
Abstract
Background
The composition of intestinal microflora in animals is affected by cross-species transmission. In a nature reserve, the foraging sites of waterbirds are relatively fixed, but frequently close to residential areas and can also be visited by domestic fowls. It is easy to result in the trans-species-flock dispersal of gut microbes between the wild birds and domestic fowls. The effects of the variable foraging site distances on the gut microbe structures of the waterbirds and the sympatric domestic fowls are currently unclear, and further research is required to evaluate the impacts of geographic location on cross-infection.
Methods
Illumina high-throughput sequencing and bioinformatics analysis software were utilized to compare and analyze the composition of gut microbes from the fecal samples of Hooded Cranes (HC; Grus monacha) and two groups of Domestic Ducks (Anas platyrhynchos domesticus) that foraged at 1 km (ducks in near areas, D-N), and 4 km (ducks in far areas, D-F) away from the habitats of the Hooded Cranes at Shengjin Lake, China.
Results
The results showed that there were significant differences in the alpha-diversity of the gut bacteria in the HC, D-N, and D-F samples under the interspecific distance factor. The dominant bacterial phyla, Cyanobacteria and Proteobacteria, showed correlations with distance for each host. The D-N group had more diverse intestinal flora than the D-F, as they were physically closer to the HC and had more indirect contact and cross-transmission of their gut microbes. More potentially pathogenic bacterial sequences, and Operational Taxonomic Units (OTUs) were found in the D-N than in HC and D-F.
Conclusions
Hooded Cranes and the Domestic Duck populations at variable distances from the cranes showed significant differences in their intestinal bacteria and potentially pathogenic bacteria. The closer the foraging sites were, the easier the intestinal flora spread across species. The results provide a basis for determining the safe distance between wild birds and domestic fowls in a nature reserve.
Similar content being viewed by others


Background
Animals have complex and dynamic microbial communities in their intestines that are mainly composed of bacteria, and the compositions of the various symbiotic and pathogenic bacteria are crucial for the survival and maintenance of the health of their host (Xu et al. 2013; Wei et al. 2013; Delgado et al. 2017). These compositions can affect the host’s functional metabolism, immune homeostasis, and even cause their risk of disease (Elmberg et al. 2017; Flandroy et al. 2018; Huang et al. 2020a; Sarkar et al. 2020). Previous studies have shown that the composition and structure of intestinal flora in animals show a flexible and stable colonization state at birth (Benskin et al. 2009; Mane et al. 2010; Grond et al. 2014; Seedorf et al. 2014). However, when exposed to complex environments and in indirect contact with other species, it may cause cross-transfer of the intestinal flora (Muegge et al. 2011; Delsuc et al. 2014; van Veelen et al 2017; Huang et al. 2020b). Many studies have been conducted on the factors that affect the structure of animal intestinal bacterial communities (e.g. diet, environmental heterogeneity) (Muegge et al. 2011; Delsuc et al. 2014; Perofsky et al. 2019); however, there have been few studies focusing on the effects of cross-species transmission of the intestinal bacterial communities in animals (Fu et al. 2020). In general, physical distance creates barriers for microbial transmission, but interspecific contact creates opportunities for cross-species communication and transmission (Moeller et al. 2017). Therefore, the cross-species transmission may have an even greater impact on the intestinal flora among host animals (Lewis et al. 2016). Some studies have indicated that wild birds are in close contact when their spatial and geographical niches overlap. Then due to cross transmission more similarities are found in intestinal microflora among sympatric birds (Ryu et al. 2014; Perofsky et al. 2019; Yang and Zhou 2021). The cross transmission of intestinal microbes might be through fecal–oral transmission (Moeller et al. 2017), mixed species foraging and by ingestion of microbes into the environment (Holman et al. 2019; Cairns et al. 2020; Liddicoat et al. 2020). Therefore, to elucidate the effect of trans-species-flock dispersal of intestinal microflora, it is important to measure the distance and degree of interspecific contact among sympatric bird species (Bunnik et al. 2014). At present, there is no consensus on whether there is a significant correlation between interspecific contact distance and changes in intestinal bacterial community composition.
Wild animals, especially migratory birds, have a high degree of mobility and dynamic intestinal microbial compositions, but the long-distance migration and high environmental selection pressure may cause the homeostasis to be broken, the immune function to be disturbed, and make birds susceptible to cross-infection by the foreign microbes (Altizer et al. 2011; Deppe et al. 2015; Leung and Koprivnikar 2016; Zou et al. 2017).
After wild waterbirds arrive at the foraging sites, they will inevitably meet the sympatric resident species (e.g. poultry, livestock) foraging at different distances (Xiang et al. 2019; Fu et al. 2020). Wild waterbirds have an indirect contact with poultry when they arrive at the shared foraging sites which might be responsible for fecal–oral transmission of microbes (Hubálek 2004) and cross-transmission of microbes through feathers contamination (Zamani et al. 2017; Huang et al. 2020a). And then the probability of cross-specie transmission of intestinal bacteria between wild waterbirds and poultry may increase (Xiang et al. 2019). In addition, as the wintering period progresses, the niche overlap of wild waterbirds and domestic fowls could be more obvious (Fu et al. 2020). The probability of cross-transmission of potential pathogens will be higher between species present at shorter distances. It is worth noting that poultry may have more potential pathogens than wild waterbirds (Liu et al. 2013) and may transmit the pathogens through indirect contact to wild waterbirds present over short distances, posing serious disease threats to populations of wild waterbirds. These addressed issues could help to shed light on the transmission mechanism of intestinal bacteria between wild waterbirds and poultry, and open interesting direction for future research.
