal digestive profiles. Probiotics Antimicrob Proteins. 2021;13(2):327–42.
Refael G, Riess HT, Levi CS, Magzal F, Tamir S, Koren O, et al. Responses of the human gut microbiota to physiologically digested insect powders or isolated chitin thereof. Future Foods. 2022. https://doi.org/10.1016/j.fufo.2022.100197.
Zhang M, Chen H, Liu L, Xu L, Wang X, Chang L, et al. The changes in the frog gut microbiome and its putative oxygen-related phenotypes accompanying the development of gastrointestinal complexity and dietary shift. Front Microb. 2020. https://doi.org/10.3389/fmicb.2020.00162.
Ding Y, Hu Y, Yao X, He Y, Chen J, Wu J, et al. Dietary essential oils improves the growth performance, antioxidant properties and intestinal permeability by inhibiting bacterial proliferation, and altering the gut microbiota of yellow-feather broilers. Poult Sci. 2022. https://doi.org/10.1016/j.psj.2022.102087.
Bui TPN, Ritari J, Boeren S, de Waard P, Plugge CM, de Vos WM. Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat Commun. 2015. https://doi.org/10.1038/ncomms10062.
Moughan PJ, Gall MPJ, Rutherfurd SM. Absorption of lysine and deoxyketosyllysine in an early-Maillard browned casein by the growing pig. J Agric Food Chem. 1996;44(6):1520–5.
Seiquer I, Díaz-Alguacil J, Delgado-Andrade C, López-Frías M, Muñoz Hoyos A, Galdó G, et al. Diets rich in Maillard reaction products affect protein digestibility in adolescent males aged 11–14 y. Am J Clin Nutr. 2006;83(5):1082–8.
Aljahdali N, Carbonero F. Impact of Maillard reaction products on nutrition and health: current knowledge and need to understand their fate in the human digestive system. Crit Rev Food Sci Nutr. 2019;59(3):474–87.
Oste RE, Dahlqvist A, Sjoestroem H, Noren O, Miller R. Effect of Maillard reaction products on protein digestion. In vitro studies. J Agric Food Chem. 1986;34(2):355–8.
Wang YS, Shelomi M. Review of black soldier fly (Hermetia illucens) as animal feed and human food. Foods. 2017. https://doi.org/10.3390/foods6100091.
Hartinger K, Greinix J, Thaler N, Ebbing MA, Yacoubi N, Schedle K, et al. Effect of graded substitution of soybean meal by Hermetia illucens larvae meal on animal performance, apparent ileal digestibility, gut histology and microbial metabolites of broilers. Animals. 2021;11(6):1628.
Proctor A, Phillips GJ. Differential effects of bacitracin methylene disalicylate (BMD) on the distal colon and cecal microbiota of young broiler chickens. Front Vet Sci. 2019. https://doi.org/10.3389/fvets.2019.00114.
Gong J, Yu H, Liu T, Gill JJ, Chambers JR, Wheatcroft R, et al. Effects of zinc bacitracin, bird age and access to range on bacterial microbiota in the ileum and caeca of broiler chickens. J Appl Microbiol. 2008;104(5):1372–82.
Neumann AP, Suen G. Differences in major bacterial populations in the intestines of mature broilers after feeding virginiamycin or bacitracin methylene disalicylate. J Appl Microbiol. 2015;119(6):1515–26.
Pedroso AA, Menten JF, Lambais MR, Racanicci AM, Longo FA, Sorbara JO. Intestinal bacterial community and growth performance of chickens fed diets containing antibiotics. Poult Sci. 2006;85(4):747–52.
Pourabedin M, Guan L, Zhao X. Xylo-oligosaccharides and virginiamycin differentially modulate gut microbial composition in chickens. Microbiome. 2015. https://doi.org/10.1186/s40168-015-0079-4.
Dolka B, Gołębiewska–Kosakowska M, Krajewski K, Kwieciński P, Nowak T, Szubstarski J, et al. Occurrence of Enterococcus spp. in poultry in Poland based on 2014–2015 data. Med Weter. 2017;73(4):220–4.
Barnes EM, Mead GC, Impey GS, Adams BW. The effect of dietary bacitracin on the incidence of Streptococcus faecalis subspecies liquefaciens and related Streptococci in the intestines of young chicks. Br Poult Sci. 1978;19(6):713–23.
