
Abstract
Interstrand DNA crosslinks (ICLs) are the link between Watson-Crick strands of DNAs with the covalent bond and prevent separation of DNA strands. Since the ICL lesion affects both strands of the DNA, the ICL repair is not simple. So far, nucleotide excision repair (NER), structure-specific endonucleases, translesion DNA synthesis (TLS), homologous recombination (HR), and factors responsible for Fanconi anemia (FA) are identified to be involved in ICL repair. Since the presence of ICL lesions causes severe defects in transcription and DNA replication, mutations in these DNA repair pathways give rise to a various hereditary disorders. NER plays an important role for the ICL recognition and removal in quiescent cells, and defects of NER causes congential progeria syndrome, such as xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. On the other hand, the ICL repair in S phase requires more complicated orchestration of multiple factors, including structure-specific endonucleases, and TLS, and HR. Disturbed this ICL repair orchestration in S phase causes genome instability resulting a cancer prone disease, Fanconi anemia. So far more than 30 factors in ICL repair have already identified. Recently, a new factor, UHRF1, was discovered as a sensor of ICLs. In addition to this, numbers of nucleases that are involved in the first incision, also called unhooking, of ICL lesions have also been identified. Here we summarize the recent studies of ICL associated disorders and repair mechanism, with emphasis in the first incision of ICLs.
Similar content being viewed by others

Background
Interstrand DNA crosslinks (ICLs) are lesions that are a covalent linkage between opposite strands of double-stranded DNA. They are formed in the presence of bifunctional alkylating agents [1–4]. Organisms are exposed to bifunctional alkylating agents, also called ICL-causing agents, as a result of endogenous metabolic processes as well as by exogenous stresses from environmental mutagens [3–5]. ICLs are extremely cytotoxic, as even a single ICL in the genome can cause severe defects in a variety of vital DNA metabolic processes, such as transcription and DNA replication [6, 7]. Particularly, the selective inhibitory effect of ICL agents on DNA replication—crucial for proliferation and cell survival—is used in both chemotherapy and phototherapy to treat various cancers and skin diseases [8]. On the other hand, the defect of ICL repair causes chromosome instability syndromes, such as Fanconi anemia. Recently, many new factors involved in ICL repair were identified from genetic studies of Fanconi anemia, and these studies suggested that ICL repair is performed in quite complicated mechanisms. In this review, we briefly summarize the recent studies of ICL associated disorders and repair mechanism, with emphasis in the first incision of ICLs.
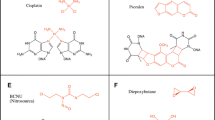
Typical ICL lesions
Chemical structures of ICL lesions have been comprehensively reviewed by Guainazzi and Schärer, and by Legerski [2, 3] and will not be discussed in detail in this review. Cisplatin and its derivatives, carboplatin and oxaliplatin, are widely used in clinical applications and can be applied to a wide variety of cancers. Cisplatin targets guanine bases in DNA, and ICLs occur at 5ʹ-GC-3ʹ sites in double-stranded DNA. The ICL formed by cisplatin shows the largest distortion of the DNA strands, compared to other ICL formed by agents described below, and it distortion is 45° of bending and 79° of unwinding [9]. Cisplatin creates not only ICL but also an intra-strand crosslink at 5′-GG-3′ sites. Similar to other bulky adducts which affect only one strand of the double-helix structures, intra-strand crosslinks caused by cisplatin are repaired by NER.
Nitrogen mustard and its derivatives also react with guanine bases, and ICL formation occurs at 5ʹ-GNC-3ʹ sites in double-stranded DNA [10]. Historically, nitrogen mustard was the first DNA damaging agent used for chemotherapy [11]. ICLs formed by nitrogen mustard show strand distortions with 14° of bending [10]. As ICL formation by nitrogen mustard is rapid (it occurs within 20 minutes of treatment), nitrogen mustard can be used in ICL repair studies in yeast [12]. Psoralen and its derivatives can form ICLs following activation with long wavelength ultraviolet radiation. Psoralen was isolated from Ammi majus. In Egypt, Ammi majus would be used in phototherapy during several millennia for treatment of psoriasis and leukoderma vulgaris [13]. Psoralen reacts with thymine bases, and ICLs occur at both 5ʹ-AT-3ʹ and 5ʹ-TA-3ʹ sites in double-stranded DNA. The ICL formed by psoralen induces 25° of unwinding and a minor local distortion of helical structure [14, 15]. As ICLs formed by psoralen are relatively stable in solution, it is often used in biochemical and cell biological studies. Mitomycin C is widely used for cell biological studies of ICL repair. Mitomycin C reacts with the guanine base in the minor groove of double-stranded DNA, and ICL formation occurs at 5ʹ-CG-3ʹ sites. The ICL formed by mitomycin C does not significantly distort the double helix [16, 17].
