
Abstract
Growing evidence suggests that damage-associated molecule patterns (DAMPs) and their receptors, pattern recognition receptors (PRRs), are associated with the progression of cardiometabolic disorders, including obesity-related insulin resistance and atherosclerosis. Cardiometabolic disorders share sterile chronic inflammation as a major cause; however, the exact mechanisms are still obscure. Toll-like receptor 9 (TLR9), one of the nucleic acid-sensing TLRs, recognizes DNA fragments derived from pathogens and contributes to self-defense by activation of the innate immune system. In addition, previous studies demonstrated that TLR9 recognizes DNA fragments released from host cells, accelerating sterile inflammation, which is associated with inflammatory diseases such as autoimmune diseases. In obese adipose tissue and atherosclerotic vascular tissue, various stresses release DNA fragments and/or nuclear proteins as DAMPs from degenerated adipocytes and vascular cells. Recent studies indicated that the activation of TLR9 in immune cells including macrophages and dendritic cells by recognition of these DAMPs promotes inflammation in these tissues, which causes cardiometabolic disorders. This review discusses recent advances in understanding the role of sterile inflammation associated with TLR9 and its endogenous ligands in cardiometabolic disorders. New insights into innate immunity may provide better understanding of cardiometabolic disorders and new therapeutic options for these major health threats in recent decades.
Similar content being viewed by others
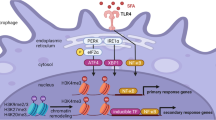


Introduction
Immune cells such as macrophages recognize the structures of pathogens through a family of pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) by detecting components referred to as pathogen-associated molecular patterns (PAMPs) and activate the innate immune system for self-defense [1]. TLRs are evolutionarily conserved proteins. Thus far, 10 functional TLRs have been identified in humans and 12 in mice. TLRs are classically categorized into two groups by their localization in/on the cell: cell surface TLRs include TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11, which mainly recognize membrane components such as lipids, lipoproteins, and proteins on bacteria; intracellular TLRs include TLR3, TLR7, TLR8, and TLR9, which recognize viral and bacterial nucleic acids. The latter group is expressed in intracellular vesicles such as endosomes, lysosomes, and endoplasmic reticulum (ER) [2]. Numerous previous studies have examined the mechanisms underlying TLR signaling and demonstrated that it requires the recruitment of several adaptor molecules, leading to the activation of the NF-κB and interferon (IFN) regulatory factor (IRF) pathways, which accelerate inflammatory responses (Fig. 1). In addition, emerging evidence has revealed that TLR signaling is involved in not only innate immune systems but also in the pathogenesis of various diseases such as autoimmune diseases and lifestyle-associated diseases. Among them, the role of TLRs in the pathogenesis of cardiometabolic disorders, one of the health threats for humans in recent decades, has attracted much attention. TLRs whose roles in cardiometabolic disorders have been most studied are TLR2 and TLR4 [3,4,5,6,7,8,9], while recent studies have suggested the participation of TLR9, originally known as a sensor for exogenous DNA fragments, in these diseases (Table 1). This review briefly summarizes the role of TLR9 in the pathogenesis of inflammatory diseases and describes recent findings, including our own, on the potential participation of TLR9 in the development of cardiometabolic disorders.

Overview of TLR signaling. TLRs are classified into cell surface TLRs and intracellular TLRs. Each TLR recognizes their specific ligands and promotes gene expression of inflammatory molecules mainly via NF-κB and IRF pathway in immune cells
DNA sensors in innate immunity
Nucleic acids are indispensable for life; however, exogenous nucleic acids, especially those from exogenous organisms such as bacteria and viruses, strongly induce inflammation. There are several sensors for nucleic acids. Generally, TLR3, TLR7/8, and TLR9 recognize double-stranded (ds) RNA, RNA, and DNA, respectively. Other than these TLRs, nucleic acids are also detected by other groups of molecules, such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) by detecting single-stranded (ss) RNA and dsRNA, respectively [31, 32]. In addition to endosomal DNA-sensing proteins, cytoplasmic DNA sensors, such as the cGAMP synthase-cGAMP-stimulator of interferon genes (STING), have been reported [33].
Among these, TLR9 is one of the most studied sensors for nucleic acids. TLR9 recognizes DNA fragments that contain unmethylated CpG DNA and plays a role in innate immunity [34, 35]. TLR9 localizes in the ER in multiple cell types, including macrophages, B cells, dendritic cells, and plasma cells [34]. After uptake of the ligands by phagocytosis, TLR9 immediately redistributes from the ER to the CpG DNA-containing structures. TLR9 activation leads to the production of type I IFN through myeloid differentiation primary response 88 (MyD88)-IRF7 or of inflammatory cytokines through MyD88-NF-κB, accelerating inflammatory responses [2, 36,37,38].
