
Abstract
Background
The congenital long QT syndrome type 2 is caused by mutations in KCNH2 gene that encodes the alpha subunit of potassium channel Kv11.1. The carriers of the pathogenic variant of KCNH2 gene manifest a phenotype characterized by prolongation of QT interval and increased risk of sudden cardiac death due to life-threatening ventricular tachyarrhythmias.
Results
A family composed of 17 members with a family history of sudden death and recurrent syncopes was studied. The DNA of proband with clinical manifestations of long QT syndrome was analyzed using a massive DNA sequencer that included the following genes: KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, ANK2, KCNJ2, CACNA1, CAV3, SCN1B, SCN4B, AKAP9, SNTA1, CALM1, KCNJ5, RYR2 and TRDN. DNA sequencing of proband identified a novel pathogenic variant of KCNH2 gene produced by a heterozygous frameshift mutation c.46delG, pAsp16Thrfs*44 resulting in the synthesis of a truncated alpha subunit of the Kv11.1 ion channel. Eight family members manifested the phenotype of long QT syndrome. The study of family segregation using Sanger sequencing revealed the identical variant in several members of the family with a positive phenotype.
Conclusions
The clinical and genetic findings of this family demonstrate that the novel frameshift mutation causing haploinsufficiency can result in a congenital long QT syndrome with a severe phenotypic manifestation and an elevated risk of sudden cardiac death.
Similar content being viewed by others


Background
The congenital long QT syndrome (LQTS) is characterized by prolongation of QT interval on the electrocardiogram (ECG) and increased risk of sudden cardiac death due to life-threatening ventricular tachyarrhythmias like torsades de pointes and ventricular fibrillation [1,2,3].
Since 1995, mutations have been identified in 20 different genes responsible for congenital LQTS. Most of them are clinically known as Romano–Ward syndrome inherited with autosomal dominant pattern. In contrast, those included in Jervell and Lange-Nielsen syndrome show autosomal recessive inheritance pattern and are associated with sensorineural hearing loss [4,5,6,7].
The LQTS type 2 represents 30–45% of all cases of LQTS [8]. It is caused by mutations in the KCNH2 gene that encodes the alpha subunit of voltage dependent potassium channel known as Kv11.1 (or hERG) responsible for rectifier current IKr (OMIM # 613688).
We report a novel pathogenic variant of KCNH2 in a family whose members manifest LQTS type 2 with high penetrance and severe phenotype characterized by long QT interval, recurrent syncopes and multiple family antecedents of sudden death.
Methods
Study participants
A family composed of 17 members with a family history of sudden death and recurrent syncopes was studied.
DNA isolation and sequencing
After signing the informed consent, peripheral blood samples were obtained from the proband and first and second degree relatives. Initially, the proband’s DNA sample was sent to Molecular Biology and Pathology Laboratory of High Medical Group (Buenos Aires, Argentina). Sequencing was carried out on a NextSeq500 instrument (Illumina, San Diego, CA, USA) as 151-bp paired-end runs. Exome analysis of the proband yielded a mean coverage of 149.89X. The most common genes associated with LQTS were selected from ClinVar, OMIM (Online Mendelian Inheritance in Man) databases and the international public literature [9]. The exonic and adjacent intronic regions of the following 17 genes were sequenced: KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, ANK2, KCNJ2, CACNA1, CAV3, SCN1B, SCN4B, AKAP9, SNTA1, CALM1, KCNJ5, RYR2 and TRDN.
Read alignment and variant calling were performed with Burrows-Wheeler Aligner and FreeBayes. The variants have been annotated according to reference sequences cited in the Human Gene Mutation Database. For the filtering of variants, the quality of the genotype, the allelic frequency and the coverage of the position were considered. For variant validation and family segregation analysis, Sanger sequencing was performed. Genomic DNA (gDNA) was extracted using the EasyPure Blood Genomic DNA Kit (Trans, China, Cat. No. EE121), and then quantified using a Qubit fluorometer (Invitrogen, California, USA). Amplifications corresponding to exon 1 were carried out on a Verity® thermal cycler (Applied Biosystems, Massachussets, USA). The PCR products were purified using AccuPrep®PCR Purification Kit (Bioneer, cat. No. K3034). Finally, the Sanger-sequencing was performed on an ABI PRISM 3730XL Analyzer 96 capillary type (Applied Biosystems, Massachussets, USA) and subsequently analyzed with Variant Reporter Software Version 1.1 at the next-generation sequencing facility of Macrogen (Seoul, Republic of Korea).