ICL removal in quiescent G0/G1 phase cells
Understanding the molecular mechanisms of ICL repair is exceptionally challenging because an ICL lesion affects both DNA strands. When a DNA lesion is located on only one DNA strand, the DNA fragment with lesion is excised by the introduction of two single-stranded breaks on either side of the lesion. This principle is common among various excision repair pathways, including nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR). However, in the case of ICLs, repair mechanisms involving a simple excision followed by template resynthesis are not sufficient [18]. In quiescent cells (cells in G0/G1 phase), HR is not essential for ICL repair [19]. Therefore, in all eukaryotes from Saccharomyces cerevisiae to humans, both the first and second rounds of ICL incisions occur by NER (Fig. 1a) [19, 20]. The ICL lesion with the oligonucleotide on the single-stranded gap produced by the first round of NER is bypassed with translesion DNA polymerases, such as DNA polymerases η, ι, κ, and ζ, and REV1 (Fig. 1) [19, 21–23]. In particular, DNA polymerases κ, and ζ, and REV1 seem to be important for this step [19, 21, 22, 24].

Models of ICL repair. a Model of ICL repair in quiescent cells (G0/G1 phase). An ICL on DNA is recognized by NER machinery. In the case of ICL-blocked transcription, two specific factors for transcription-coupled NER, CSA and CSB, are required to load the incision complex. In contrast, for ICLs in non-transcribed regions, the XPC-HHR23B complex is responsible for loading of incision complex of NER. The first incision is introduced by the incision complex composed of XPA-RPA, TFIIH, XPF-ERCC1 and XPG. After the first incision, the ICL lesion with the oligonucleotide is bypassed by a TLS polymerase such as DNA polymerase κ, DNA polymerase ζ, or REV1. The second incision is the introduced by another NER incision complex. b Model of ICL repair in S phase. ICL lesions cause stalling of DNA replication forks. The FANCM-FAAP24-MHF complex binds to a stalled replication fork and recruits both the FA core complex and the BLM-TOP3α-RMI1 complex. Activated FA core complex mono-ubiquitinates both FANCD2 and FANCI, which permits incisions of the ICL using structure-specific endonucleases such as XPF/FANCQ-ERCC1, SLX4/FANCP-SLX1, MUS81-EME1 and FAN1. The incision introduces a DSB which is repaired by. HR. Both RAD51 paralogs (RAD51B, RAD51C/FANCO, RAD51D, XRCC2 and XRCC3) and BRCA complexes (BRCA1, BRCA2/FANCD1, PALB2/FANCN, and BRIP1/FANCJ) are required for the formation of RAD51 filaments at damage sites. c Models of ICL incisions. An ICL lesion causes a stalled DNA replication fork that must be resolved by ICL incision. Three models for this process have been suggested. One model suggests that the first incision involves cleavage of the leading strand at a single stalled replication fork. The second model suggests that the first incision involves cleavage of the lagging strand at a single stalled replication fork. The third model suggests cleavage at two converged replication forks. After incision, the oligonucleotide with an ICL lesion is bypassed by a TLS polymerase, such as DNA polymerase κ, DNA polymerase ζ, or REV1, The DSB end is subsequently repaired by homologous recombination
ICL recognition in proliferating S phase cells
The process of ICL repair in S phase is quite complicated. Many studies have observed that treatment with ICL-causing agents introduces double-stranded DNA breaks (DSBs) in S phase cells [7, 25, 26]. One curious phenomenon about ICL-induced DSBs is that they are repaired by HR and not by non-homologous end joining (NHEJ) [12, 25]. Such phenomena strongly indicate that ICL-induced DSBs are associated with DNA replication forks. In S. cerevisiae, ICLs are mostly recognized by NER and complete NER function is responsible for the incisions. Therefore, all NER-mutants show hyper sensitive to ICL agents [25, 27]. In contrast, only XPF- and ERCC1-deficient cells are extremely hypersensitive to ICL agents, such as mitomycin C and nitrogen mustard in mammalian cells [25, 27]. The gene products of XPF and ERCC1 form a hetero-dimeric endonuclease that specifically recognizes and cleaves single-stranded branched structures [28]. Interestingly, the homologous structure-specific endonucleases MUS81-EME1 and XPF-ERCC1, are also involved in the repair process of ICL removal [7, 29]. MUS81-EME1 preferentially binds double-stranded branched structures, 3ʹ-flaps, and Holliday junctions [30]. Both XPF-ERCC1 and MUS81-EME1 are involved in ICL-induced