Role of TLR9 in inflammatory diseases
TLR9 activation plays a central role in self-defense against exogenous organisms as a sensor for exogenous DNA fragments; however, accumulating evidence has revealed that TLR9 also recognizes self-derived DNA and promotes inflammation improperly in certain disease contexts such as autoimmune diseases [39, 40]. The pathogenesis of autoimmune diseases is not clear. The initiating stimuli are often unidentified, and the reasons why the mechanisms that ordinarily handle the immune response fail are unknown. However, it is clear that these diseases are characterized by an extraordinarily destructive tissue environment. Hence, the DAMP level is elevated locally and/or systemically in these conditions. To avoid unwanted activation of TLR9 by endogenous DNA fragments, its level is thought to be maintained under a certain threshold. However, disease conditions in which an abundant supply of DNA fragments overwhelms the removal mechanisms, or the removal mechanisms of DNA fragments deteriorate even if the supply is maintained, cause elevation of the levels of endogenous DNA fragments. The existence of extracellular DNA in human plasma, also known as cell-free DNA (cfDNA), has been described almost from the 1940s [41], whereas recent studies demonstrated elevation of cfDNA level and its association with the pathophysiology of several inflammatory diseases [42]. For example, circulating cfDNA level is higher in systemic sclerosis [43], experimental pulmonary thromboembolism [44], end-stage renal disease [45], and sepsis [46]. Of note, the role of cfDNA has attracted much attention in the pathogenesis of autoimmune diseases including systemic lupus erythematosus (SLE) and rheumatoid arthritis [47,48,49,50,51,52]. In fact, autoantibodies against dsDNA and nucleosomes represent a feature of SLE [39, 53, 54]. In general, TLR9 activates IRFs and/or NF-κB, which produces interferons and cytokines, leading to the acceleration of inflammatory responses in these diseases [2, 36,37,38]. In addition, other studies demonstrated that TLR9 promotes p38 mitogen-activated protein kinase (MAPK) activation and the subsequent NF-κB activation, stimulating inflammation [19, 55]. On the other hand, a few studies reported that ligation of TLR9 with its ligand has beneficial effects on some disease context such as cerebral ischemia/reperfusion injury by activation of PI3K/Akt signaling [56]. These results indicated that the role of TLR9 in inflammatory diseases and the underlying mechanisms are context-dependent and signaling systems under TLR9 might not be fully understood. TLR9-mediated signaling and following response suggested in the inflammatory diseases are summarized in Fig. 2. A number of studies have suggested a link between TLR9 and inflammatory diseases; however, the role of TLR9 in the development of cardiometabolic diseases in which chronic sterile inflammation takes part as an underlying mechanism remains not fully investigated.

TLR9 signaling in inflammatory diseases. Activation of TLR9 signaling in immune cells leads to release various cytokines and interferons in cardiometabolic organs and other tissues, participating in the pathogenesis of both infectious and sterile inflammatory diseases
Role of TLR9 in metabolic diseases
Because of the change in our lifestyle, the prevalence of obesity is increasing all over the world. Obesity is closely associated with multiple metabolic abnormalities including insulin resistance, hyperglycemia, dyslipidemia, hepatic steatosis, and hypertension. In the pathobiology of obesity and obesity-related complications, chronic sterile inflammation in metabolic organs plays a central role. The mechanisms by which obesity promotes inflammation in metabolic organs are still unknown, although recent studies suggested the contribution of TLRs [7, 57,58,59]. For example, adipose tissue is an energy-storing organ, in which interaction of immune cells, hypertrophy and proliferation of adipocytes, and angiogenesis are highly coordinated [60,61,62,63]. However, obesity-related conditions, such as higher oxidative stress [62], lower oxygen pressure [63], and enhanced inflammation [60, 64], disturb this balance, leading to the induction of cellular degeneration and enhancement of cellular turnover in adipose tissue [65,66,67]. A previous study demonstrated that local and/or systemic adipocyte-derived factors contribute to multiple pathological states associated with obesity, including adipose tissue inflammation [68]. Here, TLR2 and TLR4 recognize their ligands from obese and degenerated adipose tissue, mediating adipose tissue inflammation [59, 69,70,71,72]. Importantly, clinical studies showed that TLR2/TLR4 expression was increased in adipose tissue and monocytes in obese or diabetic patients, which is correlated with the severity of insulin resistance [5, 73].
In addition to other types of endogenous ligands for TLRs, such as saturated fatty acids and heat shock protein (HSP), self-derived DNA fragments are thought to be released. However, the role of self-derived DNA fragments and the contribution of TLR9 to the development of adipose tissue inflammation have not been studied. We previously demonstrated that high-fat feeding increased the level of plasma ssDNA in mice. Also, plasma ssDNA level was higher in patients with visceral obesity diagnosed by computed tomography compared with the non-obese population. We further found that the level of ssDNA positively correlated with HOMA-IR, a parameter of insulin resistance, in humans [10]. The increase in plasma cfDNA in obese subjects was significant but modest compared with previous research on patients with other inflammatory diseases, including cancer and SLE [45, 51, 74]. Importantly, obesity is associated with chronic, low-grade inflammation. This might be one of the explanations for the lower level of cfDNA in obese individuals. Next, we investigated the roles of TLR9 in adipose tissue inflammation. In in vitro studies, a TLR9 agonist, ODN1826, promoted the expression of inflammatory molecules such as tumor necrosis factor-α (TNF-α) and monocyte chemoattractant protein-1 (MCP-1), important molecules for adipose tissue inflammation, in macrophages. Also, cfDNA collected from degenerated adipocytes activated macrophages through TLR9 and stimulated the expression of inflammatory molecules [10]. Obesity induced by high-fat diet (HFD) feeding promoted TLR9 expression in adipose tissue in addition to cfDNA in animal studies [10, 12]. Genetic deletion of TLR9 decreased the accumulation of macrophages in obese adipose tissue (Fig. 3) and inhibited the development of obesity-induced adipose tissue inflammation and insulin resistance. Furthermore, bone marrow-specific expression of TLR9 worsened insulin resistance under HFD feeding compared with that in mice lacking TLR9 in their body. On the other hand, administration of an inhibitory oligonucleotide for TLR9, iODN2088, to HFD-fed wild-type mice attenuated inflammation in adipose tissue and improved insulin resistance. These results suggest a link between TLR9 and obesity-associated insulin resistance, and the potential of cfDNA-TLR9 signaling as a therapeutic target. In addition, in the development of obesity-associated metabolic disorders, several reports suggest the role of another endogenous ligand of TLR9 beside self-derived DNA fragments. In a clinical study of obese individuals, Guzmán-Ruiz et al. showed an elevated level of high mobility group box protein-1 (HMGB1) in the plasma as well as increased expression in visceral adipose tissue, which correlated with markers of adipose tissue inflammation [75].