Gebhard S, Fang C, Shaaly A, Leslie DJ, Weimar MR, Kalamorz F, et al. Identification and characterization of a bacitracin resistance network in Enterococcus faecalis. Antimicrob Agents Chemother. 2014;58(3):1425–33.
Shaaly A, Kalamorz F, Gebhard S, Cook GM. Undecaprenyl pyrophosphate phosphatase confers low-level resistance to bacitracin in Enterococcus faecalis. J Antimicrob Chemother. 2013;68(7):1583–93.
Manson JM, Keis S, Smith JMB, Cook GM. Acquired bacitracin resistance in Enterococcus faecalis is mediated by an ABC transporter and a novel regulatory protein. BcrR. 2004;48(10):3743–8.
Yu L, Petros AM, Schnuchel A, Zhong P, Severin JM, Walter K, et al. Solution structure of an rRNA methyltransferase (ErmAM) that confers macrolide-lincosamide-streptogramin antibiotic resistance. Nat Struct Biol. 1997;4(6):483–9.
Chen MY, Lira F, Liang HQ, Wu RT, Duan JH, Liao XP, et al. Multilevel selection of bcrABDR-mediated bacitracin resistance in Enterococcus faecalis from chicken farms. Sci Rep. 2016. https://doi.org/10.1038/srep34895.
Han X, Du XD, Southey L, Bulach DM, Seemann T, Yan XX, et al. Functional analysis of a bacitracin resistance determinant located on ICECp1, a novel Tn916-like element from a conjugative plasmid in Clostridium perfringens. Antimicrob Agents Chemother. 2015;59(11):6855–65.
Charlebois A, Jalbert L-A, Harel J, Masson L, Archambault M. Characterization of genes encoding for acquired bacitracin resistance in Clostridium perfringens. PLoS ONE. 2012. https://doi.org/10.1371/journal.pone.0044449.
Wang X-M, Li X-S, Wang Y-B, Wei F-S, Zhang S-M, Shang Y-H, et al. Characterization of a multidrug resistance plasmid from Enterococcus faecium that harbours a mobilized bcrABDR locus. J Antimicrob Chemother. 2014;70(2):609–11.
Kameoka S, Motooka D, Watanabe S, Kubo R, Jung N, Midorikawa Y, et al. Benchmark of 16S rRNA gene amplicon sequencing using Japanese gut microbiome data from the V1–V2 and V3–V4 primer sets. BMC Genom. 2021. https://doi.org/10.1186/s12864-021-07746-4.
Wagner J, Coupland P, Browne HP, Lawley TD, Francis SC, Parkhill J. Evaluation of PacBio sequencing for full-length bacterial 16S rRNA gene classification. BMC Microbiol. 2016. https://doi.org/10.1186/s12866-016-0891-4.
Callahan BJ, Wong J, Heiner C, Oh S, Theriot CM, Gulati AS, et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019. https://doi.org/10.1093/nar/gkz569.
Matsuo Y, Komiya S, Yasumizu Y, Yasuoka Y, Mizushima K, Takagi T, et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION™ nanopore sequencing confers species-level resolution. BMC Microbiol. 2021. https://doi.org/10.1186/s12866-021-02094-5.
Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL. Analysis of the microbiome: advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun. 2016;469(4):967–77.
Robinson K, Xiao Y, Johnson TJ, Chen B, Yang Q, Lyu W, et al. Chicken intestinal mycobiome: initial characterization and its response to bacitracin methylene disalicylate. Appl Environ Microbiol. 2020. https://doi.org/10.1128/AEM.00304-20.
Yan T, Li G, Zhou D, Hu L, Hao X, Li R, et al. Long read sequencing revealed proventricular virome of broiler chicken with transmission viral proventriculitis. BMC Vet Res. 2022. https://doi.org/10.1186/s12917-022-03339-9.
Saengkerdsub S, Herrera P, Woodward CL, Anderson RC, Nisbet DJ, Ricke SC. Detection of methane and quantification of methanogenic archaea in faeces from young broiler chickens using real-time PCR. Lett Appl Microbiol. 2007;45(6):629–34.
Saengkerdsub S, Anderson RC, Wilkinson HH, Kim W-K, Nisbet DJ, Ricke SC. Identification and quantification of methanogenic archaea in adult chicken ceca. Appl Environ Microbiol. 2007;73(1):353–6.
Aviagen W. Ross 308 broiler nutrition specifications. Aviagen: Huntsville, AL, USA. 2014.