DSB formation. However, further investigation is required to confirm whether DSB formation is directly involved in the removal of ICLs (Fig. 1b) [7, 31]. As many nucleases involved in the incision of ICLs have been recently identified, understanding the mechanisms of ICL incision is relevant for DNA repair. Here, we summarise our current understanding of ICL repair mechanisms in S phase. DSBs induced by ICLs in S phase are repaired by HR. In S. cerevisiae, hypersensitivity to ICL-causing agents is observed in rad51, rad52, rad54, rad59, and mre11 mutants, but not in yku70 mutants, and the hypersensitivity of rad52 yku70 double mutants to ICLs is comparable to that of rad52 mutants [12]. In fact, increased accumulation of DSBs after treatment with ICL-causing agents and defects in DSB repair are observed in HR-deficient strains, suggesting that NHEJ is not required for the repair of DSBs induced by ICLs [12]. A similar phenomenon is observed in mammalian cells [25]. Hypersensitivity to ICLs is seen in HR-deficient cells, such as cells carrying mutations in RAD51 paralogs, RAD54, RAD54B, and BRCA2, but not in NHEJ-deficient cells [32–34]. It is likely that HR plays a role in not only repairing DSBs but also in restarting stalled DNA replication forks [7, 35]. In higher eukaryotes, genes responsible for Fanconi anemia (FA) play important roles in ICL repair. Although the biological roles of FA gene products are not entirely characterized [36], they are known to control HR at DNA replication forks [4]. We will describe the roles of FA gene products in the next section, emphasizing the regulation of HR at stalled DNA replication forks.
ICL repair genes and human disorders
Proteins implicated in the repair of ICLs have a critical role in the pathophysiology of several hereditary disorders, known as FA, xeroderma pigmentosum (XP), Cockayne syndrome (CS), cerebro-oculo-facio-skeletal syndrome (COFS), and trichothyodistrophy (TTD, Table 1) [37]. FA is a genetic disorder characterized by aplastic anaemia, bone marrow failure, and cancers (typically acute myelogenous leukemia) [38, 39]. Mutations in one of the FANC genes cause severe sensitivity to ICL agents and genomic instability [38]. So far, at least 18 genes have been implicated in FA, and all the genes products act on the ICLs repair in S phase [4, 40]. On the other side, defects in NER pathways, which have a role in G0/G1 phase, result in also rare autosomal-recessive diseases, XP, CS, COFS syndrome, and TTD [41]. Mutations in eleven genes have been associated with these NER disorders [41]. Characteristics of XP include a photosensitivity, pigmentation, and frequent skin cancers. CS is an inherited syndrome characterized by short stature, mental deficiency, photosensitivity, disproportionately large hands, feet, and ears, ocular defects, and extensive demyelination [37]. CS has wide spectrum clinical features, and the most severely affected patients are included in a category of COFS syndrome [42]. TTD has a distinct sulfur-deficient brittle hair and neuroectodermal symptoms [41, 43, 44]. These NER disorders are distinguished from each other by these physical characteristics, including cutaneous malignancies (Table 2). Interestingly only ERCC1 and XPF gene products play a role in both S phase and G0/G1 phase pathways.
Given the established role of DNA repair factors as a genome keeper against a mutagenesis, it is not surprising that some of ICL genes have a strong linkage with cancer. Recent next-generation sequencing revealed the hereditary breast and/or ovary cancer syndrome (HBOC) related genes, and BRCA1, BRCA2, BRIP1, PALB2, and RAD51C genes are associated with HBOC in ICL repair pathways [4]. From the view of preventive medicine, early detection strategy is required in the social framework. Especially in the patients having the mutations in BRCA1 and BRCA2 genes, reasonable interventions are strongly recommended.
Many DNA cross-liker agents, such as cisplatin, psoralen, mitomycin C, and so on, were investigated in the long history of ICL repair fields. However none of these agents are produced in mammal internal organs. What is the pathophysiological accelerator of ICL repair defected patients? Recent study suggested the interesting story that aldehyde, one of the endogenous reactive metabolites, at least partially leads to genotoxic of FA patients [45]. Of course it is forbidden to forget that careful choice of medicine could prevent the incidental adverse event on these patients, who are apt to be given an anti cancer agents, such as cisplatin.