Genetic deletion of TLR9 attenuated obesity-induced adipose tissue inflammation. Representative figures of Mac3 staining of visceral adipose tissue from HFD-fed wild-type or TLR9-deficient mice. Genetic deletion of TLR9 reduced the accumulation of macrophages in adipose tissue, indicating less inflammation. Bar, 100 μm
A role of TLR9 in the pathogenesis of non-alcoholic steatohepatitis (NASH) has also been reported. In mice, a TLR7/9 antagonist (IRS954) was effective for improving hepatic steatosis and NASH [15]. TLR9-deficient mice had less insulin resistance than wild-type mice on a choline-deficient amino acid-defined diet [17]. Liver damage including mitochondrial stress promotes mitochondrial DNA (mtDNA) leakage. Similar to bacterial DNA, mtDNA contains a predominantly unmethylated CpG motif [76, 77] and can act on macrophage TLR9, leading to a strong induction of inflammatory responses. Saito et al. demonstrated mtDNA-mediated activation of the TLR9/IFN-β signal pathway accelerates non-apoptotic hepatocyte death and liver fibrosis [16]. In addition to animal studies, a clinical study demonstrated that the number of mtDNA copies was 3.2-fold higher in NASH patients than in healthy controls [78]. Of note, recent studies have established that mtDNA triggers various inflammatory or degenerative diseases as an important DAMP [79]. Also, an animal study using anti-HMGB1 antibody revealed that neutralization of HMGB1 attenuates weight gain and liver inflammation, but not adipose tissue inflammation, under HFD feeding [14].
Another study, however, showed that TLR9 deficiency promoted insulin resistance in response to a HFD, suggesting anti-inflammatory roles of TLR9 in macrophage activation [13]. Several differences in the study design, including the diet and duration of feeding, might account for these discrepancies (Table 2). Further studies are required to elucidate the role of cfDNA-TLR9 signaling in the pathogenesis of metabolic disorders.
Role of TLR9 in cardiovascular diseases
Chronic inflammation in the vasculature, initiated by endothelial dysfunction under risk factors such as hypertension, diabetes mellitus, and dyslipidemia, causes atherosclerosis [80]. Controlling these risk factors reduces cardiovascular events; however, considerable residual risk remains and is a clinical issue. This also indicates that the mechanisms that cause vascular inflammation and atherosclerosis are not fully understood.
Accumulating evidence indicates that the innate immune system plays a role in the development of vascular inflammation despite it being multifactorial in etiology [81]. Many types of PRRs are expressed in multiple cell types present in arterial lesions, including endothelial cells and infiltrated monocytes, macrophages, and dendritic cells. TLRs are essentially associated with the process of atherosclerosis [82]. Both in murine and human lipid-rich atherosclerotic lesions, macrophages show TLR4 expression preferentially, which is upregulated by oxidized low-density lipoprotein (oxLDL) [83]. Other studies showed that TLR2 expression is enhanced in patients with diabetes, and TLR2/TLR4 stimuli promote inflammation in obese patients with atherosclerosis [84, 85].
In addition to TLR2 and TLR4, recent studies have suggested the contribution of TLR9 to the development of vascular inflammation and atherogenesis. In vitro studies demonstrated that the activation of TLR9 accelerates the shift from macrophages into foam cells via the NF-κB and IRF7 pathways [23, 24]. Also, we found that a TLR9 agonist, ODN1826, markedly promoted the pro-inflammatory activation of apolipoprotein E-deficient (Apoe−/−) macrophages, partially through p38 MAPK signaling [19]. Another study demonstrated the activation of plasmacytoid dendritic cells (pDCs) through the TLR9 pathway, leading to the development of vascular lesions [25]. Previous studies have shown degeneration of vascular cells including endothelial cells and macrophages in atherosclerotic lesions [86,87,88], suggesting the release of cellular debris that contains various endogenous ligands for TLRs [89]. Therefore, we hypothesized that TLR9 plays a role in the development of atherosclerosis through the recognition of DNA fragments released by vascular damage. To address this hypothesis, we employed three different mouse models. Genetic deletion of TLR9 in subcutaneous angiotensin II (Ang II)-infused Apoe−/− mice on a Western-type diet (WTD) reduced the development of atherosclerotic lesions (Fig. 4). Pharmacological blockade of TLR9 using iODN2088, one of the inhibitory oligodeoxynucleotides specific to TLR9, attenuated atherogenesis in Ang II-infused Apoe−/− mice compared with control oligodeoxynucleotide. Genetic deletion and pharmacological inhibition of TLR9 also decreased macrophage and lipid accumulation and the expression of inflammatory molecules at both the RNA and protein levels in this mouse model [19], while restoration of TLR9 in the bone marrow in Tlr9−/−Apoe−/− mice accelerated atherogenesis in the aortic arch. These findings indicate proatherogenic roles of TLR9 [19]. Furthermore, Ma et al. showed that the inactivation of TLR9 by employing IRS869, another type of inhibitory oligodeoxynucleotide for TLR9, reduced plaque burden and shifted macrophage polarization to the anti-inflammatory M2 population [90]. Krogmann et al. also showed that intravenous administration of ODN1826 to Apoe−/− mice impaired reendothelialization of an acute vascular injury and increased subsequent atherosclerotic plaque development [26]. Similarly, we have reported the contribution of TLR9 activation to neointima formation after mechanical vascular injury, which was blocked by the administration of an anti-HMGB1 antibody [21]. In addition, we also demonstrated that TLR9 activation impaired blood flow recovery in an ischemic hind limb model by the promotion of TNF-α expression [91]. All of these studies suggested TLR9 activation promotes inflammation and accelerates atherosclerotic and/or vascular diseases.

Genetic deletion of TLR9 attenuated the development of atherosclerosis. Representative figures of Sudan IV staining of the aortic arch of Ang II-infused Apoe−/− or Tlr9−/−Apoe−/− mice. Genetic deletion of TLR9 attenuated the development of atherosclerosis. Bar, 1 mm
Recent clinical studies also indicated the contribution of the cfDNA-TLR9 axis in the development of atherosclerosis in humans. Borissoff et al. demonstrated that patients with severe coronary artery disease diagnosed by coronary computed tomographic angiography have elevated plasma dsDNA and nucleosome levels [18]. We also measured the level of cfDNA in the plasma collected from the target vessel of patients with acute myocardial infarction and examined the correlation between the concentration and plaque morphology of the target lesion assessed by optical coherence tomography. The plasma level of cfDNA in the target vessel was positively correlated with lipid deposition, macrophage content, and ruptured plaque cavity length/area in the target lesion, all of which are associated with plaque inflammation [19]. Several studies have shown the expression of TLR9 in vascular lesions, although its expression may be lower than that of other TLRs such as TLR4 and TLR2 [82]. However, these studies suggested that the cfDNA-TLR9 axis participates in the pathogenesis of vascular inflammation and atherogenesis.
