
Abstract
Burkholderia dilworthii strain WSM3556T is an aerobic, motile, Gram-negative, non-spore-forming rod that was isolated from an effective N2-fixing root nodule of Lebeckia ambigua collected near Grotto Bay Nature Reserve, in the Western Cape of South Africa, in October 2004. This plant persists in infertile and deep sandy soils with acidic pH, and is therefore an ideal candidate for a perennial based agriculture system in Western Australia. WSM3556T thus represents a potential inoculant quality strain for L. ambigua for which we describe the general features, together with genome sequence and annotation. The 7,679,067 bp high-quality permanent draft genome is arranged in 140 scaffolds of 141 contigs, contains 7,059 protein-coding genes and 64 RNA-only encoding genes, and is part of the GEBA-RNB project proposal.
Similar content being viewed by others


Introduction
Over the last decade, agricultural scientists have sought to discover perennial legumes from a wide range of natural environments to develop new plants for grazing systems [1]. It is thought that these plants might be more resilient to changing rainfall patterns, such as in the target environments of Western Australia. Here, winter rainfall has declined by 20 % in the last two decades [2], although more frequent summer rainfall events have been experienced. In the fynbos biome of South Africa, several species that offer potential for domestication have been discovered [1, 3]. These legumes are frequently nodulated by Burkholderia bacteria in the class Betaproteobacteria [3, 4]. The symbiosis between these Burkholderia and legumes from the genera Lebeckia and Rhynchosia fix atmospheric nitrogen to enable their cultivation on infertile soils [4–7]. Lebeckia ambigua is proving well adapted to Western Australia [1] because in areas where it is naturally found in South Africa the soil and climatic conditions approximate those of Western Australia.
Nodules and seeds of L. ambigua were collected in four expeditions to the Western Cape of South Africa between 2002 and 2007. The isolation of bacteria from these nodules gave rise to a collection of 23 strains that were identified as Burkholderia [3]. Unlike most of the previously studied nodulating Burkholderia strains, this South African group appears to associate with papilionoid forage legumes, rather than Mimosa species. WSM3556T belongs to a subgroup of strains that were isolated in 2004 from nodules collected south west of Darling, in a natural rangeland site on the southern border of the Grotto Bay Nature Reserve [3]. The soil at the site of collection was deep sand with a pH of 6. Burkholderia dilworthii strain WSM3556T was isolated from those nodules and is effective at fixing nitrogen with L. ambigua and L. sepiaria. The nodules formed by these symbioses are crotaloid and indeterminate [3].
WSM3556T thus represents a potential inoculant quality strain for L. ambigua, which is being developed as a grazing legume adapted to infertile soils that receive 250–400 mm annual rainfall in southern Australia and is therefore of special interest to the RNB chapter of the GEBA project. Here we present a summary classification and a set of general features for Burkholderia dilworthii strain WSM3556T together with the description of the permanent draft genome sequence and annotation.
Organism information
Classification and features
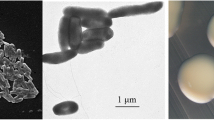
Burkholderia dilworthii strain WSM3556T is a motile, Gram-negative, non-spore-forming rod (Fig. 1 Left, Center) in the order Burkholderiales of the class Betaproteobacteria . The rod-shaped form varies in size with dimensions of 0.9–2 μm in width and 0.4–3.0 μm in length (Fig. 1 Left). It is fast growing, forming 0.4–2 mm diameter colonies after 24 h when grown on half Lupin Agar [8] and TY [9] at 28 °C. Colonies on ½LA are white-opaque, slightly domed, moderately mucoid with smooth margins (Fig. 1 Right). Additional physiological properties of this strain were previously published [5].

Images of Burkholderia dilworthii strain WSM3556T using scanning (Left) and transmission (Center) electron microscopy and the appearance of colony morphology on solid media (Right)
Figure 2 shows the phylogenetic relationship of Burkholderia dilworthii strain WSM3556T in a 16S rRNA gene sequence based tree. This strain is most similar to Burkholderia rhynchosiae WSM3937T and Burkholderia phytofirmans PsJNT based on the 16S rRNA with sequence identities of 98.50 % and 98.11 %, respectively, as determined using the EzTaxon-e server [10]. Burkholderia rhynchosiae WSM3937T has been isolated from Rhynchosia ferulifolia , a herbaceous legume from the fynbos biome in South Africa [7]. Burkholderia phytofirmans PsJNT was isolated from surface sterilized onion roots and has plant growth promoting properties on various plants, however it has not been reported in association with legumes [11]. Minimum Information about the Genome Sequence of WSM3556T is provided in Table 1.