ICL incisions at stalled DNA replication sites
As an ICL lesion inhibits the unwinding of DNA strands, the progression of replicative DNA helicases are completely blocked at the site of the lesion. Previously, two distinct models of incision were proposed to explain stalling of a single replication fork [26]. One model suggests that the first incision cleaves the leading strand [46]. In this case, cleavage of either the 3ʹ-end of splayed arms or the 3ʹ-flap structure is required and XPF-ERCC1 and MUS81-EME1 are potentially involved in the process (Fig. 1c) [31]. The major shortcoming of this mechanism is the loss of MCM proteins that act as replicative DNA helicases. Another model is that the first incision occurs on the lagging strand [46]. This also requires cleavage of either the 3ʹ-end of splayed arms or the 3ʹ-flap structure. SLX1 and FAN1 may be the endonucleases involved in this process (Fig. 1c). This cleavage requires reloading of RNA primase-DNA polα complexes to restart DNA replication. Loading of MCM2-7 and RNA primase-DNA polα complexes at origins of DNA replication is strictly regulated in eukaryotic cells. Although it was suggested that MCM8-9 is involved in ICL repair to promote HR, the mechanism through which the complete DNA replication machinery is reconstructed at the recombination sites has not yet been characterized [47].
Recently, a dual fork convergence model was proposed based on biochemical studies of Xenopus egg extracts [48–51]. In this case, even if a single fork collapses because of ICLs, the stalled replication fork remains at the lesion site until another replication fork reaches the ICL from the other side. As the incision occurs at the DNA replication termination site, reloading of the complete DNA replication machinery is not required after incision. SLX4 is involved in the selection of structure-specific endonucleases and introduces incisions in one strand of the DNA on both sides of the ICL lesion (Fig. 1c). For cleavage of ICLs, it seems that XPF-ERCC1 and SLX1 are preferentially selected, but all the components of the entire incision complex have not been identified.
A new component that recognizes ICL lesions in mammalian cells
Recently, two groups independently discovered that a ubiquitin-like protein with both PHD and RING finger domains, UHRF1, directly recognizes ICL lesions (Fig. 2) [52, 53]. UHRF1 recognizes specific forms of histones and hemi-methylated DNA, and recruits DNMT1 [54, 55]. It is known that UHRF1-deficient ES cells exhibit hypersensitivity to DNA damaging agents such as ionizing radiation, UV light, N-methyl-Nʹ-nitro-N-nitrosoguanidine, and hydroxyurea [56]. UHRF1 strongly responds to ICLs formed by trimethyl psoralen and mitomycin C, and exhibits a weaker response to those formed by cisplatin [53]. This is because UHRF1 preferentially recognizes ICLs that cause minor distortions of the DNA helix, such as those formed by either trimethyl psoralen or mitomycin C. In contrast, ICLs formed by cisplatin cause a major distortion [2]. Both groups suggested that the likely role of UHRF1 is the recruitment of structure-specific endonucleases such as XPF-ERCC1 and MUS81-EME1 (Fig. 2) [52, 53]. However, Tian et al. argued that recruitment of nucleases is required for FA functions [52], while Liang et al. suggested that recruitment of nucleases by UHRF1 is independent of FA pathway components [53]. The mechanism of ICL recognition remains unclear and further investigation is required to precisely elucidate the mechanism.

UHRF1-dependent recruitment of structure-specific endonucleases to ICLs. UHRF-1 is involved in the recruitment of FANCD2 and other DNA repair factors including XPF-ERCC1 and MUS81-EME1. Detailed mechanisms how UHRF1 recruits ICL repair factors are unclear
Perspective
Recently, many factors involved in ICL repair have been identified, particularly in mammalian cells. In quiescent eukaryotic cells from species including yeasts and higher eukaryotes, both the first and second rounds of ICL incisions are performed by NER. In contrast, not all NER factors are involved in ICL repair in S phase cells in higher eukaryotes. Instead, a variety of structure-specific endonucleases, TLS, HR, and FA pathways are required for ICL repair in S phase cells. One open question is whether ICL repair is error-free repair or not. ICL usually occurs between purine residues, however, it may also happen between pyrimidine residues. Because of such complexity, TLS bypass have no grantee to maintain error-free bypass for all ICL sites. To understand the accuracy of ICL repair requires further investigation.
In addition, very recent studies suggest that UHRF1 recognizes ICL lesions independent of the FA pathway, which is activated by recognition of stalled DNA replication forks. In ICL repair, the role of chromatin reorganization is poorly understood relative to other DNA repair pathways. One important phenomenon related to ICL repair is that NHEJ is not required for the DSB repair after ICL incision. Single DNA ends, which are produced by DSB formation at DNA replication forks, are selectively repaired by HR. In contrast, two DNA ends are predominantly repaired by NHEJ in mammalian cells. However, the dual fork incision model produces two DNA ends. If broken ends produced by dual fork incision are selectively repaired by HR, chromatin structures might be a key factor in selecting a DSB repair pathway. Certainly, many factors involved in chromatin reorganization will be identified in the future as co-factors of UHRF1 that participate in ICL repair. This will provide an engaging challenge for researchers in this field.