On the other hand, several groups have reported incongruous findings which suggested protective roles of TLR9 in atherosclerosis. In vitro stimulation of TLR9 triggered IL-10 production in B cells in humans, which in turn inhibited CD4+CD25+ T cell proliferation [92, 93]. Also, Koulis et al. demonstrated antiatherogenic roles of TLR9 using a genetic deletion model of TLR9 in HFD-fed Apoe−/− mice [27]. In that study, TLR9-deficient Apoe−/− mice showed increased macrophage, DC, and CD4+ T cell in the plaque. Also, the administration of CpG-ODN1668, a TLR9 agonist, attenuated atherosclerotic lesion development in Apoe−/− mice under HFD feeding. Of note, the study of Koulis et al. showed that TLR9 deletion increased blood lipid levels by an undetermined mechanism. This might have affected the results and suggested that TLR9 is associated with lipid metabolism in addition to the innate immune system. Thus, both pro- and anti-atherosclerotic roles of TLR9 have been described. Interestingly, a previous study mentioned conflicting roles of TLR9 activation due to the concentration of its ligand [94]. Therefore, the difference in the study design such as mouse model, mouse strain, food, duration of treatment, and types of agonist or antagonist might cause the difference in the levels of ligands, which results in these discrepancies observed in previous studies (Table 2). Further experiments are needed to determine the effect of TLR9 in atherosclerotic diseases.
Accumulating evidence suggests that cardiac inflammation contributes to promoting heart failure (HF). In HF patients, levels of circulating cytokines including TNF-α, interleukin (IL)-1β, and IL-6 are elevated, which is associated with the severity and outcome of these patients [95]. In most cases, microbial infection is not involved in the development of HF, indicating that there is a state of sterile inflammation. However, the complex mechanisms underlying cardiac inflammation are unclear [96]. In HF, multiple endogenous DAMPs such as HMGB1, HSP, and mtDNA are released and recognized by TLRs, stimulating NF-κB-dependent inflammatory responses [97]. Recently, a study demonstrated that intracellular mtDNA escaping degradation induces cardiac inflammation signaling through TLR9 in an animal model of pressure overload-induced HF [28]. Endogenous mtDNA in the extracellular space activates NF-κB signaling through TLR9 in cardiomyocytes, resulting in its detrimental effects [29]. In addition, inhibition of TLR9 attenuated the development of pressure overload-induced HF [98].
Thus, recently, the role of TLR9 is expanding to the cardiovascular field as well as the innate immune field. Investigating the role of the cfDNA-TLR9 axis would increase the understanding of the pathogenesis and generation of new therapeutic approaches for these diseases.
TLR9 as a therapeutic target for inflammatory diseases
Because recent advances of immunology indicated the role of TLRs in various inflammatory diseases, TLRs are receiving increased attention as the therapeutic target. Previously, two phase III clinical trials using TAK-242, a small molecule which targets TLR4, were carried out [99]. The first trial (NCT00143611) resulted in not enough satisfaction because of failure to effectively decrease serum cytokine levels (IL-6, IL-8, and TNF-α) compared to controls, in spite of its well-tolerance [100]. Another trial (NCT00633477) ended because of a business decision. Since then, TAK-242 has not been developed clinically.
In contrast, drugs targeting endosomal TLR attract much attention. A group of antimalarial drugs, such as chloroquine (CQ), hydroxychloroquine sulfate (HCQ), and quinacrine, have been used to treat autoimmune diseases in clinical practice [101]. These drugs are weak bases that accumulate in the acidic intracellular compartment such as endosomes and lysosomes, and modulate the pH in these vesicles, leading to the suppression of autoantigen presentation and inhibition of endosomal TLR signaling (TLR7, 8, and 9) [102]. In addition to the effects for autoimmune diseases, pre-clinical studies suggested beneficial effects of these drugs on cardiovascular diseases. For example, pretreatment with CQ improved cerebral ischemia symptoms in a transient global cerebral ischemia rat model animal [103], and long-term treatment with HCQ attenuated hypertension and endothelial dysfunction in a lupus animal model as well [104]. Oligonucleotides with specific sequences also function as antagonists of endosomal TLRs because endosomal TLRs recognize nucleic acid structures. These oligonucleotides can block the TLR signal transduction by inhibiting the binding of TLRs to their ligands. Because of this background, variety types of these oligonucleotides have been developed for the treatment of autoimmune diseases including SLE and plaque psoriasis in both basic and clinical researches [105, 106].
Thus, controlling TLR9 signaling might have a potential to inhibit cardiometabolic diseases. In fact, previous studies have suggested that several types of oligonucleotides for TLR9 have inhibitory effects on the development of cardiometabolic diseases in animal models (Table 2). In addition, other studies have suggested that antibodies targeting HMGB1 or CD4 attenuate TLR9-mediated atherogenesis [27, 107]. Targeting immune responses mediated by TLR9 has a potential as a therapeutic strategy to control unwanted, disease-associated inflammation; however, further studies are needed to develop therapeutic strategies targeting immune systems clinically.