Phylogenetic tree highlighting the position of Burkholderia dilworthii strain WSM3556T (shown in blue print), relative to other strains in the Burkholderia genus using a 1,322 bp internal region of the 16S rRNA gene. Cupriavidus taiwanensis LMG 19424T was used as an outgroup. All sites were informative and there were no gap-containing sites. Phylogenetic analyses were performed using MEGA, version 5.05 [31]. The tree was build using the maximum likelihood method with the General Time Reversible model. Bootstrap analysis with 500 replicates was performed to assess the support of the clusters. Type strains are indicated with a superscript T. Strains with a genome sequencing project registered in GOLD [14] are in bold print and the GOLD ID is provided after the NCBI accession number. Published genomes are designated with an asterisk
Symbiotaxonomy
Burkholderia dilworthii strain WSM3556T belongs to a group of Burkholderia strains that nodulate papilionoid forage legumes rather than the classical Mimosa host species (Mimosoideae) described for other Burkholderia microsymbionts [12]. Burkholderia dilworthii strain WSM3556T was assessed for nodulation and nitrogen fixation on three separate L. ambigua genotypes (CRSLAM-37, CRSLAM-39 and CRSLAM-41) [3]. It could nodulate and fix effectively on CRSLAM-41 but was partially effective on CRSLAM-37 and CRSLAM-39 [3]. Moreover, WSM3556T also nodulates and fixes nitrogen in association with Lebeckia sepiaria .
Genome sequencing information
Genome project history
This organism was selected for sequencing on the basis of its environmental and agricultural relevance to issues in global carbon cycling, alternative energy production, and biogeochemical importance, and is part of the Genomic Encyclopedia of Bacteria and Archaea, The Root Nodulating Bacteria chapter project at the U.S. Department of Energy, Joint Genome Institute for projects of relevance to agency missions [13]. The genome project is deposited in the Genomes OnLine Database [14] and the high-quality permanent draft genome sequence in IMG [15]. Sequencing, finishing and annotation were performed by the JGI using state of the art sequencing technology [16]. A summary of the project information is shown in Table 2.
Growth conditions and genomic DNA preparation
Burkholderia dilworthii strain WSM3556T was grown on TY solid medium [9] for 3 days, a single colony was selected and used to inoculate 5 ml TY broth medium. The culture was grown for 48 h on a gyratory shaker (200 rpm) at 28 °C. Subsequently 1 ml was used to inoculate 60 ml TY broth medium and grown on a gyratory shaker (200 rpm) at 28 °C until OD 0.6 was reached. DNA was isolated from 60 mL of cells using a CTAB bacterial genomic DNA isolation method [17]. Final concentration of the DNA was 0.5 mg/ml.
Genome sequencing and assembly
The genome of Burkholderia dilworthii strain WSM3556T was sequenced at the DOE Joint Genome Institute using state of the art technology [18]. For this genome, an Illumina standard shotgun library was constructed and sequenced using the Illumina HiSeq 2000 platform, which generated 9,394,768 reads totalling 2,818.4 Mbp of Illumina data. All general aspects of library construction and sequencing performed at the JGI can be found on the JGI web site [16]. All raw Illumina sequence data was passed through DUK, a filtering program developed at JGI, which removes known Illumina sequencing and library preparation artifacts (Mingkun L, Copeland A, Han J. unpublished). The following steps were then performed for assembly: (1) filtered Illumina reads were assembled using Velvet, version 1.1.04 [19], (2) 1–3 Kbp simulated paired end reads were created from Velvet contigs using wgsim [20], (3) Illumina reads were assembled with simulated read pairs using Allpaths (version r37348) [21]. Parameters for assembly steps were: 1) Velvet -exp_cov 90 -cov_cutoff 20 -exportFiltered yes -very_clean yes), 2) wgsim (−e 0–1 76–2 76 -r 0 -R 0 -X 0 -d 3000 -s 300 -N 1266735), 3) Allpaths–LG (PrepareAllpathsInputs: PHRED_64 = 1 PLOIDY = 1 JUMP_COVERAGE = 25 FRAG_COVERAGE = 125, RunAllpathsLG: RUN = 125std + 25xfakedpairs TARGETS = standard VAPI_WARN_ONLY = True OVERWRITE = True). The final draft assembly contained 141 contigs in 140 scaffolds. The total size of the genome is 7.7 Mbp and the final assembly is based on 2,818.4 Mbp of Illumina draft data, which provides an average of 367x coverage of the genome.
