
Abstract
Background
Previous phylogenetic studies that include the four recognized species of Gallus have resulted in a number of distinct topologies, with little agreement. Several factors could lead to the failure to converge on a consistent topology, including introgression, incomplete lineage sorting, different data types, or insufficient data.
Methods
We generated three novel whole genome assemblies for Gallus species, which we combined with data from the published genomes of Gallus gallus and Bambusicola thoracicus (a member of the sister genus to Gallus). To determine why previous studies have failed to converge on a single topology, we extracted large numbers of orthologous exons, introns, ultra-conserved elements, and conserved non-exonic elements from the genome assemblies. This provided more than 32 million base pairs of data that we used for concatenated maximum likelihood and multispecies coalescent analyses of Gallus.
Results
All of our analyses, regardless of data type, yielded a single, well-supported topology. We found some evidence for ancient introgression involving specific Gallus lineages as well as modest data type effects that had an impact on support and branch length estimates in specific analyses. However, the estimated gene tree spectra for all data types had a relatively good fit to their expectation given the multispecies coalescent.
Conclusions
Overall, our data suggest that conflicts among previous studies probably reflect the use of smaller datasets (both in terms of number of sites and of loci) in those analyses. Our results demonstrate the importance of sampling large numbers of loci, each of which has a sufficient number of sites to provide robust estimates of gene trees. Low-coverage whole genome sequencing, as we did here, represents a cost-effective means to generate the very large data sets that include multiple data types that enabled us to obtain a robust estimate of Gallus phylogeny.
Similar content being viewed by others

Background
The Domestic Chicken (Gallus gallus domesticus) is among the best-studied bird species, with many studies examining the genetics, physiology, and development of various chicken breeds. Due to its economic and agricultural importance, the wild progenitor of the Domestic Chicken, Gallus gallus (Red Junglefowl), was the first avian species whose genome was completely sequenced (ICGSC 2004). Since then, genomes of many of the hundreds of different breeds of Gallus gallus have been sequenced (e.g., Imsland et al. 2012; Yi et al. 2014; Wang et al. 2015; Lawal et al. 2018; Li et al. 2019; Yang et al. 2019), resulting in a large number of available chicken genomes.
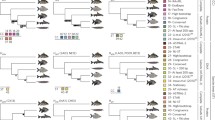
In addition to G. gallus, the genus Gallus has three additional species (Gill and Donsker 2019): Gallus lafayettii (Sri Lanka Junglefowl), G. sonneratii (Grey Junglefowl), and G. varius (Green Junglefowl). These species are relatively closely related, with the earliest divergence approximately 4‒5 million years ago (Mya; Hosner et al. 2017; Wang et al. 2017). Males of these species all have combs and wattles, traits that are involved in sexual selection (Ligon et al. 1998), although there are some differences among species in the plumage as well as the color and structure of the combs and wattles (Fig. 1). All molecular studies (e.g., Kimball et al. 1999; Armstrong et al. 2001; all studies in Fig. 2) have consistently shown that Bambusicola, the Bamboo Partridges, is the sister genus to Gallus, but in contrast to the high sexual dimorphism in the four Gallus species, Bambusicola is monomorphic, being relatively drab in color in both sexes (Fig. 1). The estimated divergence time between the two genera is approximately 15‒16 Mya (Hosner et al. 2017; Wang et al. 2017).

Appearance of each focal species. aBambusicola thoracicus. bGallus gallus. cG. varius. dG. sonneratii. eG. lafayettii. For Gallus, only males are shown; Bambusicola is sexually monomorphic. Sources of each photograph are available in the Additional file 1

Published molecular studies that included all four species of Gallus. Analyses based just on nuclear data are in bold, and those just on mitochondrial data are in italics. Specific figures for each study are noted (as some studies included multiple analyses with different topologies)
Despite the intensive focus on the genomes of Domestic Chickens, genomes for close relatives on G. gallus have not yet been sequenced. Recently, Tiley et al. (2018) published a genome description from Bambusicola thoracicus, the Chinese Bamboo Partridge, along with genomic comparisons to G. gallus. However, genomes of the additional species of Gallus have the potential to provide a more in-depth understanding of the evolution of the genus as a whole. Indeed, the phylogenetic relationships among the four Gallus species remain unresolved. Earlier studies, using mitochondrial and/or small numbers of nuclear markers, resulted in six alternative topologies (Fig. 2). More recently, studies using over 4800 ultra-conserved element (UCE) markers found strong support for a seventh topology (Hosner et al. 2016, 2017). However, analyses of different types of genomic data can yield alternative topologies (e.g., Jarvis et al. 2014; Chen et al. 2017; Reddy et al. 2017). Therefore, it is not clear whether a broader sampling of different data types from throughout the genome will result in the same species tree estimate as Hosner et al. (2016).