The Hooded Crane (Grus monacha) is a large migratory waterbird categorized as a vulnerable (VU) species on the red list of the International Union for Conservation of Nature (IUCN) (IUCN 2020), and national first-class key protected animal in China. It mainly breeds in eastern Siberia in Russia, and one of the populations winters in the middle and lower Yangtze River floodplain in China (Zheng et al. 2015; Xiang et al. 2019; Zhang et al. 2020). During winter season, the Hooded Cranes feed in variable groups based on their families in mudflats, grasslands, and rice fields, where their main foraging habitats are relatively fixed (Wei et al. 2020).
The Domestic Duck (Anas platyrhynchos domesticus) is one of the most common poultries in China. In the middle and lower Yangtze River floodplain, large populations of them occur free-range in paddy fields and mudflats (Zhang et al. 2007). Shengjin Lake is a typical river-connected lake in the middle of the Yangtze River floodplain in China and a Ramsar site (Zhang et al. 2018), which provides an important wintering ground for the waterbirds on the East Asian-Australasian flyway (Wang et al. 2016, 2017). It is also the largest wintering site for Hooded Cranes on the mainland. Previous studies have verified that Domestic Ducks forage at different distances around the Hooded Crane habitats (Xiang et al. 2019; Fu et al. 2020). The reduction in food resources of wild birds at the lake is gradually causing niche overlap between the Hooded Cranes and Domestic Ducks, which is responsible for the increase in contact between the two sympatric species (Xiang et al. 2019; Fu et al. 2020). It provides an opportunity to compare the intestinal bacteria and potentially pathogenic bacterial communities of the two hosts in different foraging locations. This study could assist in improving strategies to manage the wild waterbirds and poultry in the nature reserve, and in assessing the risk of outbreaks of pathogenic bacteria disease in poultry at important wintering sites.
In this study, we evaluated the gut microbial communities of Hooded Cranes and sympatric Domestic Ducks, and tested that: (1) inter-specific transmission resulting from inter-specific contact distance is one of the main driving factors for the change of intestinal bacterial community composition; (2) there are structural similarities between the intestinal flora of wild birds and domestic poultry present at closer distance; and (3) the common pathogens of cranes and poultry change with the contact distance.
Methods
Ethics statement
This investigation involved non-invasive fecal sample collection and consequently did not involve the hunting of any experimental animals (Knutie and Gotanda 2018). Fecal samples of the Hooded Cranes and Domestic Ducks were collected after their foraging activities, to avoid excessive human disturbance. Administrative permission was obtained from the management office of Anhui Shengjin Lake National Nature Reserve.
Site selection and sample collection
The study site was at Anhui Shengjin Lake National Nature Reserve (116°55′–117°15′ E, 30°15′–30°30′ N), which is a river-collected lake in the middle and lower Yangtze River floodplain in China (Cao and Fox 2009; Zhou et al. 2020). The average annual temperature of the lake is 14–19 °C, and the annual precipitation is 1000–1450 mm. The lake is in a dry season from November to May of each year, exposing a large expanse of grass and mudflat, and provides plentiful food resources for the migratory waterbirds (Zheng et al. 2015).
In recent decades, exploiting aquaculture has resulted in degradation of submerged vegetation in tidal flat and loss of suitable foraging habitats for waterbirds. Consequently, the waterbirds that depend on submerged plants for food, such as the cranes, will gradually shift their foraging sites to paddy habitats (Zhao et al. 2013; Wang et al. 2016; Nilsson et al. 2018; Zhang et al. 2018). In the middle wintering period (December to February), Hooded Cranes shift their foraging habitat from the mud land and grasslands to the rice paddy fields, resulting in high foraging niche overlap with domestic fowls (Dong et al. 2019).
We chose the middle wintering period to collect the samples. The fecal samples were collected from the Hooded Cranes, and groups of Domestic Ducks at two distances from the cranes in the paddy fields at Shengjin Lake on January 10–15, 2019 (Fig. 1). Before collecting fecal samples, we used a monocular and binoculars to focus on the foraging groups of the Hooded Cranes and Domestic Ducks. The population of Hooded Cranes was about 100 individuals, and the duck population was over 250. The birds chosen for sampling foraged for more than one hour without being disturbed and intermingling with other bird species. The samples were collected at more than 5 m intervals to avoid sample repetition (Xiang et al. 2019). The fresh fecal samples were collected using sterile polyethylene (PE) gloves, discarding the contaminated peripheral parts, and each sample was kept in bioclean zipper bags (Liu et al. 2020). All fecal samples were immediately placed on dry ice in a pre-sterilized cooler after collection. Samples were taken to the laboratory as soon as possible and stored at ‒ 80 °C. A total of 60 samples were collected, including 20 fecal samples from the foraging flock of Hooded Cranes (HC), 20 samples from the foraging flock of Domestic Ducks within 1 km (ducks in near areas, D-N), and 20 samples more than 4 km (ducks in far area, D-F) away from the foraging Hooded Cranes.

Fecal sampling sites for the Hooded Cranes and Domestic Ducks at Shengjin Lake
Sample pretreatment
Fecal DNA extraction, bird species determination, polymerase chain reaction (PCR), and amplicon library preparation are described in the Additional file 1: Supplementary information.