Genome annotation
Genes were identified using Prodigal [22], as part of the DOE-JGI genome annotation pipeline [23, 24] followed by a round of manual curation using GenePRIMP [25] for finished genomes and Draft genomes in fewer than 10 scaffolds. The predicted CDSs were translated and used to search the NCBI non-redundant database, UniProt, TIGRFam, Pfam, KEGG, COG, and InterPro databases. The tRNAScanSE tool [26] was used to find tRNA genes, whereas ribosomal RNA genes were found by searches against models of the ribosomal RNA genes built from SILVA [27]. Other non–coding RNAs such as the RNA components of the protein secretion complex and the RNase P were identified by searching the genome for the corresponding Rfam profiles using INFERNAL [28]. Additional gene prediction analysis and manual functional annotation was performed within the Integrated Microbial Genomes-Expert Review system [29] developed by the Joint Genome Institute, Walnut Creek, CA, USA.
Genome properties
The genome is 7,679,067 nucleotides with 61.77 % GC content (Table 3) and comprised of 140 scaffolds and 141 contigs. From a total of 7,123 genes, 7,059 were protein encoding and 64 RNA only encoding genes. The majority of genes (76.25 %) were assigned a putative function whilst the remaining genes were annotated as hypothetical. The distribution of genes into COG functional categories is presented in Table 4.
Conclusion
Burkholderia dilworthii WSM3556T belongs to a group of Beta-rhizobia isolated from Lebeckia ambigua from the fynbos biome in South Africa [3]. WSM3556T is phylogeneticaly most closely related to Burkholderia rhynchosiae WSM3937T and Burkholderia phytofirmans PsJNT. Of these strains only WSM3556T and WSM3937T are legume microsymbionts. Out of 13 Burkholderia strains that are known legume microsymbionts, only four (WSM3556T , WSM4176, WSM5005T , STM678T) nodulate South African papilionoid species. A comparison of these nodulating strains reveals that WSM3556T has the smallest genome (7.7 Mbp), the smallest KOG count (1295) and the lowest GC (61.77 %) percentage in this group. These four genomes share the nitrogenase-RXN MetaCyc pathway catalyzed by a multiprotein nitrogenase complex. Strains WSM3556T , WSM4176, WSM5005T [30] have been shown to fix nitrogen with Lebeckia ambigua provenances with varying degrees of effectiveness. WSM3556T is partially effective on two out of three L. ambigua provenances, WSM4176 is partially effective on only one L. ambigua provenance and WSM5005T is effective on all three L. ambigua provenances. The genome sequences of these fynbos bacteria provides an unprecedented opportunity to reveal the genetic determinants required for effective nitrogen fixation with Lebeckia .
Abbreviations
- GEBA-RNB:
-
Genomic Encyclopedia of Bacteria and Archaea – Root Nodule Bacteria
- JGI:
-
Joint Genome Institute
- TY:
-
Trypton yeast
- CTAB:
-
Cetyl trimethyl ammonium bromide
- WSM:
-
Western Australian Soil Microbiology
References
Howieson JG, Yates RJ, Foster K, Real D, Besier B. Prospects for the future use of legumes. In: Dilworth MJ, James EK, Sprent JI, Newton WE, editors. Leguminous Nitrogen-Fixing Symbioses. London, UK: Elsevier; 2008. p. 363–94.
George RJ, Speed RJ, Simons JA, Smith RH, Ferdowsian R, Raper GP, et al. Long-term groundwater trends and their impact on the future extent of dryland salinity in Western Australia in a varibale climate. In Salinity Forum. 2008.
Howieson JG, De Meyer SE, Vivas-Marfisi A, Ratnayake S, Ardley JK, Yates RJ. Novel Burkholderia bacteria isolated from Lebeckia ambigua - A perennial suffrutescent legume of the fynbos. Soil Biol Biochem. 2013;60:55–64.
Garau G, Yates RJ, Deiana P, Howieson JG. Novel strains of nodulating Burkholderia have a role in nitrogen fixation with papilionoid herbaceous legumes adapted to acid, infertile soils. Soil Biol Biochem. 2009;41:125–34.
De Meyer SE, Cnockaert M, Ardley JK, Van Wyk B-E, Vandamme PA, Howieson JG. Burkholderia dilworthii sp. nov., isolated from Lebeckia ambigua root nodules. Int J Syst Evol Microbiol. 2014;64:1090–5.