Conclusions
From human genetic studies, many factors involved in ICL repair were identified. FA core complex play important roles to conduct ICL-recognition as well as DSB repair by HR. As unhooking enzymes, several structure-specific endonucleases, SLX4-SLX1, FAN1, and XPF-ERCC1, were also identified. Based on these, several models of ICL repair in S phase were proposed. In this review, we summarized the resent achievements of ICL repair. However, to discuss the entire mechanism of ICL repair, many factors are still missing. Therefore, many new factors will be discovered in the near future. In addition, understanding the molecular mechanisms of ICL repair also contribute to studies of genome instabilities and mutagenesis caused by ICL agents. Studies of ICL repair will certainly attract attention of researchers in this field for a while.
Abbreviations
- BER:
-
base excision repair
- CS:
-
Cockayne syndrome
- COFS:
-
cerebro-oculo-facio-skeletal syndrome
- DSB:
-
double-strand break
- FA:
-
Fanconi anemia
- HBOC:
-
hereditary breast and/or ovary cancer syndrome
- HR:
-
homologous recombination
- ICL:
-
interstand crosslink
- MMR:
-
mismatch repair
- NER:
-
nucleotide excision repair
- NHEJ:
-
non-homologous end-joining
- TLS:
-
translesion DNA synthesis
- TTD:
-
trichothyodistrophy
- XP:
-
xeroderma pigmentosum
References
Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486(4):217–47.
Guainazzi A, Scharer OD. Using synthetic DNA interstrand crosslinks to elucidate repair pathways and identify new therapeutic targets for cancer chemotherapy. Cell Mol Life Sci. 2010;67(21):3683–97. doi:10.1007/s00018-010-0492-6.
Legerski RJ. Repair of DNA interstrand cross-links during S phase of the mammalian cell cycle. Environ Mol Mutagen. 2010;51(6):540–51. doi:10.1002/em.20566.
Clauson C, Scharer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Biol. 2013;5(10):a012732. doi:10.1101/cshperspect.a012732.
Liu X, Lao Y, Yang IY, Hecht SS, Moriya M. Replication-coupled repair of crotonaldehyde/acetaldehyde-induced guanine-guanine interstrand cross-links and their mutagenicity. Biochemistry. 2006;45(42):12898–905. doi:10.1021/bi060792v.
Zhu G, Song L, Lippard SJ. Visualizing inhibition of nucleosome mobility and transcription by cisplatin-DNA interstrand crosslinks in live mammalian cells. Cancer Res. 2013;73(14):4451–60. doi:10.1158/0008-5472.CAN-13-0198.
Hanada K, Budzowska M, Modesti M, Maas A, Wyman C, Essers J, et al. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 2006;25(20):4921–32. doi:10.1038/sj.emboj.7601344.
Stern RS. Psoralen and ultraviolet a light therapy for psoriasis. N Engl J Med. 2007;357(7):682–90. doi:10.1056/NEJMct072317.
Huang H, Zhu L, Reid BR, Drobny GP, Hopkins PB. Solution structure of a cisplatin-induced DNA interstrand cross-link. Science. 1995;270(5243):1842–5.
Rink SM, Hopkins PB. A mechlorethamine-induced DNA interstrand cross-link bends duplex DNA. Biochemistry. 1995;34(4):1439–45.
Gilman A. The initial clinical trial of nitrogen mustard. Am J Surg. 1963;105:574–8.
McHugh PJ, Sones WR, Hartley JA. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20(10):3425–33.
Mutzhas MF, Holzle E, Hofmann C, Plewig G. A new apparatus with high radiation energy between 320-460 nm: physical description and dermatological applications. J Invest Dermatol. 1981;76(1):42–7.
Haran TE, Crothers DM. Phased psoralen cross-links do not bend the DNA double helix. Biochemistry. 1988;27(18):6967–71.
Hwang GS, Kim JK, Choi BS. The solution structure of a psoralen cross-linked DNA duplex by NMR and relaxation matrix refinement. Biochem Biophys Res Commun. 1996;219(1):191–7. doi:10.1006/bbrc.1996.0204.
Norman D, Live D, Sastry M, Lipman R, Hingerty BE, Tomasz M, et al. NMR and computational characterization of mitomycin cross-linked to adjacent deoxyguanosines in the minor groove of the d(T-A-C-G-T-A).d(T-A-C-G-T-A) duplex. Biochemistry. 1990;29(11):2861–75.
Rink SM, Lipman R, Alley SC, Hopkins PB, Tomasz M. Bending of DNA by the mitomycin C-induced, GpG intrastrand cross-link. Chem Res Toxicol. 1996;9(2):382–9. doi:10.1021/tx950156q.
McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2(8):483–90. doi:10.1016/S1470-2045(01)00454-5.
Sarkar S, Davies AA, Ulrich HD, McHugh PJ. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J. 2006;25(6):1285–94. doi:10.1038/sj.emboj.7600993.