Results
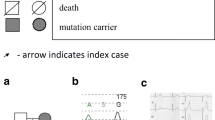
The proband was a 42-year-old Caucasian woman with family history of sudden death. Her father (46 years old) and 1 brother (28 years old) died while sleeping at night. Another brother (17 years old) died suddenly after doing physical activity. Since puberty, she had repeated episodes of syncope. The ECG showed a QTc interval (Bazett’s formula: QTc = QT/RR1/2) of 510 ms (Fig. 1a). The 24-h Holter monitoring did not show significant cardiac arrhythmias. The tilt test was normal. Transthoracic echocardiogram and cardiac nuclear magnetic resonance showed no structural abnormalities. In the absence of a history of drug use that could prolong the QT interval, a diagnosis of LQTS was made according to diagnostic criteria of international guidelines (score of Schwartz: 4.5 points. QT > 480 ms = 3 points; syncope without stress = 1 point; and unexplained sudden cardiac death at age < 30 years in immediate family = 0.5 points) [3, 10,11,12].

a The ECG of proband. The QTc interval was prolonged: 510 ms. b Family pedigree. Seventeen individuals with their respective ages and QTc interval values (between parentheses). Squares depict male subjects; circle, female subject; open symbols, unaffected members; and solid symbol, affected member. Arrow (P) denotes proband. Her father and 2 brothers died suddenly at a young age (cross). Three family members had recurrent syncope refractory to beta-blockers and/or non-sustained ventricular tachycardia requiring an implantable cardiac defibrillator (ICD). The genetic study was carried out on 7 family members (asterisk). The novel pathogenic variant was detected in five of them that manifested the LQTS phenotype
She received beta-blocker treatment with nadolol 40 to 80 mg/day or propranolol 120 to 240 mg/day orally. Despite beta-blocker therapy that did not result in a substantial reduction in the QTc interval, she had several recurrent episodes of syncope and non-sustained ventricular tachycardia. Because the patient had high-risk features of LQTS (QTc > 500 ms, women with type 2 genotype long QT syndrome and recurrent syncope), an implantable cardiac defibrillator (ICD) was indicated [3, 13]. No electrical shocks were observed for ventricular arrhythmias after ICD.
Subsequent family study revealed that 8 of the proband’s relatives, including her 12-year-old daughter, 3 siblings (2 women and 1 male), 2 nephews and 2 nieces manifested LQTS phenotype. Two of them (1 sister and 1 nephew) presented recurrent episodes of refractory syncopes and non-sustained ventricular tachycardia requiring an implantable cardiac defibrillator (Fig. 1b).
The proband’s DNA analysis identified the presence of a heterozygous variant occurred by deletion of guanine nucleotide from position 46 of exon 1 of KCNH2 gene (c.46delG) that replaces aspartic acid with threonine at position 16 of the protein product. The reading frame shift generates the appearance of a premature stop codon in exon 2 after encoding 44 amino acids from mutation site (p.Asp16Thrfs*44) and produces a truncated protein with a loss of 90% of amino acids (Fig. 2). Subsequent Sanger-sequencing to study familial segregation revealed the identical variant in 4 other family members with positive phenotype (daughter, 1 sister, 1 nephew and 1 niece).

a Electropherogram of the sequence surrounding the heterozygous mutation in KCNH2 from the proband’s genomic DNA. b Scheme of cDNA region involved in c.46delG variant. Nucleotides are separated into triplets (codon) with their corresponding amino acid. The deletion of guanine located at position 46 (asterisk) in exon 1 (codons marked in white) of KCNH2 gene produces a reading frame shift in the mutant allele that generates a premature stop codon in exon 2 (codons marked in gray) resulting in a truncated protein
Discussion
The congenital LQTS is characterized by prolongation of the ventricular myocardial action potential due to the increase in sodium and calcium input currents (INa and ICaL) or the decrease in potassium outflow currents (IKs, IKr and IK1). Molecular genetic studies identified mutations in 20 different genes that encode the subunits of cardiac ion channels and/or modulator proteins that directly or indirectly intervene in the formation of these currents [7, 14].
The KCNH2 (potassium voltage-gated channel subfamily H member 2) gene also known as hERG (human Ether-à-go-go-Related Gene) which is located on chromosome 7 (locus 7q35–36) is composed of 15 exons that encode the alpha subunit of the voltage-dependent potassium channel called Kv11.1 of 1159 amino acids. Four alpha subunits of Kv11.1 assemble a tetrameric ionic channel that generates the rapidly activating delayed rectifier potassium current (IKr) during phase 3 of the repolarization of myocardial cells. The loss of ion channel function results in increase in the duration of myocardial action potentials manifested as prolonged QT intervals in the ECG of patients with LQTS type 2 [15, 16].