Conclusion
Chronic low-grade inflammation plays a central role in the pathophysiology of cardiometabolic disorders, in which various cellular and molecular mechanisms participate. Recent studies have suggested that activation of innate immune systems by DAMPs contributes to the development of chronic inflammation [89]. The present review focused on the role of TLR9, which was originally known as a sensor for exogenous DNA fragments, in pro-inflammatory activation of immune cells and in the pathogenesis of cardiometabolic disorders. Innate immune systems are essential for survival. Originally, inflammation induced by sensing of DNA fragments has a protective role, although emerging evidence demonstrated that this immune system also has harmful effects. The prevalence and incidence of metabolic disorders associated with aging, obesity, and nutritional excess have dramatically increased worldwide in recent decades. This change may induce a shift in what are usually favorable for physiological processes to pathological events. Recent studies including our own demonstrated that the cfDNA-TLR9 pathway plays a pivotal role in the pathogenesis of adipose tissue inflammation and vascular inflammation via pro-inflammatory activation of macrophages, leading to the development of cardiometabolic disorders including obesity-related insulin resistance and atherosclerosis (Fig. 5). Regardless of accumulating research these days, there is still limited knowledge about the cfDNA-TLR9 pathway in cardiometabolic fields. For example, the origin of elevated DNA fragments in obese and atherosclerotic conditions is still not clear. The release of nucleic acids as cellular debris from degraded cells/tissues in metabolic organs together with other TLR agonists is considered to be one of them. However, recent studies have shown a link between the gut microbiota and metabolic disorders [17, 108, 109]. These studies suggested the translocation of bacterial components (such as DNA) and inflammatory factors (including lipopolysaccharide) in the host circulation under certain circumstances, accompanied by intestinal epithelial dysfunction caused by obesity or other metabolic disorders.

Role of TLR9 in the development of cardiometabolic disorders. DNA fragments and/or nuclear proteins released from damaged cells/tissues activate immune cells such as macrophages and DCs through TLR9, leading to the development of inflammation in these tissues, which is central in the pathogenesis of cardiometabolic disorders
In summary, the cfDNA-TLR9 pathway contributes to the pathogenesis of cardiometabolic disorders. This pathway might be a potential therapeutic target and a possible biomarker for this health threat. However, further studies are required to define the possible clinical application of this pathway.
Availability of data and materials
The datasets during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Ang II:
-
Angiotensin II
- Apoe−/− :
-
Apolipoprotein E-deficient
- cfDNA:
-
Cell-free DNA
- CQ:
-
Chloroquine
- DAMPs:
-
Damage-associated molecular patterns
- dsDNA:
-
Double-stranded DNA
- ER:
-
Endoplasmic reticulum
- HCQ:
-
Hydroxychloroquine
- HF:
-
Heart failure
- HFD:
-
High-fat diet
- HMGB1:
-
High mobility group box protein-1
- HSP:
-
Heat shock protein
- IFN:
-
Interferon
- IL:
-
Interleukin
- IRF:
-
Interferon regulatory factor
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Monocyte chemoattractant protein-1
- MDA5:
-
Melanoma differentiation-associated gene 5
- mtDNA:
-
Mitochondrial DNA
- MyD88:
-
Myeloid differentiation primary response
- NASH:
-
Non-alcoholic steatohepatitis
- oxLDL:
-
Oxidized low-density lipoprotein
- PAMPs:
-
Pathogen-associated molecular patterns
- pDCs:
-
Plasmacytoid dendritic cells
- PRR:
-
Pattern recognition receptor
- RIG-I:
-
Retinoic acid-inducible gene I
- SLE:
-
Systemic lupus erythematosus
- ssDNA:
-
Single-stranded DNA
- STING:
-
Stimulator of interferon genes
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumor necrosis factor-α
- WTD:
-
Western-type diet
References
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801.
Falck-Hansen M, Kassiteridi C, Monaco C. Toll-like receptors in atherosclerosis. Int J Mol Sci. 2013;14:14008–23.
Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4:444–54.
Jialal I, Huet BA, Kaur H, Chien A, Devaraj S. Increased Toll-like receptor activity in patients with metabolic syndrome. Diabetes Care. 2012;35:900–4.
Konner AC, Bruning JC. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metab. 2011;22:16–23.
Fresno M, Alvarez R, Cuesta N. Toll-like receptors, inflammation, metabolism and obesity. Arch Physiol Biochem. 2011;117:151–64.
Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105:1158–61.
Velloso LA, Folli F, Saad MJ. TLR4 at the crossroads of nutrients, gut microbiota, and metabolic inflammation. Endocr Rev. 2015;36:245–71.
Nishimoto S, Fukuda D, Higashikuni Y, Tanaka K, Hirata Y, Murata C, Kim-Kaneyama JR, Sato F, Bando M, Yagi S, Soeki T, Hayashi T, Imoto I, Sakaue H, Shimabukuro M, Sata M. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci Adv. 2016;2:e1501332.
Revelo XS, Ghazarian M, Chng MHY, Luck H, Kim JH, Zeng K, Shi SY, Tsai S, Lei H, Kenkel J, Liu CL, Tangsombatvisit S, Tsui H, Sima C, Xiao C, Shen L, Li X, Jin T, Lewis GF, Woo M, Utz PJ, Glogauer M, Engleman E, Winer S, Winer DA. Nucleic acid-targeting pathways promote inflammation in obesity-related insulin resistance. Cell Rep. 2016;16:717–30.
Kim SJ, Choi Y, Choi YH, Park T. Obesity activates Toll-like receptor-mediated proinflammatory signaling cascades in the adipose tissue of mice. J Nutr Biochem. 2012;23:113–22.
Hong C-P, Yun CH, Lee G-W, Park A, Kim Y-M, Jang MH. TLR9 regulates adipose tissue inflammation and obesity-related metabolic disorders. Obesity. 2015;23:2199–206.
Montes VN, Subramanian S, Goodspeed L, Wang SA, Omer M, Bobik A, Teshigawara K, Nishibori M, Chait A. Anti-HMGB1 antibody reduces weight gain in mice fed a high-fat diet. Nutr Diabet. 2015;5:e161.
Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, Mehal WZ. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. 2016;126:859–64.
Saito Y, Hikita H, Nozaki Y, Kai Y, Makino Y, Nakabori T, Tanaka S, Yamada R, Shigekawa M, Kodama T, Sakamori R, Tatsumi T, Takehara T. DNase II activated by the mitochondrial apoptotic pathway regulates RIP1-dependent non-apoptotic hepatocyte death via the TLR9/IFN-β signaling pathway. Cell Death Differ. 2019;26:470–86.
Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology. 2010;139:323–334.e7.
Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, Gallant M, Martinod K, Cate H, Hofstra L, Crijns HJ, Wagner DD, Kietselaer BLJH. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. 2013;33:2032–40.
Fukuda D, Nishimoto S, Aini K, Tanaka A, Nishiguchi T, Kim-Kaneyama J-R, Lei X-F, Masuda K, Naruto T, Tanaka K, Higashikuni Y, Hirata Y, Yagi S, Kusunose K, Yamada H, Soeki T, Imoto I, Akasaka T, Shimabukuro M, Sata M. Toll-like receptor 9 plays a pivotal role in angiotensin II-induced atherosclerosis. J Am Heart Assoc. 2019;8:e010860.
Clement M, Charles N, Escoubet B, Guedj K, Chauveheid MP, Caligiuri G, Nicoletti A, Papo T, Sacre K. CD4+CXCR3+ T cells and plasmacytoid dendritic cells drive accelerated atherosclerosis associated with systemic lupus erythematosus. J Autoimmun. 2015;63:59–67.
Hirata Y, Kurobe H, Higashida M, Fukuda D, Shimabukuro M, Tanaka K, Higashikuni Y, Kitagawa T, Sata M. HMGB1 plays a critical role in vascular inflammation and lesion formation via Toll-like receptor 9. Atherosclerosis. 2013;231:227–33.
Karper JC, Ewing MM, Habets KL, de Vries MR, Peters EA, van Oeveren-Rietdijk AM, de Boer HC, Hamming JF, Kuiper J, Kandimalla ER, La Monica N, Jukema JW, Quax PH. Blocking toll-like receptors 7 and 9 reduces postinterventional remodeling via reduced macrophage activation, foam cell formation, and migration. Arterioscler Thromb Vasc Biol. 2012;32:e72–80.
Sorrentino R, Morello S, Chen S, Bonavita E, Pinto A. The activation of liver X receptors inhibits toll-like receptor-9-induced foam cell formation. J Cell Physiol. 2010;223:158–67.
Lee J-G, Lim E-J, Park D-W, Lee S-H, Kim J-R, Baek S-H. A combination of Lox-1 and Nox1 regulates TLR9-mediated foam cell formation. Cell Signal. 2008;20:2266–75.
Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-α. Circulation. 2006;114:2482–9.
Krogmann AO, Lüsebrink E, Steinmetz M, Asdonk T, Lahrmann C, Lütjohann D, Nickenig G, Zimmer S. Proinflammatory stimulation of Toll-like receptor 9 with high dose CpG ODN 1826 impairs endothelial regeneration and promotes atherosclerosis in mice. PLoS ONE. 2016;11:e0146326.
Koulis C, Chen Y-C, Hausding C, Ahrens I, Kyaw TS, Tay C, Allen T, Jandeleit-Dahm K, Sweet MJ, Akira S, Bobik A, Peter K, Agrotis A. Protective role for Toll-like receptor-9 in the development of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2014;34:516–25.
Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–5.
Bliksøen M, Mariero LH, Torp MK, Baysa A, Ytrehus K, Haugen F, Seljeflot I, Vaage J, Valen G, Stensløkken K-O. Extracellular mtDNA activates NF-κB via toll-like receptor 9 and induces cell death in cardiomyocytes. Basic Res Cardiol. 2016;111:42.
Ye W, Tang X, Yang Z, Liu C, Zhang X, Jin J, Lyu J. Plasma-derived exosomes contribute to inflammation via the TLR9-NF-κB pathway in chronic heart failure patients. Mol Immunol. 2017;87:114–21.
Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–7.
Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–8.
Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–80.
Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5.
Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–60.
Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–8.
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461.
Kaisho T, Tanaka T. Turning NF-κB and IRFs on and off in DC. Trends Immunol. 2008;29:329–36.
Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–35.
Gilliet M, Cao W, Liu Y-J. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606.
Gould TJ, Lysov Z, Liaw PC. Extracellular DNA and histones: double-edged swords in immunothrombosis. J Thromb Haemost. 2015;13:S82–91.
Swarup V, Rajeswari MR. Circulating (cell-free) nucleic acids--a promising, non-invasive tool for early detection of several human diseases. FEBS Lett. 2007;581:795–9.
Mosca M, Giuliano T, Cuomo G, Doveri M, Tani C, Curcio M, Abignano G, De Feo F, Bazzichi L, Della Rossa A, Valentini G, Bombardieri S. Cell-free DNA in the plasma of patients with systemic sclerosis. Clin Rheumatol. 2009;28:1437–40.
Uzuelli JA, Dias-Junior CA, Izidoro-Toledo TC, Gerlach RF, Tanus-Santos JE. Circulating cell-free DNA levels in plasma increase with severity in experimental acute pulmonary thromboembolism. Clin Chim Acta. 2009;409:112–6.
Atamaniuk J, Kopecky C, Skoupy S, Säemann MD, Weichhart T. Apoptotic cell-free DNA promotes inflammation in haemodialysis patients. Nephrol Dial Transplant. 2011;27:902–5.
Saukkonen K, Lakkisto P, Pettila V, Varpula M, Karlsson S, Ruokonen E, Pulkki K, Finnsepsis Study G. Cell-free plasma DNA as a predictor of outcome in severe sepsis and septic shock. Clin Chem. 2008;54:1000–7.
Arneth B. Systemic lupus erythematosus and DNA degradation and elimination defects. Front Immunol. 2019;10:1697.
Duvvuri B, Lood C. Cell-free DNA as a biomarker in autoimmune rheumatic diseases. Front Immunol. 2019;10:502.
Rykova E, Sizikov A, Roggenbuck D, Antonenko O, Bryzgalov L, Morozkin E, Skvortsova K, Vlassov V, Laktionov P, Kozlov V. Circulating DNA in rheumatoid arthritis: pathological changes and association with clinically used serological markers. Arthritis Res Ther. 2017;19:85.