Processing of sequence data
The original sequencing data obtained by pretreatment with QIIME (V.1.9) were mainly used to eliminate the inferior sequences (Caporaso et al. 2012) with a sequence length ≤ 250 bp or an average quality score of 30. High quality sequences were grouped into operational taxonomic units (OTUs) with a similarity of 97% using UCLUST (Edgar 2010). The chimerism and singleton OTUs were then removed to ensure that the most abundant sequence in each OTU was selected as a representative sequence and was identified by the ribosome database item classifier (Wang et al. 2007). The representative sequences were then aligned using PyNAST (Caporaso et al. 2012). We selected 12,500 sequence subsets (minimum sequence read depth; repeat 20 times) randomly to equally rarefy samples for comparison of the bacterial community compositions. All identified bacterial species were searched on the Web of Science to identify them as pathogens or potential pathogens. Bacteria that were confirmed to be pathogenic to humans, animals, or plants, were classified as OTUs for special analysis.
Potentially pathogenic species determination
The name of the bacteria was entered as a keyword into the Web of Science to investigate if the species of bacteria had been documented to be pathogenic to humans or animals. A total of 29 potentially pathogenic bacteria and their pathogenicity were identified in this study (Additional file 2: Table S1).
Statistical analysis
We used R software (Version 2.0–2) to analyze most of the statistical data. Linear discriminant analysis (LDA) and linear discriminant analysis effect size (LEfSe) were used to evaluate the bacterial flora responsible for the differences between the species. The non-parametric Kruskal–Wallis rank sum with default settings was used to determine and assess the biomarkers (Segata et al. 2011). Non-metric multidimensional scaling (NMDS) and ANOSIM (analysis of similarity; permutations = 999) were implemented (Anderson and Walsh 2013). To test the alpha diversity and relative abundance of the bacteria and potential pathogens, a one-way analysis of variance (ANOVA: P < 0.05) was used. Using indicator bacteria analysis, the indicator bacteria that caused the differences between the species were analyzed. The Mann–Whitney-Wilcoxon test was used to analyze the relative abundance of potentially pathogenic species with non-normal distributions.
Results
Intestinal bacterial alpha diversity
A total of 1,576,131 high-quality bacterial sequences were obtained for analysis in this study, with 15,299 to 40,556 sequences obtained from each sample (Additional file 2: Table S2). A total of 4752 bacterial OTUs were identified, ranging from 77 to 1123 in all samples (97% similarity), among them, there were 1180 (24.8%) shared OTUs between HC and D-N, and 1120 (23.5%) shared OTUs between HC and D-F. However, the proportion shared by D-N and D-F was the highest at 1790 (37.7%) (Fig. 2).

Venn diagram showing the unique and shared intestinal bacterial operational taxonomic units (OTUs) among HC, D-N and D-F samples. HC Hooded Cranes, D-N Domestic Ducks in near areas, D-F Domestic Ducks in far areas. The explanation to the abbreviations apply also to Figs. 3–6
The Shannon index (ANOVA: P = 0.637), Pielou index (ANOVA: P = 0.137), OTU richness, and phylogenetic diversity were used to comprehensively evaluate the alpha diversity of the intestinal bacteria. The one-way ANOVA showed that the alpha diversity of the intestinal microflora in D-N was higher than that of HC and D-F, as indicated by OTU richness (ANOVA: P = 0.004), and phylogenetic diversity (ANOVA: P = 0.018) (Fig. 3).

Gut bacterial alpha diversity (a OTU richness; b Phylogentic diversity) of HC, D-N and D-F. Different letters in the graphs represent significant differences from one-way ANOVA by Tukey’s HSD comparisons (P < 0.05) and Kruskal-Wallis (P < 0.05); Bar represent means; error bars denote standard deviations. The one with the highest alpha diversity is marked as “a”. The alpha diversity is compared with other values, and the difference is significant, marked as “b”
Intestinal bacterial community structure
The dominant intestinal phyla of HC, D-N, and D-F were Firmicutes (51.56%), Proteobacteria (26.41%), Actinobacteria (8.69%), Cyanobacteria (8.41%), and Bacteroidetes (2.07%) (Additional file 3: Fig. S1a). Within the Firmicutes, the dominant bacterial classes were Bacilli (44.53%) and Clostridia (5.81%), while within the Proteobacteria the dominant classes were Alphaproteobacteria (14.13%) and Gammaproteobacteria (9.56%) (Additional file 3: Fig. S1b). However, the distribution of the dominant phylum and bacterial classes was uneven between the Hooded Cranes and the Domestic Ducks in the different areas (Additional file 3: Fig. S1c, d).
For HC, compared with D-N and D-F, Cyanobacteria (19.41%) was the dominant phylum, and Chloroplast (19.18%) was the dominant bacterial class. For D-N, the dominant bacterial phyla were Proteobacteria (30.74%) and Actinobacteria (10.91%), and the dominant bacteria classes were Alphaproteobacteria (24.59%) and Actinobacteria (9.62%). The Proteobacteria (31.38%) was also dominant for D-F, whose dominant bacterial class was Gammaproteobacteria (21.02%) (Additional file 3: Fig. S1c, d).
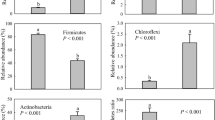
The relative abundance of the Proteobacteria was highest in D-F and lowest in HC. In contrast, the relative abundance of Cyanobacteria was highest in HC and lowest in D-F. The relative abundance of the two dominant bacteria in each host were correlated with distance (Fig. 4). In addition, no significant differences in relative abundance were found for the Firmicutes and Actinobacteria in the Hooded Cranes and Domestic Ducks from the different areas (Fig. 4).