Approximately 40% of KCNH2 gene mutations related to LQTS type 2 are: (a) nonsense mutations (point mutation in a sequence of DNA that results in a premature stop codon) or (b) frameshift mutation caused by the insertion or deletion of nucleotides in a DNA sequence and results a completely different reading frame from the original [15]. These mutations alter protein synthesis and generate defective alpha subunits of Kv11.1 channel. The remaining 60% of the mutations of KCNH2 gene are missense mutations, where a single nucleotide change alters an amino acid codon causing loss of channel function by disrupting the intracellular traffic of Kv11.1 to the cell membrane, altering channel gating or negatively affecting ion permeability [15, 17].
In heterozygous mutations of KCNH2 encoding truncated alpha subunits of Kv11.1 or disrupting intracellular traffic, the defective proteins are degraded by proteasomes. If alpha subunits encoded from the wild-type allele are still homomerized to form functional channels, the affected individuals manifest a less severe phenotype (haploinsufficiency). In contrast, individuals with point mutations that alter channel permeability or gating can assemble Kv11.1 heteromeric dysfunctional channels with alpha subunits encoded by both alleles (wild-type and mutant) and manifest a more severe phenotype (negative dominance) [18, 19].
In the sudden death risk stratification of patients with long QT syndrome, all patients with a mutation at the KCNQ1 gene (LQT1 locus) who have a QTc of 500 ms or more, male patients with a mutation at the KCNH2 gene (LQT2 locus) who have a QTc of 500 ms or more, all female patients with a mutation at the LQT2 locus irrespective of the QTc, and all patients with a mutation at the SCN5A gene (LQT3 locus) are considered high risk [20]. Most members of proband’s family, irrespective of the sex, had QT interval of 500 ms or more (Fig. 1b) and recurrent episodes of syncope.
At present, the beneficial effect of beta blockers in the treatment of patients with LQTS is clear. There is consensus that nadolol, unfortunately not available in all countries (including Argentina since 2016), is probably one of the most effective drugs. The efficacy of other more commonly prescribed beta-blockers such as propranolol, metoprolol, and atenolol is in dispute, especially in symptomatic patients [21, 22].
For patients with symptomatic LQTS in whom the beta-blocker is ineffective or poorly tolerated, left cardiac sympathetic denervation (LCSD) and/or an ICD are recommended [3, 13]. LCSD may be more effective in patients with long QT syndrome type 1. Although a marked reduction in the incidence of aborted cardiac arrest and syncope is usually seen after LCSD, 20% to 50% of patients with high-risk LQTS have experienced at least 1 recurrent arrhythmic event after LCSD [23, 24]. Therefore, LCSD is an important therapeutic option for the management of patients with a first episode of syncope that occurs despite beta-blocker therapy, but it should not be considered as a curative or alternative to ICD in patients with high risk of sudden cardiac death [25].
The genetic variant identified in this family is due to a heterozygous mutation resulting in a premature stop codon. According to the literature, these mutations would lead to haploinsufficiency causing a mild phenotype because there would be up to 50% loss of function, while the remaining wild-type Kv11.1 channels function normally [15, 18, 19].
However, the clinical and genetic findings of this family demonstrate that the mutations that cause haploinsufficiency can result in LQTS with a severe phenotypic manifestation and risk of arrhythmic events.
Conclusions
The novel pathogenic variant of the KCNH2 gene identified in this family produces a heterozygous frameshift mutation that results in a premature stop codon and causes LQTS with a severe phenotypic manifestation and an elevated risk of sudden cardiac death.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The ClinVar accession numbers for the DNA variant data are reported in this manuscript (dbSNP: rs1584885912) is VCV000638156.1 [26].
Abbreviations
- LQTS:
-
Long QT syndrome
- ECG:
-
Electrocardiogram
- KCNH2:
-
Potassium voltage-gated channel subfamily H member 2
- Kv11.1:
-
Voltage-dependent potassium channel 11.1
- DNA:
-
Deoxyribonucleic acid
- OMIM:
-
Online Mendelian Inheritance in Man
- PCR:
-
Polymerase chain reaction
- LQT1:
-
Long QT syndrome type 1
- LQT2:
-
Long QT syndrome type 2
- LQT3:
-
Long QT syndrome type 3
- IKr:
-
Rapidly activating delayed rectifier potassium current
- ICD:
-
Implantable cardiac defibrillator
- LCSD:
-
Left cardiac sympathetic denervation
References
Schwartz PJ, Periti M, Melliani A. The long QT syndrome. Am Heart J. 1975;89:378–90.