Truszewska A, Foroncewicz B, Paczek L. The role and diagnostic value of cell-free DNA in systemic lupus erythematosus. Clin Exp Rheumatol. 2017;35:330–6.
Zhang S, Lu X, Shu X, Tian X, Yang H, Yang W, Zhang Y, Wang G. Elevated plasma cfDNA may be associated with active lupus nephritis and partially attributed to abnormal regulation of neutrophil extracellular traps (NETs) in patients with systemic lupus erythematosus. Intern Med. 2014;53:2763–71.
Xu Y, Song Y, Chang J, Zhou X, Qi Q, Tian X, Li M, Zeng X, Xu M, Zhang W, Cram DS, Liu J. High levels of circulating cell-free DNA are a biomarker of active SLE. Eur J Clin Investig. 2018;48:e13015.
Tan EM, Schur PH, Carr RI, Kunkel HG. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J Clin Invest. 1966;45:1732–40.
Celhar T, Magalhães R, Fairhurst A-M. TLR7 and TLR9 in SLE: when sensing self goes wrong. Immunol Res. 2012;53:58–77.
Lim E-J, Lee S-H, Lee J-G, Kim J-R, Yun S-S, Baek S-H, Lee C. Toll-like receptor 9 dependent activation of MAPK and NF-kB is required for the CpG ODN-induced matrix metalloproteinase-9 expression. Exp Mol Med. 2007;39:239–45.
Lu C, Ha T, Wang X, Liu L, Zhang X, Kimbrough EO, Sha Z, Guan M, Schweitzer J, Kalbfleisch J, Williams D, Li C. The TLR9 ligand, CpG-ODN, induces protection against cerebral ischemia/reperfusion injury via activation of PI3K/Akt signaling. J Am Heart Assoc. 2014;3:e000629.
Engin AB. Adipocyte-macrophage cross-talk in obesity. Adv Exp Med Biol. 2017;960:327–43.
Jin C, Flavell RA. Innate sensors of pathogen and stress: linking inflammation to obesity. J Allergy Clin Immunol. 2013;132:287–94.
Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor α. Arterioscler Thromb Vasc Biol. 2005;25:2062–8.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7.
Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8:923–34.
Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–61.
Sung H-K, Doh K-O, Son Joe E, Park Jin G, Bae Y, Choi S, Nelson Seana Mary L, Cowling R, Nagy K, Michael Iacovos P, Koh Gou Y, Adamson SL, Pawson T, Nagy A. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab. 2013;17:61–72.
Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801.
Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW 2nd, DeFuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56:2910–8.
Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, Cinti S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res. 2008;49:1562–8.
Rigamonti A, Brennand K, Lau F, Cowan CA. Rapid cellular turnover in adipose tissue. PLoS One. 2011;6:e17637.
Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005;11:797–803.
Davis JE, Braucher DR, Walker-Daniels J, Spurlock ME. Absence of Tlr2 protects against high-fat diet-induced inflammation and results in greater insulin-stimulated glucose transport in cultured adipocytes. J Nutr Biochem. 2011;22:136–41.
Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45.
Suganami T, Ogawa Y. Adipose tissue macrophages: their role in adipose tissue remodeling. J Leukoc Biol. 2010;88:33–9.
Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–71.
Ahmad R, Al-Mass A, Atizado V, Al-Hubail A, Al-Ghimlas F, Al-Arouj M, Bennakhi A, Dermime S, Behbehani K. Elevated expression of the Toll like receptors 2 and 4 in obese individuals: its significance for obesity-induced inflammation. J Inflamm. 2012;9:48.
van der Vaart M, Pretorius PJ. Is the role of circulating DNA as a biomarker of cancer being prematurely overrated? Clin Biochem. 2010;43:26–36.
Guzmán-Ruiz R, Ortega F, Rodríguez A, Vázquez-Martínez R, Díaz-Ruiz A, Garcia-Navarro S, Giralt M, Garcia-Rios A, Cobo-Padilla D, Tinahones FJ, López-Miranda J, Villarroya F, Frühbeck G, Fernández-Real JM, Malagón MM. Alarmin high-mobility group B1 (HMGB1) is regulated in human adipocytes in insulin resistance and influences insulin secretion in β-cells. Int J Obes. 2014;38:1545–54.
Pollack Y, Kasir J, Shemer R, Metzger S, Szyf M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 1984;12:4811–24.
Cardon LR, Burge C, Clayton DA, Karlin S. Pervasive CpG suppression in animal mitochondrial genomes. Proc Natl Acad Sci. 1994;91:3799–803.
Kamfar S, Alavian SM, Houshmand M, Yadegarazari R, Seifi Zarei B, Khalaj A, Shabab N, Saidijam M. Liver mitochondrial DNA copy number and deletion levels may contribute to nonalcoholic fatty liver disease susceptibility. Hepat Mon. 2016;16:e40774.
Grazioli S, Pugin J. Mitochondrial damage-associated molecular patterns: from inflammatory signaling to human diseases. Front Immunol. 2018;9:832.
Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–51.
Hansson GK, Libby P, Schönbeck U, Yan Z-Q. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–91.
Roshan MHK, Tambo A, Pace NP. The role of TLR2, TLR4, and TLR9 in the pathogenesis of atherosclerosis. Int J Inflamm. 2016;2016:1532832.
Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, Luthringer D, Xu X-P, Rajavashisth TB, Yano J, Kaul S, Arditi M. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–8.
Scholtes Vincent PW, Versteeg D, de Vries J-Paul PM, Hoefer Imo E, Schoneveld Arjan H, Stella Pieter R, Doevendans Pieter AFM, van Keulen Karlijn JK, de Kleijn Dominique PV, Moll Frans L, Pasterkamp G. Toll-like receptor 2 and 4 stimulation elicits an enhanced inflammatory response in human obese patients with atherosclerosis. Clin Sci. 2011;121:205–14.