Relative abundances of the dominant bacteria in the Hooded Crane and Domestic Duck samples. Bar represents means; error bars denote standard deviations; The lower case letters above error bar represents the statistically significant differences, as determined with the Kruskal-Wallis test (P < 0.05). The one with the highest alpha diversity is marked as “a”. The alpha diversity is compared with other values, and the difference is significant, marked as “b”. Significant with all three, marked as “c”
LEfSe analysis revealed specific that intestinal bacterial taxa of HC, D-N and D-F were different across the three hosts. The results showed that one phylum (i.e. Cyanobacteria), two classes (i.e. Zetaproteobacteria and Chloroplast), three orders (i.e. Turicibacterales, Streptophyta, and Mariprofundales), and six genera (Planococcaceae, Turicibacteraceae, Methylocystaceae, etc.) were found in HC. Three phyla (Thermi, Gemmatimonadetes, and Chloroflexi), three classes (Deinococci, Thermomicrobia, and Gemm_1), eight orders (Legionellales, MND1, Roseiflexales, etc.), and twenty families (Streptococcaceae, Eubacteriaceae, etc.) were abundant in D-N. However, only two families (Carnobacteriaceae and Williamsiaceae) were significantly different in the D-F group. Thus, HC and D-N both had more diverse flora compared with D-F, and D-N had the most diverse flora (Fig. 5).

LEfSe analysis (ranked by effect size LDA > 2, P < 0.05) of Hooded Crane gut bacteria across Hooded Cranes and Domestic Ducks present in near and far areas. The cladogram represents the taxonomic hierarchical structure of identified biomarkers among HC, D-N and D-F; blue, phylotypes overrepresented for HC; green, phylotypes overrepresented for D-N; and red, phylotypes overrepresented for D-F samples. Yellow color indicates that they are less significant among HC, D-N and D-F
The intestinal bacterial species compositions differed between HC, D-N, and D-F (ANOSIM: R = 0.815, P = 0.001) (Additional file 3: Fig. S2). Furthermore, the intestinal bacterial community of HC was significantly different from that of D-N and D-F (Table 1). A significant difference was also found between D-N and D-F (Table 1). However, HC and D-N communities tended to be more similar in composition (Table 1; Additional file 3: Fig. S2). According to the indicator analysis, a total of 33 OTUs were identified, including 12 for HC, 13 for D-N, and 8 for D-F (Additional file 2: Table S3). We found OTU 2704 (s_acidipiscis) and OTU 34 (o_Streptophyta) had the highest relative abundance (10.69%) in HC. OTU 35 (g_Martelella) with OTU 1075 (s_acidipiscis) were the most abundant in D-N (7.80%), and OTU 6 (s_acidipiscis) and OTU 759 (s_fragi) were the indicators and were most abundant in D-F (21.21%) (Additional file 2: Table S3). The relative abundance of Lactobacillus acidipiscis and Martelella were responsible for the differences in the intestinal community structures between the Hooded Cranes and the Domestic Ducks at different distances.
Intestinal potential pathogenic bacteria
A total of 31,811 potentially pathogenic sequences were identified in this study, accounting for 2.02% of the total bacterial sequences, with each sample ranging from 7 to 3675 (Additional file 2: Table S2). HC and D-N shared more OTUs (approximately 24.61%), and D-F had more unique OTUs (26.92%) (Fig. 6).

Venn diagram indicating the shared and unique gut bacterial pathogenic operational taxonomic units (OTUs) among HC, D-N and D-F samples
There were 29 pathogenic species selected from HC, D-N, and D-F host intestinal microorganisms (22 from HC, 23 from D-N, and 19 from D-F) (Additional file 2: Table S1, S4). The potential pathogen OTU richness of the Domestic Ducks was higher than that of Hooded Cranes (one-way ANOVA; P < 0.001) (Additional file 3: Fig. S3). Enterococcus cecorum and Clostridium botulinum were found in all samples with high relative abundance. The other potential pathogens mostly caused disease in human and other species (Additional file 2: Table S1).
Discussion
In this study, we found that HC shared OTUs with D-N (24.8%) and D-F (23.5%), but unique OTUs were also present in gut of HC (16.3%), D-N (20%), and D-F (12.2%) (Fig. 2). Therefore, it indicates that different species may face niche overlap and cooccur which results in shared OTUs between HC, D-N and D-F. The phylogenetic difference between the species is also responsible for the occurrence of unique OTUs. The proportion of OTUs that HC shared with D-N was slightly higher than that shared with D-F and D-N, and D-F shared the highest proportion of OTUs (Fig. 2). It can be inferred that similar bacterial OTUs are still maintained in the intestines of Domestic Ducks in the different distance groups, and that cross-species transmission under close distance may only affect one (or some) bacterial species. The alpha diversity of the gut bacteria in D-N was significantly higher than in HC and D-F, and distance was the casual factor of the significant changes in the phylogenetic diversity and richness of their host intestinal bacterial communities (Fig. 3). This indicates that the cross-species transmission between the Hooded Cranes and the Domestic Ducks was responsible for their intestinal bacterial structures and the exchange of their intestinal microbes in the nearby area. This strongly implies that distance between populations might be a major factor directly affecting and shaping the population structure of gut bacteria. However, previous studies have shown that endogenous factors may not be the only factors to consider when assessing intestinal bacterial communities (Hills et al. 2019).