Acunzo RS. Polymorphic ventricular tachycardias and long QT syndrome. In: Elizari MV, Chiale PA, editors. Cardiac arrhythmias. Cellular and molecular bases, diagnosis and treatment. 2nd ed. Buenos Aires: Editorial Panamericana; 2003. p. 671–98.
Priori SG, Blomström-Lundqvist C, Mazzanti A, et al. ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2015;36:2793–867.
Romano C, Gemme G, Pongiglione R. Rare cardiac arrythmias of the pediatric age. II. Syncopal attacks due to paroxysmal ventricular fibrillation (Presentation of 1st case in Italian pediatric literature). Clin Pediatr. 1963;45:656–83.
Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103–6.
Jervell A, Lange-Nielsen F. Congenital deaf mutism, functional heart disease with prolongation of the QT interval and sudden death. Am Heart J. 1956;54:59–68.
García-Elias A, Bergoña B. Ion channel disorders and sudden cardiac death. Int J Mol Sci. 2018;19:692.
Tester DJ, Ackerman MJ. Genetics of long QT syndrome. Methodist Debakey Cardiovasc J. 2014;10:29–33.
Giudicessi JR, Wilde AAM, Ackerman MJ. The genetic architecture of long QT syndrome: a critical reappraisal. Trends Cardiovasc Med. 2018;28:453–64.
Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88:782–4.
Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med. 2006;259:39–47.
Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011;124:2181–4.
Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2018;72:1760.
Schwartz PJ, Ackerman MJ, George AL, et al. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013;62:169–80.
Smith JL, Anderson CL, Burgess DE, et al. Molecular pathogenesis of long QT syndrome type 2. J Arrhythm. 2016;32:373–80.
Trudeau MC, Warmke JW, Ganetzky B, et al. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–5.
Anderson CL, Kuzmicki CE, Childs RR, et al. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat Commun. 2014;5:5535.
Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–77.
Bohnen MS, Peng G, Robey SH, et al. Molecular pathophysiology of congenital long QT syndrome. Physiol Rev. 2017;97:89–134.
Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–74.
Wilde AA, Ackerman MJ. Beta-blockers in the treatment of congenital long QT syndrome: is one beta-blocker superior to another? J Am Coll Cardiol. 2014;64:1359–61.
Ahn J, Kim HJ, Choi JI, Lee KN, Shim J, Ahn HS, Kim YH. Effectiveness of beta-blockers depending on the genotype of congenital long-QT syndrome: a meta-analysis. PLoS ONE. 2017;12:e0185680.
Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation. 2004;109:1826–33.
Bos JM, Bos KM, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation in long QT syndrome: analysis of therapeutic nonresponders. Circ Arrhythm Electrophysiol. 2013;6:705–11.
Dusi V, De Ferrari GM, Pugliese L, Schwartz PJ. Cardiac sympathetic denervation in channelopathies. Front Cardiovasc Med. 2019;6:27.
National Center for Biotechnology Information. ClinVar [VCV000638156.1]. https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000638156.1.
Acknowledgements
The authors thank Dr. Marcelo Kauffmann of Neurogenetics Laboratory of Hospital Ramos Mejía; Macrogen; Laboratory of Molecular Biology and Pathology of High Medic Group; Einthoven Cardiological Research Foundation; Dr. Omar Scapin and Dr. Matías Feidman of Roemmers Laboratory for providing technical assistance and suggestions on the manuscript.
Funding
This research received no grant from any funding agency in the public, commercial or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
YHS has drafted the manuscript; MN performed the processing of the DNA sample; vWMA and CN acquired and analyzed the patient data; GHA contributed to interpretation of data and diagnosis; PA and PMB contributed to design of the work; ARS and CJ have substantively revised the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the Hospital Dr. José María Ramos Mejía, according to the Declaration of Helsinki. All individuals signed the informed consent before participating in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoo, H.S., Medina, N., von Wulffen, M.A. et al. A novel KCNH2 frameshift mutation (c.46delG) associated with high risk of sudden death in a family with congenital long QT syndrome type 2. Int J Arrhythm 22, 1 (2021). https://doi.org/10.1186/s42444-020-00029-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42444-020-00029-1