Wood RD. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ Mol Mutagen. 2010;51(6):520–6. doi:10.1002/em.20569.
McHugh PJ, Sarkar S. DNA interstrand cross-link repair in the cell cycle: a critical role for polymerase zeta in G1 phase. Cell Cycle. 2006;5(10):1044–7.
Williams HL, Gottesman ME, Gautier J. Replication-independent repair of DNA interstrand crosslinks. Mol Cell. 2012;47(1):140–7. doi:10.1016/j.molcel.2012.05.001.
Klug AR, Harbut MB, Lloyd RS, Minko IG. Replication bypass of N2-N2 deoxyguanosine interstrand cross-links by human DNA polymerases eta and iota. Chem Res Toxicol. 2012;25(3):755–62. doi:10.1021/tx300011w.
Minko IG, Harbut MB, Kozekov ID, Kozekova A, Jakobs PM, Olson SB, et al. Role for DNA polymerase kappa in the processing of N2-N2-guanine interstrand cross-links. J Biol Chem. 2008;283(25):17075–82. doi:10.1074/jbc.M801238200.
De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20(21):7980–90.
Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24(13):5776–87. doi:10.1128/MCB.24.13.5776-5787.2004.
De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defects in interstrand cross-link uncoupling do not account for the extreme sensitivity of ERCC1 and XPF cells to cisplatin. Nucleic Acids Res. 2002;30(17):3848–56.
Enzlin JH, Scharer OD. The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J. 2002;21(8):2045–53. doi:10.1093/emboj/21.8.2045.
Abraham J, Lemmers B, Hande MP, Moynahan ME, Chahwan C, Ciccia A, et al. Eme1 is involved in DNA damage processing and maintenance of genomic stability in mammalian cells. EMBO J. 2003;22(22):6137–47. doi:10.1093/emboj/cdg580.
Chen XB, Melchionna R, Denis CM, Gaillard PH, Blasina A, Van de Weyer I, et al. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol Cell. 2001;8(5):1117–27.
Rothfuss A, Grompe M. Repair kinetics of genomic interstrand DNAcross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2004;24(1):123–34.
Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, et al. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol. 2001;21(8):2858–66. doi:10.1128/MCB.21.8.2858-2866.2001.
Kraakman-van der Zwet M, Overkamp WJ, van Lange RE, Essers J, van Duijn-Goedhart A, Wiggers I, et al. Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol Cell Biol. 2002;22(2):669–79.
Wesoly J, Agarwal S, Sigurdsson S, Bussen W, Van Komen S, Qin J, et al. Differential contributions of mammalian Rad54 paralogs to recombination, DNA damage repair, and meiosis. Mol Cell Biol. 2006;26(3):976–89. doi:10.1128/MCB.26.3.976-989.2006.
Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol. 2010;11(10):683–7. doi:10.1038/nrm2974.
Bakker ST, de Winter JP, te Riele H. Learning from a paradox: recent insights into Fanconi anaemia through studying mouse models. Dis Model Mech. 2013;6(1):40–7. doi:10.1242/dmm.009795.
Vermeij WP, Hoeijmakers JH, Pothof J. Aging: not all DNA damage is equal. Curr Opin Genet Dev. 2014;26:124–30. doi:10.1016/j.gde.2014.06.006.
Taniguchi T, D’Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107(11):4223–33. doi:10.1182/blood-2005-10-4240.
Wilson DB, Link DC, Mason PJ, Bessler M. Inherited bone marrow failure syndromes in adolescents and young adults. Ann Med. 2014;46(6):353–63. doi:10.3109/07853890.2014.915579.
Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet. 2015;96(6):1001–7. doi:10.1016/j.ajhg.2015.04.022.
Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–85. doi:10.1056/NEJMra0804615.
Laugel V. Cockayne syndrome: the expanding clinical and mutational spectrum. Mech Ageing Dev. 2013;134(5-6):161–70. doi:10.1016/j.mad.2013.02.006.
Hashimoto S, Egly JM. Trichothiodystrophy view from the molecular basis of DNA repair/transcription factor TFIIH. Hum Mol Genet. 2009;18(R2):R224–30. doi:10.1093/hmg/ddp390.
Sepe S, Payan-Gomez C, Milanese C, Hoeijmakers JH, Mastroberardino PG. Nucleotide excision repair in chronic neurodegenerative diseases. DNA Repair (Amst). 2013;12(8):568–77. doi:10.1016/j.dnarep.2013.04.009.
Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, Patel KJ. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489(7417):571–5. doi:10.1038/nature11368.
Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst). 2014;19:135–42. doi:10.1016/j.dnarep.2014.03.018.
Nishimura K, Ishiai M, Horikawa K, Fukagawa T, Takata M, Takisawa H, et al. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol Cell. 2012;47(4):511–22. doi:10.1016/j.molcel.2012.05.047.
Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134(6):969–80. doi:10.1016/j.cell.2008.08.030.
Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326(5960):1698–701. doi:10.1126/science.1182372.
Long DT, Raschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333(6038):84–7. doi:10.1126/science.1204258.
Budzowska M, Graham TG, Sobeck A, Waga S, Walter JC. Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link. EMBO J. 2015;34(14):1971–85. doi:10.15252/embj.201490878.
Tian Y, Paramasivam M, Ghosal G, Chen D, Shen X, Huang Y, et al. UHRF1 Contributes to DNA damage repair as a lesion recognition factor and nuclease scaffold. Cell Rep. 2015;10(12):1957–66. doi:10.1016/j.celrep.2015.03.038.
Liang CC, Zhan B, Yoshikawa Y, Haas W, Gygi SP, Cohn MA. UHRF1 Is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi Anemia pathway. Cell Rep. 2015;10(12):1947–56. doi:10.1016/j.celrep.2015.02.053.
Liu X, Gao Q, Li P, Zhao Q, Zhang J, Li J, et al. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat Commun. 2013;4:1563. doi:10.1038/ncomms2562.
Nishiyama A, Yamaguchi L, Sharif J, Johmura Y, Kawamura T, Nakanishi K, et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013;502(7470):249–53. doi:10.1038/nature12488.
Muto M, Kanari Y, Kubo E, Takabe T, Kurihara T, Fujimori A, et al. Targeted disruption of Np95 gene renders murine embryonic stem cells hypersensitive to DNA damaging agents and DNA replication blocks. J Biol Chem. 2002;277(37):34549–55. doi:10.1074/jbc.M205189200.
Tachibana A, Kato T, Ejima Y, Yamada T, Shimizu T, Yang L, et al. The FANCA gene in Japanese Fanconi anemia: reports of eight novel mutations and analysis of sequence variability. Hum Mutat. 1999;13(3):237–44. doi:10.1002/(SICI)1098-1004(1999)13:3<237::AID-HUMU8>3.0.CO;2-F.
Fei P, Yin J, Wang W. New advances in the DNA damage response network of Fanconi anemia and BRCA proteins. FAAP95 replaces BRCA2 as the true FANCB protein. Cell Cycle. 2005;4(1):80–6.
Strathdee CA, Gavish H, Shannon WR, Buchwald M. Cloning of cDNAs for Fanconi’s anaemia by functional complementation. Nature. 1992;356(6372):763–7. doi:10.1038/356763a0.
Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297(5581):606–9. doi:10.1126/science.1073834.
Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, et al. Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell. 2001;7(2):241–8.
Waisfisz Q, Saar K, Morgan NV, Altay C, Leegwater PA, de Winter JP, et al. The Fanconi anemia group E gene, FANCE, maps to chromosome 6p. Am J Hum Genet. 1999;64(5):1400–5. doi:10.1086/302385.
de Winter JP, Leveille F, van Berkel CG, Rooimans MA, van Der Weel L, Steltenpool J, et al. Isolation of a cDNA representing the Fanconi anemia complementation group E gene. Am J Hum Genet. 2000;67(5):1306–8. doi:10.1016/S0002-9297(07)62959-0.
de Winter JP, Rooimans MA, van Der Weel L, van Berkel CG, Alon N, Bosnoyan-Collins L, et al. The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nat Genet. 2000;24(1):15–6. doi:10.1038/71626.
de Winter JP, Waisfisz Q, Rooimans MA, van Berkel CG, Bosnoyan-Collins L, Alon N, et al. The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nat Genet. 1998;20(3):281–3. doi:10.1038/3093.
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald 3rd ER, Hurov KE, Luo J, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129(2):289–301. doi:10.1016/j.cell.2007.03.009.
Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, et al. Identification of the Fanconi anemia complementation group I gene. FANCI Cell Oncol. 2007;29(3):211–8.
Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J Nat Genet. 2005;37(9):934–5. doi:10.1038/ng1625.
Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8(3):255–65. doi:10.1016/j.ccr.2005.08.004.
Meetei AR, Yan Z, Wang W. FANCL replaces BRCA1 as the likely ubiquitin ligase responsible for FANCD2 monoubiquitination. Cell Cycle. 2004;3(2):179–81.
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39(2):165–7. doi:10.1038/ng1959.
Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39(2):162–4. doi:10.1038/ng1947.
Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet. 2010;42(5):406–9. doi:10.1038/ng.570.
Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42(5):410–4. doi:10.1038/ng.569.
Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat Genet. 2011;43(2):142–6. doi:10.1038/ng.750.