Data type is not the only explanation for the observed differences among studies; the different topologies may result from introgression. Crosses among all Gallus species are known to produce viable offspring (reviewed by Johnsgard 1999; McCarthy 2006). There is molecular evidence for recent introgression among multiple Gallus species (Nishibori et al. 2005), as well as a nuclear gene from G. sonneratii (yellow skin) having introgressed into Domestic Chickens (Eriksson et al. 2008). Another alternative may be the random sampling of gene trees. Gene trees may exhibit multiple evolutionary histories, even without introgression, due to incomplete lineage sorting (Maddison 1997). Thus, any relatively small sample of gene trees will represent a somewhat unpredictable set of topologies. Finally, insufficient data from individual loci can also be problematic, since it would not include enough informative sites to accurately resolve relationships (e.g., Saitou and Nei 1986; Chojnowski et al. 2008; Kimball and Braun 2014).
Here we present whole genome sequences for the three remaining species of Gallus and use those sequences to obtain a better understanding of Gallus phylogeny and molecular evolution. We accomplished this by combining the new genomic data with the published genome assemblies available for G. gallus and B. thoracicus (as an outgroup), allowing us to generate a more rigorous estimate of the relationships among Gallus species. The use of whole genomes allowed us to evaluate alternative explanations for conflicts among previous studies. To examine data type effects we estimated independent phylogenies using protein coding exons, introns, UCEs, and conserved non-exonic elements (CNEEs). Finally, we also tested whether our gene tree results were consistent with expectations given the multispecies coalescent or whether some degree of historical introgression was necessary to explain our data.
Methods
Genome sequencing, assembly, and annotation
The genomes of Gallus lafayettii, Gallus sonneratii, and Gallus varius were sequenced using methods described in Tiley et al. (2018) for Bambusicola thoracicus. The individuals were all captives, sampled from breeders in the United States. Two of the samples were from males (G. lafayettii and G. varius), while the other was from a female (G. sonneratii). Briefly, DNA was isolated using the Gentra PureGene DNA Isolation Kit (Qiagen) following manufacturer’s instructions. DNA was then sent to the University of Florida Interdisciplinary Center for Biotechnology Research for library preparation, fragment selection, and sequencing. For each of the three genomes, a single insert library was prepared using Illumina’s NEBNext Ultra DNA Library Prep Kit following the manufacturer’s instructions. A single insert of approximately 500 base pairs (bp) was selected for the NextSeq 500 sample preparation protocol. All three libraries, as well as libraries for Bambusicola thoracicus, were combined and sequenced across four lanes on a single flow cell using paired-end 150 bp reads on an Illumina NextSeq 500.
The three Gallus genomes were assembled using MaSuRCA v2.3.2 (Zimin et al. 2013), which automatically trims Illumina reads with QuorUM (Marcais et al. 2015) and determines the optimal kmer size with Jellyfish (Marcais and Kingsford 2011). Genomes were masked and repeat content estimated with RepeatMasker v4.0.5 based on the “Aves” repeat library (Smit et al. 2015). We annotated protein-coding gene models from our de novo assemblies using MAKER v2.31.8 (Cantarel et al. 2008). We used AUGUSTUS v3.2.1 (Stanke et al. 2006) for ab intio gene prediction based on a library of Gallus gallus RefSeq sequences. For gene prediction, we also used protein homology evidence. To do this we downloaded protein sequences for Gallus gallus (ftp.ensembl.org/pub/release-84/fasta/gallus_gallus/pep/; accessed 21 December 2015), Melagris gallopavo (ftp.ensembl.org/pub/release-83/fasta/meleagris_gallopavo/pep/; accessed 21 December 2015), and Taeniopygia guttata (ftp.ensembl.org/pub/release-83/fasta/taeniopygia_guttata/pep/; accessed 21 December 2015) from Ensembl (Cunningham et al. 2015).