De Meyer SE, Cnockaert M, Ardley JK, Maker G, Yates R, Howieson JG, et al. Burkholderia sprentiae sp. nov., isolated from Lebeckia ambigua root nodules. Int J Syst Evol Microbiol. 2013;63:3950–7.
De Meyer SE, Cnockaert M, Ardley JK, Trengove RD, Garau G, Howieson JG, et al. Burkholderia rhynchosiae sp. nov., isolated from Rhynchosia ferulifolia root nodules. Int J Syst Evol Microbiol. 2013;63:3944–9.
Howieson JG, Ewing MA, D'antuono MF. Selection for acid tolerance in Rhizobium meliloti. Plant Soil. 1988;105:179–88.
Beringer JE. R factor transfer in Rhizobium leguminosarum. J Gen Microbiol. 1974;84:188–98.
Kim O-S, Cho Y-J, Lee K, Yoon S-H, Kim M, Na H, et al. Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol. 2012;62:716–21.
Sessitsch A, Coenye T, Sturz AV, Vandamme P, Barka EA, Salles JF, et al. Burkholderia phytofirmans sp. nov., a novel plant-associated bacterium with plant-beneficial properties. Int J Syst Evol Microbiol. 2005;55:1187–92.
Elliott GN, Chou J-H, Chen W-M, Bloemberg GV, Bontemps C, Martínez-Romero E, et al. Burkholderia spp. are the most competitive symbionts of Mimosa, particularly under N-limited conditions. Environ Microbiol. 2009;11:762–78.
Reeve W, Ardley J, Tian R, Eshragi L, Yoon J, Ngamwisetkun P, et al. A genomic encyclopedia of the root nodule bacteria: Assessing genetic diversity through a systematic biogeographic survey. Stand Genomic Sci. 2015;10:14.
Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, et al. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40:D571–579.
Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Pillay M, et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42:D560–7.
JGI Website. [http://www.jgi.doe.gov]
CTAB DNA extraction protocol. [http://jgi.doe.gov/collaborate-with-jgi/pmo-overview/protocols-sample-preparation-information/]
Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS ONE. 2012;7, e48837.
Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9.
wgsim. [https://github.com/lh3/wgsim]
Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 2011;108:1513–8.
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119.
Chen IM, Markowitz VM, Chu K, Anderson I, Mavromatis K, Kyrpides NC, et al. Improving microbial genome annotations in an integrated database context. PLoS ONE. 2013;8, e54859.
Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI Standard Operating Procedure for the annotations of microbial genomes. Stand Genomic Sci. 2009;1:63–7.
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, et al. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods. 2010;7:455–7.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–96.
INFERNAL. Inference of RNA alignments. [http://infernal.janelia.org]
Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–8.
Reeve W, De Meyer S, Terpolilli J, Melino V, Ardley J, Rui T, et al. Genome sequence of the Lebeckia ambigua - nodulating Burkholderia sprentiae strain WSM5005T. Stand Genomic Sci. 2013;9:385–94.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. Towards a richer description of our complete collection of genomes and metagenomes "Minimum Information about a Genome Sequence" (MIGS) specification. Nat Biotechnol. 2008;26:541–7.
Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, et al. The Genomic Standards Consortium. PLoS Biol. 2011;9, e1001088.
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–9.
Chen WX, Wang ET, Kuykendall LD. The Proteobacteria. New York: Springer - Verlag; 2005.
Garrity GM, Bell JA, Lilburn TE. Class II. Betaproteobacteria. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. Volume 2. Secondth ed. New York: Springer - Verlag; 2005.
List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol 2006, 56:1–6.
Garrity GM, Bell JA, Lilburn TE. Order 1. Burkholderiales. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. Volume 2. Secondth ed. New York: Springer - Verlag; 2005.
Garrity GM, Bell JA, Lilburn TE. Family I. Burkholderiaceae. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. Volume 2. Secondth ed. New York: Springer - Verlag; 2005.
Palleroni NJ. Genus I. Burkholderia. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. Volume 2. Secondth ed. New York: Springer - Verlag; 2005.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat Genet. 2000;25:25–9.
Acknowledgements
This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JH and RY supplied the strain and background information for this project, TR supplied DNA to JGI and performed all imaging, SDM and WR drafted the paper, JH provided financial support and all other authors were involved in sequencing the genome and editing the final manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
De Meyer, S.E., Tian, R., Seshadri, R. et al. High-quality permanent draft genome sequence of the Lebeckia - nodulating Burkholderia dilworthii strain WSM3556T . Stand in Genomic Sci 10, 64 (2015). https://doi.org/10.1186/s40793-015-0048-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-015-0048-3