Amplicon PCR. P.C.R. Clean-up, and P.C.R. index. 16S metagenomic sequencing library preparation. Illumina:SanDiego, CA, USA. 2013.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–7.
Marti R, Scott A, Tien Y-C, Murray R, Sabourin L, Zhang Y, et al. Impact of manure fertilization on the abundance of antibiotic-resistant bacteria and frequency of detection of antibiotic resistance genes in soil and on vegetables at harvest. Appl Environ Microbiol. 2013;79(18):5701–9.
Team RC. R: A language and environment for statistical computing [Internet]. Vienna, Austria.: R Foundation for Statistical Computing. (2021). Accessed 2023.
Bisanz JE. Importing QIIME2 artifacts and associated data into R sessions. 2018. https://github.com/jbisanz/qiime2R.
McMurdie PJ, Holmes S. Phyloseq: an R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013. https://doi.org/10.1371/journal.pone.0061217.
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016.
Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018. https://doi.org/10.1186/s40168-018-0605-2.
Oksanen JBF, Friendly M, Kindt R, Legendre P, McGlinn D et al. vegan: community ecology package. http://CRAN.R-project.org/package=vegan (2020).
A K. Pipe-friendly framework for basic statistical tests in R. http://CRAN.R-project.org/package=rstatix (2021).
Paradis E, Schliep K. Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2018. https://doi.org/10.1093/bioinformatics/bty633.
Martinez Arbizu P, pairwiseAdonis. Pairwise multilevel comparison using adonis. R package version 0.4. (2020).
CCAC. Canadian care on animal care (CCAC) guidelines on: The care and use of farm animals in research, teaching and testing. 2009.
Acknowledgements
We thank the staff at the Arkell Poultry Research Station (University of Guelph, Guelph, ON, Canada) for helping with the chicken feeding trial. We also thank the bioinformatics unit of the Research and Development Section at the Carling laboratory of CFIA for the technical assistance provided in analyzing the 16S rRNA sequencing data.
Funding
Research funding was awarded through AAFC and the Canadian Federal Genomics Research and Development Initiative on AMR II (GRDI-AMR2) to M. Fruci, E. Topp, and M. Diarra. This research was also supported with funding awarded through the Ontario Agri-Food Innovation Alliance [grant number: 27320] and Canada First Research Excellence [grant number: 499129] to E. Kiarie.
Author information
Authors and Affiliations
Contributions
M.F. conceived the study. M.F., E.K., and M.D. designed the study. Sample collection was performed by M.F., M.K., and H.K. L.V. completed cecal digesta DNA extraction and quality analyses. Y.T. completed litter DNA extraction and quality analyses, and qPCR assays. E.T. and Y.T. analyzed qPCR data. C.L. and S.C. performed 16S rRNA gene amplicon sequencing and bioinformatic analyses. M.F. and C.L. wrote the main manuscript, prepared figures and tables, and supplementary files. M.F., C.L., E.K., M.D., M.K., and E.T. contributed to editing the manuscript. All authors read and approved the submitted manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All experimental procedures performed were approved (AUP #3521) by the Animal Care Committee of the University of Guelph Animal Ethics Committee according to the guidelines of the Canadian Council of Animal Care [104].
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Additional file 1: Supplementary Material 1. Table S1.
16S rRNA gene sequencing output statistics derived from QIIME2-enabled DADA2 plugin; Table S2. Two-factor permutational analysis of variance (PERMANOVA) results for the broiler chicken cecal bacterial community composition; Table S3. Primers and probes used for quantitative PCR in detection of antimicrobial resistance genes.
Additional file 2: Supplementary Material 2. Figure S1.
Box and whisker plots comparing the effects of age on the alpha diversity metrics of the cecal bacterial community for a given diet; Figure S2. Genus-level PCoA based on the Bray-Curtis dissimilarity comparing the effects of age on the cecal bacterial community composition of chickens fed different experimental diets; Figure S3. Genus-level PCoA based on the Bray-Curtis dissimilarity comparing the effects of diet on the cecal bacterial community composition of chickens at each growth phase.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lau, C.HF., Capitani, S., Tien, YC. et al. Dynamic effects of black soldier fly larvae meal on the cecal bacterial microbiota and prevalence of selected antimicrobial resistant determinants in broiler chickens. anim microbiome 6, 6 (2024). https://doi.org/10.1186/s42523-024-00293-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-024-00293-9