Thompson LH, Brookman KW, Weber CA, Salazar EP, Reardon JT, Sancar A, et al. Molecular cloning of the human nucleotide-excision-repair gene ERCC4. Proc Natl Acad Sci U S A. 1994;91(15):6855–9.
Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444(7122):1038–43. doi:10.1038/nature05456.
Osorio A, Bogliolo M, Fernandez V, Barroso A, de la Hoya M, Caldes T, et al. Evaluation of rare variants in the new fanconi anemia gene ERCC4 (FANCQ) as familial breast/ovarian cancer susceptibility alleles. Hum Mutat. 2013;34(12):1615–8. doi:10.1002/humu.22438.
Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M. University of Washington Centre for Mendelian G et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5(2):135–42. doi:10.1158/2159-8290.CD-14-1156.
Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM, Kennedy JA, et al. Deficiency of UBE2T, the E2 Ubiquitin Ligase necessary for FANCD2 and FANCI Ubiquitination, causes FA-T subtype of Fanconi anemia. Cell Rep. 2015;12(1):35–41. doi:10.1016/j.celrep.2015.06.014.
Jaspers NG, Raams A, Silengo MC, Wijgers N, Niedernhofer LJ, Robinson AR, et al. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet. 2007;80(3):457–66. doi:10.1086/512486.
Tanaka K, Miura N, Satokata I, Miyamoto I, Yoshida MC, Satoh Y, et al. Analysis of a human DNA excision repair gene involved in group A xeroderma pigmentosum and containing a zinc-finger domain. Nature. 1990;348(6296):73–6. doi:10.1038/348073a0.
Koken MH, Vreeken C, Bol SA, Cheng NC, Jaspers-Dekker I, Hoeijmakers JH, et al. Cloning and characterization of the Drosophila homolog of the xeroderma pigmentosum complementation-group B correcting gene, ERCC3. Nucleic Acids Res. 1992;20(21):5541–8.
Weeda G, Eveno E, Donker I, Vermeulen W, Chevallier-Lagente O, Taieb A, et al. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet. 1997;60(2):320–9.
Oh KS, Khan SG, Jaspers NG, Raams A, Ueda T, Lehmann A, et al. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum Mutat. 2006;27(11):1092–103. doi:10.1002/humu.20392.
Legerski R, Peterson C. Expression cloning of a human DNA repair gene involved in xeroderma pigmentosum group C. Nature. 1992;359(6390):70–3. doi:10.1038/359070a0.
Arrand JE, Bone NM, Johnson RT. Molecular cloning and characterization of a mammalian excision repair gene that partially restores UV resistance to xeroderma pigmentosum complementation group D cells. Proc Natl Acad Sci U S A. 1989;86(18):6997–7001.
Broughton BC, Steingrimsdottir H, Weber CA, Lehmann AR. Mutations in the xeroderma pigmentosum group D DNA repair/transcription gene in patients with trichothiodystrophy. Nat Genet. 1994;7(2):189–94. doi:10.1038/ng0694-189.
van Hoffen A, Kalle WH, de Jong-Versteeg A, Lehmann AR, van Zeeland AA, Mullenders LH. Cells from XP-D and XP-D-CS patients exhibit equally inefficient repair of UV-induced damage in transcribed genes but different capacity to recover UV-inhibited transcription. Nucleic Acids Res. 1999;27(14):2898–904.
Chu G, Chang E. Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science. 1988;242(4878):564–7.
Vermeulen W, Jaeken J, Jaspers NG, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum complementation group G associated with Cockayne syndrome. Am J Hum Genet. 1993;53(1):185–92.
Nouspikel T, Clarkson SG. Mutations that disable the DNA repair gene XPG in a xeroderma pigmentosum group G patient. Hum Mol Genet. 1994;3(6):963–7.
Henning KA, Li L, Iyer N, McDaniel LD, Reagan MS, Legerski R, et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell. 1995;82(4):555–64.
Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell. 1992;71(6):939–53.
Coin F, Proietti De Santis L, Nardo T, Zlobinskaya O, Stefanini M, Egly JM. p8/TTD-A as a repair-specific TFIIH subunit. Mol Cell. 2006;21(2):215–26. doi:10.1016/j.molcel.2005.10.024.
Acknowledgements
SH is funded by the Inamori Foundation, and by Grant-in-Aid for Scientific Research (C) (15 K06758), Japan Society for the Promotion of Science (JSPS), The Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. KH is funded by Grant-in-Aid for Young Scientists (A) (25710010), JSPS. No conflict of interest statement is declared.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All three authors were contributed to write this manuscript, and approved the final manuscript. KH is responsible for biochemical part of this manuscript, and SH and HA summarized clinical part.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hashimoto, S., Anai, H. & Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes and Environ 38, 9 (2016). https://doi.org/10.1186/s41021-016-0037-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41021-016-0037-9