The dominant bacteria with the highest relative abundances in this investigation were Proteobacteria, Firmicutes, Cyanobacteria, and Actinobacteria (Kong et al. 2020; Liu et al. 2020; Yang and Zhou 2021). However, shifts in the relative abundance of these phyla were found in HC, D-N and D-F (Additional file 3: Fig. S1). Cyanobacteria were most abundant in HC (Fig. 4). However, due to the influence of distance, Domestic Ducks far from the Hooded Crane feeding areas had the least amount of Cyanobacteria in their intestines. Cyanobacteria mostly enter the intestinal tract when animals feed on plants (Friedland et al. 2005). The diet structure of the Hooded Cranes as wild birds is different from that of the poultry (Madden et al. 2020), which mainly feed on rice and plant tubers. The feeding range of Domestic Ducks is wider, and they feed on insects as an auxiliary food in the diet, besides the weeds in the rice field. Therefore, the abundance of Cyanobacteria in Domestic Ducks feeding in close proximity to the Hooded Cranes may have been enriched via indirect contact with the cranes. Proteobacteria had the lowest relative abundance in the HC group (Fig. 4). The further the Domestic Ducks were from HC, the higher the relative abundance of Proteobacteria they hosted. Proteobacteria is the most abundant phylum that contains pathogenic bacteria, and its presence is regarded as an indicator of the state of the intestinal health of mammals (Shin et al. 2015). Large populations of free-range Domestic Ducks have a close relationship with human livestock, with frequent contact, and mainly sleep in narrow cages or gather in rice fields for a rest. Compared with Hooded Cranes living as a family unit, there were more potential pathogens belonging to phylum Proteobacteria in the intestines of Domestic Ducks. It also supports some studies that indicate poultry are a major carrier of potentially disease-causing bacteria (Revolledo et al. 2006; Dziva and Stevens 2008; Fu et al. 2020). In addition, these two dominant bacterial groups (Cyanobacteria and Proteobacteria) confirmed that spatial distance causes cross-species transmission of intestinal bacteria and affects the intestinal bacterial community structure of the host (Fig. 4).
The study also found that the relative abundance of probiotics (e.g. Lactobacillus acidipiscis and Martelella) in the intestines of Hooded Cranes and Domestic Ducks was higher (Additional file 2: Table S3), while the relative abundance of probiotics in the host intestines of D-N and HC was lower, and the structure of the intestinal flora was not coordinated (which was different from D-F). Interestingly, the relative abundance of D-F intestinal probiotics was generally higher, and the intestines were healthier (Additional file 2: Table S3). This suggests that Hooded Cranes and Domestic Ducks disturb the population structure of each other’s mutual intestinal bacteria.
In this study, 146 potential pathogenic OTUs were found in all samples. The overlap ratio of HC and D-N potential pathogenic OTUs was higher than that of HC and D-F, and D-F had more unique OTUs (Fig. 6). This indicates that mutual transmission and pathogenic infection may occur between Hooded Cranes and Domestic Ducks in nearby areas. It is important to note that the poultry (D-N and D-F) had proportionately more potential pathogenic bacterial OTUs than Hooded Cranes, which confirmed findings from previous research that poultry may have more pathogenic groups than wild birds (Kang et al. 2018; Fu et al. 2020) (Additional file 2: Table S2). Due to the complex social environment of Domestic Ducks, they can move from the residential areas (in close proximity to humans and livestock) to the open rice fields or grass land areas in lakes. In addition, the omnivorous feeding nature of Domestic Ducks increase the probability of more diversity of intestinal pathogenic bacterial OTUs. Thus, domestic fowls have been previously found to be responsible for the transmission of pathogenic bacteria and are regarded as “Trojan horses” (Iacob and Iacob 2019) that can cause diseases. It could explain why HC and D-N share more unique pathogen OTUs (6.15%) than HC and D-F (Fig. 6). Therefore, it is suspected that the Domestic Ducks present in close proximity are responsible for the cross-infection in cranes.
The abundance of the potential pathogens Flavobacterium succinicans, Enterococcus cecorum, and Clostridium botulinum was relatively high in HC samples (Additional file 2: Table S1). According to previous studies, the two main pathogenic bacteria (E. cecorum and C. botulinum) are concentrated in the intestinal tract of the poultry and are present in small amounts in birds (Rossetto et al. 2014; Warnke et al. 2015; Jung et al. 2018). The F. succinicans potential pathogen may cause diseases in humans and other species (Good et al. 2015). These pathogens mainly cause bacterial gill disease, host tissue necrosis, muscle paralysis, and poisoning (Rossetto et al. 2014; Good et al. 2015; Warnke et al. 2015). The results showed that Enterococcus cecorum, Clostridium botulinum, and Capnocytophaga ochracea were abundant in D-N. Hooded Cranes and Domestic Ducks in the nearby areas shared some pathogenic bacterial species, which indicated that there might be a process of mutual infection. The relative abundance of D-F pathogens was similar to that reported in previous studies (Rouger et al. 2017). Domestic Ducks near the feeding grounds of the Hooded Cranes contained more potential bacterial pathogens and transmitted these pathogens to the Hooded Cranes. We speculate that the population of Hooded Cranes may be physically weak after their long-distance migration, and young and weak birds, in particular, may be more susceptible to infection (Leung and Koprivnikar 2016). In addition, the direct or indirect transmission of these pathogens to livestock, humans and animals may occur and could also cause infections in humans (Reed et al. 2003; Hansen et al. 2015) (Additional file 2: Table S1).