Snodgrass RG, Huang S, Choi I-W, Rutledge JC, Hwang DH. Inflammasome-mediated secretion of IL-1β in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol. 2013;191:4337–47.
Isner JM, Kearney M, Bortman S, Passeri J. Apoptosis in human atherosclerosis and restenosis. Circulation. 1995;91:2703–11.
Littlewood TD, Bennett MR. Apoptotic cell death in atherosclerosis. Curr Opin Lipidol. 2003;14:469–75.
Martinet W, Schrijvers DM, De Meyer GRY. Necrotic cell death in atherosclerosis. Basic Res Cardiol. 2011;106:749–60.
Zheng Y, Gardner SE, Clarke MCH. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2011;31:2781–6.
Ma C, Ouyang Q, Huang Z, Chen X, Lin Y, Hu W, Lin L. Toll-like receptor 9 inactivation alleviated atherosclerotic progression and inhibited macrophage polarized to M1 phenotype in ApoE-/- mice. Dis Markers. 2015;2015:909572.
Nishimoto S, Aini K, Fukuda D, Higashikuni Y, Tanaka K, Hirata Y, Yagi S, Kusunose K, Yamada H, Soeki T, Shimabukuro M, Sata M. Activation of Toll-like receptor 9 impairs blood flow recovery after hind-limb ischemia. Front Cardiovasc Med. 2018;5:144.
Bouaziz J-D, Calbo S, Maho-Vaillant M, Saussine A, Bagot M, Bensussan A, Musette P. IL-10 produced by activated human B cells regulates CD4+ T-cell activation in vitro. Eur J Immunol. 2010;40:2686–91.
Waibler Z, Anzaghe M, Konur A, Akira S, Müller W, Kalinke U. Excessive CpG 1668 stimulation triggers IL-10 production by cDC that inhibits IFN-α responses by pDC. Eur J Immunol. 2008;38:3127–37.
Wu J, Cui H, Dick AD, Liu L. TLR9 Agonist regulates angiogenesis and inhibits corneal neovascularization. Am J Pathol. 2014;184:1900–10.
Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the studies of left ventricular dysfunction (SOLVD). J Am Coll Cardiol. 1996;27:1201–6.
Nakayama H, Otsu K. Translation of hemodynamic stress to sterile inflammation in the heart. Trends Endocrinol Metab. 2013;24:546–53.
Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-κB dependent inflammatory response. Cardiovasc Res. 2006;72:384–93.
Ueda H, Yamaguchi O, Taneike M, Akazawa Y, Wada-Kobayashi H, Sugihara R, Yorifuji H, Nakayama H, Omiya S, Murakawa T, Sakata Y, Otsu K. Administration of a TLR9 inhibitor attenuates the development and progression of heart failure in mice. JACC Basic Transl Sci. 2019;4:348–63.
Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol. 2011;79:34–41.
Rice TW, Wheeler AP, Bernard GR, Vincent J-L, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K and Cohen J. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis*. Read Online: Critical Care Medicine | Society of Critical Care Medicine. 2010;38.
Lee S-J, Silverman E, Bargman JM. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat Rev Nephrol. 2011;7:718–29.
Kužnik A, Benčina M, Švajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol. 2011;186:4794–804.
Cui G, Ye X, Zuo T, Zhao H, Zhao Q, Chen W, Hua F. Chloroquine pretreatment inhibits Toll-like receptor 3 signaling after stroke. Neurosci Lett. 2013;548:101–4.
Gómez-Guzmán M, Jiménez R, Romero M, Sánchez M, Zarzuelo MJ, Gómez-Morales M, O’Valle F, López-Farré AJ, Algieri F, Gálvez J, Pérez-Vizcaino F, Sabio JM, Duarte J. Chronic hydroxychloroquine improves endothelial dysfunction and protects kidney in a mouse model of systemic lupus erythematosus. Hypertension. 2014;64:330–7.
Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37:3582–6.
Balak DMW, van Doorn MBA, Arbeit RD, Rijneveld R, Klaassen E, Sullivan T, Brevard J, Thio HB, Prens EP, Burggraaf J, Rissmann R. IMO-8400, a toll-like receptor 7, 8, and 9 antagonist, demonstrates clinical activity in a phase 2a, randomized, placebo-controlled trial in patients with moderate-to-severe plaque psoriasis. Clin Immunol. 2017;174:63–72.
Kanellakis P, Agrotis A, Kyaw TS, Koulis C, Ahrens I, Mori S, Takahashi HK, Liu K, Peter K, Nishibori M, Bobik A. High-mobility group box protein 1 neutralization reduces development of diet-induced atherosclerosis in apolipoprotein E deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:313–9.
Konrad D, Wueest S. The gut-adipose-liver axis in the metabolic syndrome. Physiology. 2014;29:304–13.
Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez-Humarán LG, Smirnova N, Bergé M, Sulpice T, Lahtinen S, Ouwehand A, Langella P, Rautonen N, Sansonetti PJ, Burcelin R. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med. 2011;3:559–72.
Acknowledgements
Not applicable
Funding
This work was partially supported by JSPS Kakenhi Grants (Number 19K08584 to D.F. and Number 19H03654 to M.S.), a Japan Heart Foundation Research Grant (S.N.), Bristol-Myers Squibb Research Grants (D.F.), Takeda Science Foundation (M.S.), and the Vehicle Racing Commemorative Foundation (M.S.). The funders had no role in the study design, data collection and analysis, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the preparation of the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental procedures conformed to the guidelines for animal experimentation of Tokushima University. The protocol was reviewed and approved by our institutional ethics committee.
Consent for publication
Not applicable
Competing interests
The Department of Cardio-Diabetes Medicine, Tokushima University Graduate School, is supported in part by unrestricted research grants from Boehringer Ingelheim. The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nishimoto, S., Fukuda, D. & Sata, M. Emerging roles of Toll-like receptor 9 in cardiometabolic disorders. Inflamm Regener 40, 18 (2020). https://doi.org/10.1186/s41232-020-00118-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41232-020-00118-7