However, this study also has some limitations. Only fecal sample analysis was conducted and no soil samples were analyzed. The time of observation was short which might be not enough for bird digesting and excreting feces. In addition, the impact of environmental microbes was not studied and discussed, and the sources of OTUs were not distinguished clearly. In future, these limitations can be overcome by applying new microbiological analysis methods and technologies.
Conclusions
Our results identified that the foraging site distance between Hooded Cranes and Domestic Ducks plays a crucial role in cross species transmission of intestinal bacteria and potential pathogenic infection. The Domestic Ducks present in close contact with Hooded Cranes are more prone to spread cross infection in Hooded Cranes and other organisms and it may have more potential pathogens in proportion to Hooded Cranes, which could threaten the survival of Hooded Cranes, and cause zoonotic diseases. This study clears that the shifts in intestinal bacterial communities are due to cross species transmission between sympatric birds present at close distance near 1 km. And it provides a theoretical basis for determining the safe stocking distance of the protected area and establishing a free-range area.
Availability of data and materials
The raw data were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (BioProject ID: PRJNA678619).
References
Altizer S, Bartel R, Han BA. Animal migration and infectious disease risk. Science. 2011;331:296–302.
Anderson MJ, Walsh DCI. PERMANOVA, ANOSIM, and the mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecol Monogr. 2013;83:557–74.
Benskin CMH, Wilson K, Jones K, Hartley IR. Bacterial pathogens in wild birds: a review of the frequency and effects of infection. Biol Rev Camb Philos Soc. 2009;84:349–73.
Bunnik BAD, Ssematimba A, Hagenaars TJ, Nodelijk G, Haverkate MR, Bonten MJM, et al. Small distances can keep bacteria at bay for days. Proc Natl Acad Sci U S A. 2014;119:3556–60.
Cairns J, Moerman F, Fronhofer EA, Altermatt F, Hiltunen T. Evolution in interacting species alters predator life-history traits, behaviour and morphology in experimental microbial communities. Proc Biol Sci. 2020;287:20200652.
Cao L, Fox AD. Birds and people both depend on China’s wetlands. Nature. 2009;460:173.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4.
Delgado ML, Singh P, Funk JA, Moore JA, Cannell EM, Kanesfsky J, et al. Intestinal microbial community dynamics of white-tailed deer (Odocoileus virginianus) in an agroecosystem. Microb Ecol. 2017;74:496–506.
Delsuc F, Metcalf JL, Parfrey LW, Song SJ, González A, Knight R. Convergence of gut microbiomes in myrmecophagous mammals. Mol Ecol. 2014;23:1301–17.
Deppe JL, Ward MP, Bolus RT, Diehl RH, Celis-Murillo A, Zenzal TJ Jr, et al. Fat, weather, and date affect migratory songbirds’ departure decisions, routes, and time it takes to cross the Gulf of Mexico. Proc Natl Acad Sci USA. 2015;112:e6331–8.
Dong YQ, Xiang XJ, Zhao GH, Song YW, Zhou LZ. Variations in gut bacterial communities of hooded crane (Grus monacha) over spatial-temporal scales. PeerJ. 2019;7:e7045.
Dziva F, Stevens MP. Colibacillosis in poultry: unravelling the molecular basis of virulence of avian pathogenic Escherichia coli in their natural hosts. Avian Pathol. 2008;37:355–66.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Elmberg J, Berg C, Lerner H, Waldenström J, Hessel R. Potential disease transmission from wild geese and swans to livestock, poultry and humans: a review of the scientific literature from a One Health perspective. Infect Ecol Epidemiol. 2017;7:1300450.
Flandroy L, Poutahidis T, Berg G, Clarke G, Dao MC, Decaestecker E, et al. The impact of human activities and lifestyles on the interlinked microbiota and health of humans and of ecosystems. Sci Total Environ. 2018;627:1018–38.
Friedland KD, Ahrenholz DW, Haas LW. Viable gut passage of cyanobacteria through the filter-feeding fish Atlantic menhaden, Brevoortia tyrannus. J Plankton Res. 2005;27:715–8.
Fu R, Xiang XJ, Dong YQ, Cheng L, Zhou LL. Comparing the intestinal bacterial communities of sympatric wintering Hooded Crane (Grus monacha) and Domestic Goose (Anser anser domesticus). Avian Res. 2020;11:13.
Good C, Davidson J, Wiens GD, Welch TJ, Summerfelt S. Flavobacterium branchiophilum and F. succinicans associated with bacterial gill disease in rainbow trout Oncorhynchus mykiss (Walbaum) in water recirculation aquaculture systems. J Fish Dis. 2015;38:409–13.
Grond K, Ryu H, Baker AJ, Santo Domingo JW, Buehler DM. Gastro-intestinal microbiota of two migratory shorebird species during spring migration staging in Delaware Bay, USA. J Ornithol. 2014;155:969–77.
Hansen CM, Meixell BW, Van Hemert C, Hare RF, Hueffer K. Microbial infections are associated with embryo mortality in Arctic-nesting geese. Appl Environ Microb. 2015;81:5583–92.
Hills RD, Pontefract BA, Mishcon HR, Black CA, Sutton SC, Theberge CR. Gut microbiome: profound implications for diet and disease. Nutrients. 2019;11:1613.
Holman DB, Bearson BL, Allen HK, Shippy DC, Loving CL, Kerr BJ, et al. Chlortetracycline enhances tonsil colonization and fecal shedding of multidrug-resistant Salmonella enterica serovar Typhimurium DT104 without major alterations to the porcine tonsillar and intestinal microbiota. Appl Environ Microbiol. 2019;85:e02354-e2418.
Huang R, Ju Z, Zhou PK. A gut dysbiotic microbiota-based hypothesis of human-to-human transmission of non-communicable diseases. Sci Total Environ. 2020a;745:141030.
Huang SM, Wu ZH, Li TT, Liu C, Han DD, Tao SY, et al. Perturbation of the lipid metabolism and intestinal inflammation in growing pigs with low birth weight is associated with the alterations of gut microbiota. Sci Total Environ. 2020b;719:137382.
Hubálek Z. An annotated checklist of pathogenic microorganisms associated with migratory birds. J Wildlife Dis. 2004;40:639–59.
Iacob S, Iacob DG. Infectious threats, the intestinal barrier, and its Trojan Horse: dysbiosis. Front Microbiol. 2019;10:1676.
IUCN. The IUCN red list of threatened species. Version 2019–3. 2020. https://www.iucnr.edlist.org.
Kang YF, Shen XJ, Yuan RY, Xiang B, Fang ZX, Murphy RW, et al. Pathogenicity and transmissibility of three avian influenza A (H5N6) viruses isolated from wild birds. J Infect. 2018;76:286–94.
Knutie SA, Gotanda KM. A non-invasive method to collect fecal samples from wild birds for microbiome studies. Microb Ecol. 2018;76:851–5.
Kong A, Zhang C, Cao Y, Cao Q, Liu F, Yang Y, et al. The fungicide thiram perturbs gut microbiota community and causes lipid metabolism disorder in chickens. Ecotox Environ Safe. 2020;206:111400.
Leung TLF, Koprivnikar J. Nematode parasite diversity in birds: the role of host ecology, life history and migration. J Anim Ecol. 2016;85:1471–80.
Lewis WB, Moore FR, Wang S. Characterization of the gut microbiota of migratory passerines during stopover along the northern coast of the Gulf of Mexico. J Avian Biol. 2016;47:659–68.
Liddicoat C, Sydnor H, Cando-Dumancela C, Dresken R, Liu J, Gellie NJC, et al. Naturally-diverse airborne environmental microbial exposures modulate the gut microbiome and may provide anxiolytic benefits in mice. Sci Total Environ. 2020;701:134684.
Liu D, Shi WF, Shi Y, Wang DY, Xiao HX, Li W, et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: phylogenetic, structural, and coalescent analyses. Lancet. 2013;381:1926–32.
Liu G, Gong ZZ, Li QY. Variations in gut bacterial communities between lesser white-fronted geese wintering at Caizi and Shengjin lakes in China. MicrobiologyOpen. 2020;9:e1037.
Madden JR, Santilli F, Whiteside MA. The welfare of game birds destined for release into the wild: a balance between early life care and preparation for future natural hazards. Anim Welf. 2020;29:1–18.
Mane SP, Dominguez-Bello MG, Blaser MJ, Sobral BW, Hontecillas R, Skoneczka J, et al. Host-interactive genes in Amerindian Helicobacter pylori diverge from their old world homologs and mediate inflammatory responses. J Bacteriol. 2010;192:3078–92.
Moeller AH, Suzuki TA, Lin D, Lacey EA, Wasser SK, Nachman MW. Dispersal limitation promotes the diversification of the mammalian gut microbiota. Proc Natl Acad Sci U S A. 2017;114:13768–73.
Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–4.
Nilsson L, Aronsson M, Persson J, Månsson J. Drifting space use of common cranes—is there a mismatch between daytime behaviour and management? Ecol Indic. 2018;85:556–62.
Perofsky AC, Lewis RJ, Meyers LA. Terrestriality and bacterial transfer: a comparative study of gut microbiomes in sympatric Malagasy mammals. ISME J. 2019;13:50–63.
Reed KD, Meece JK, Henkel JS, Shukla SK. Birds, migration and emerging zoonoses: west nile virus, lyme disease, influenza A and enteropathogens. Clin Med Res. 2003;1:5–12.
Revolledo L, Ferreira AJP, Mead GC. Prospects in Salmonella control: competitive exclusion, probiotics, and enhancement of avian intestinal immunity. J Appl Poultry Res. 2006;15:341–51.
Rossetto O, Pirazzini M, Montecucco C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nat Rev Microbiol. 2014;12:535–49.
Rouger A, Tresse O, Zagorec M. Bacterial contaminants of poultry meat: sources, species, and dynamics. Microorganisms. 2017;5:50.
Ryu H, Grond K, Verheijen B, Elk M, Buehler DM, Santo Domingo JW. Intestinal microbiota and species diversity of Campylobacter and Helicobacter spp. in migrating shorebirds in Delaware Bay. Appl Environ Microbiol. 2014;80:1838–47.
Sarkar A, Harty S, Johnson KVA, Moeller AH, Archie EA, Schell LD, et al. Microbial transmission in animal social networks and the social microbiome. Nat Ecol Evol. 2020;4:1020–35.
Seedorf H, Griffin NW, Ridaura VK, Reyes A, Cheng J, Rey FE, et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell. 2014;159:253–66.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33:496–503.
van Veelen HPJ, Salles JF, Tieleman BI. Multi-level comparisons of cloacal, skin, feather and nest-associated microbiota suggest considerable influence of horizontal acquisition on the microbiota assembly of sympatric woodlarks and skylarks. Microbiome. 2017;5:156.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
Wang C, Wang L, Deng DG, Zhou ZZ. Temporal and spatial variations in rotifer correlations with environmental factors in Shengjin Lake. China Environ Sci Pollut Res Int. 2016;23:8076–84.
Wang WJ, Fraser JD, Chen JK. Wintering waterbirds in the middle and lower Yangtze River floodplain: changes in abundance and distribution. Bird Conserv Int. 2017;27:167–86.
Warnke P, Köller T, Stoll P, Podbielski A. Nosocomial infection due to Enterococcus cecorum identified by MALDI-TOF MS and Vitek 2 from a blood culture of a septic patient. Eur J Microbiol Immunol. 2015;5:177–9.
Wei S, Morrison M, Yu Z. Bacterial census of poultry intestinal microbiome. Poult Sci. 2013;92:671–83.
Wei ZH, Zheng M, Zhou LZ, Xu WB. Flexible foraging response of wintering Hooded Cranes (Grus monacha) to food availability in the lakes of the Yangtze River floodplain. China Animals. 2020;10:568.
Xiang XJ, Zhang FL, Fu R, Yan SF, Zhou LZ. Significant differences in bacterial and potentially pathogenic communities between sympatric hooded crane and greater white-fronted goose. Front Microbiol. 2019;10:163.
Xu X, Xu P, Ma C, Tang J, Zhang X. Gut microbiota, host health, and polysaccharides. Biotechnol Adv. 2013;31:318–37.
Yang ZQ, Zhou LZ. Is intestinal bacterial diversity enhanced by trans-species spread in the mixed-species flock of hooded crane (Grus monacha) and bean goose (Anser fabalis) wintering in the lower and middle Yangtze River floodplain? Animals. 2021;11:233.
Zamani M, Vahedi A, Maghdouri Z, Shokri-Shirvani J. Role of food in environmental transmission of Helicobacter pylori. Caspian J Intern Med. 2017;8:146–52.
Zhang TJ, Li HF, Chen KW, Chang H, Tang QQ, Zhang JX. Genetic diversity and systematic evolution of Chinese domestic ducks along the Yangtze-Huai River. Biochem Genet. 2007;45:823–37.
Zhang Y, Fox AD, Cao L, Jia Q, Lu CH, Prins HHT, et al. Effects of ecological and anthropogenic factors on waterbird abundance at a Ramsar Site in the Yangtze River Floodplain. Ambio. 2018;48:293–303.
Zhang FL, Xiang XJ, Dong YQ, Yan SF, Song YW, Zhou LZ. Significant differences in the gut bacterial communities of Hooded crane (Grus monacha) in different seasons at a stopover site on the flyway. Animals. 2020;10:701.
Zhao FT, Zhou LZ, Xu WB. Habitat utilization and resource partitioning of wintering Hooded Cranes and three goose species at Shengjin Lake. Chinese Birds. 2013;4:281–90.
Zheng M, Zhou LZ, Zhao NN, Xu WB. Effects of variation in food resources on foraging habitat use by wintering Hooded Cranes (Grus monacha). Avian Res. 2015;6:11.
Zhou J, Zhou LZ, Xu WB. Diversity of wintering waterbirds enhanced by restoring aquatic vegetation at Shengjin Lake. China Sci Total Environ. 2020;737:140190.
Zou YA, Pan BH, Zhang H, Zhang PY, Yao Y, Liu XK, et al. Impacts of microhabitat changes on wintering waterbird populations. Sci Rep. 2017;7:13934.
Acknowledgments
We would like to thank our research team: Zhuqing Yang, Nazhong Zhang, Jingjing Gu and Dazhao Liu for their assistance in sample collection, analysis software and mapping.
Funding
The work was supported by the National Natural Science Foundation of China (Grant No. 31772485).
Author information
Authors and Affiliations
Contributions
WW and LZ conceived, designed and validated the experiments. WW, RF and LC completed the sample collection. WW and LZ performed the experiments and drafted the earlier version of this article. WW, RF and LC contributed in data analysis. LZ contributed reagents, offered funding support and reviewed the paper. NM and SY handled the DNA extraction and species identification and polished the language of the article. YS designed the feasibility test scheme and obtained reserve sample approval. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All operations were conducted in compliance with the provisions of the current Chinese laws. All animal fecal samples were collected by non-invasive methods. Our study was approved by the Shengjin Lake Nature Reserve in Anhui Province.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
Additional file 1
: Supplementary information.
Additional file 2: Table S1.
The difference in relative abundance of potentially pathogenic species present in HC, D-N and D-F samples. Table S2. The gut bacterial and pathogenic sequences across the samples. Table S3. Identified gut bacterial indicator species among HC, D-N and D-F. Table S4. The potential pathogenic species identified in HC, D-N and D-F samples.
Additional file 3: Fig. S1.
The relative abundance of gut bacteria of HC, D-N and D-F at phylum and class levels. Fig. S2. Non-metric multidimensional scaling (NMDS) indicating difference in gut bacterial community composition among HC, D-N and D-F. Fig. S3. The intestinal pathogenic bacterial characteristics in guts of HC, D-N and D-F samples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, W., Zhou, L., Fu, R. et al. Effects of foraging site distances on the intestinal bacterial community compositions of the sympatric wintering Hooded Crane (Grus monacha) and Domestic Duck (Anas platyrhynchos domesticus). Avian Res 12, 20 (2021). https://doi.org/10.1186/s40657-021-00255-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40657-021-00255-8