For our evolutionary analyses, in addition to the three genomes, we used data from the Bambuscola thoracicus assembly and annotations (Tiley et al. 2018) and the Gallus gallus version 4 reference genome assembly. For the G. gallus genome, we downloaded annotations (ftp.ensembl.org/pub/release-82/fasta/gallus_gallus/; accessed October 2015) and gene features (ftp.ensembl.org/pub/release-82/gff3/gallus_gallus/; accessed October 2015) from Ensembl (Cunningham et al. 2015).
Datasets for phylogenetic estimation
To get a genomic perspective on the phylogenetic relationships of Gallus species, we assembled various datasets from sets of orthologous genes (including exons and introns), UCEs (Faircloth et al. 2012), and CNEEs (Edwards et al. 2017). First, we used the translated amino acid sequences from annotated gene models to circumscribe orthologous groups using orthoMCL (Li et al. 2003). For phylogenetic analyses we used only the orthologous groups that had exactly one sequence represented for each of the five species in our analysis. We then extracted individual exon and intron sequences from the genome assemblies using the gene feature format files generated by MAKER, or publicly available in the case of G. gallus. To ensure we aligned orthologous regions in all cases, we created BLAST databases for exon and intron sequences and performed an all-by-all BLASTN (Altschul et al. 1990; Camacho et al. 2008) search, saving only one alignment per hit with an E-value ≤ 10‒5. Then we used single linkage clustering to recover one-to-one groups of homologous exons or introns. This strategy avoided potential complications due to absences that reflect partial gene models or even exon or intron gains and losses. We aligned each set of orthologous exons or introns with MUSCLE (Edgar 2004). Because individual exons are not guaranteed to begin on a first position of a codon, sequences were not aligned by codon. Only alignments with all five species present that were at least 100 bp in length were retained for phylogenetic analyses. We did not exclude reads from mitochondrial genome. Although this may introduce some mitochondrial data into the orthogroups we analyzed in this study, they will be a very small number compared to the total set.
We used methods described in Reddy et al. (2017) to extract UCE and CNEE sequences. Briefly, we built profile hidden Markov models (HMM) queries from the UCE and CNEE alignments generated by Jarvis et al. (2014) and Edwards et al. (2017), respectively, and then we used nhmmer (Wheeler and Eddy 2013) to search each genome. We downloaded the Jarvis et al. (2014) UCE alignments from http://gigadb.org/dataset/101041 and the Edwards et al. (2017) CNEE alignments from https://datadryad.org/resource/doi:10.5061/dryad.25f7g. The homologous regions in each genome were extracted and aligned using MAFFT v7.130b (Katoh and Standley 2013). The pipeline used for this analysis is available from GitHub (see “Exttract_seq” in the data availability section below). Some CNEEs overlap with UCEs, so we eliminated any CNEE alignments where the Gallus gallus CNEE had a BLASTN (Camacho et al. 2008) hit with an E-value ≤ 10‒4 to a UCE sequence. The procedure resulted in a total of 3660 UCE alignments and 2160 CNEE alignments.
Phylogenetic inference
For each of the data types (exons, introns, UCEs, and CNEEs), we first concatenated all loci and estimated maximum likelihood (ML) phylogenetic trees for each of the four supermatrices with RAxML v8.2.10 (Stamatakis 2014) using the GTRCAT model. For each dataset, we ran 100 non-parametric bootstrap replicates using the same methods (Felsenstein 1985). We also concatenated the supermatrices from all of the datatypes into a single comprehensive supermatrix and performed ML and ML bootstrapping analyses on it as described above.
To conduct species tree inference based on a coalescent model, we used ASTRAL-III v.5.6.3 (Zhang et al. 2018). This requires gene trees from individual partitions (e.g., a single exon, intron, UCE, or CNEE). Since including poorly supported trees can mislead summary tree coalescence methods (Patel et al. 2013; Mirarab et al. 2014; Xi et al. 2015; Meiklejohn et al. 2016), we only considered individual intron, exon, UCE or CNEE alignments with at least 10 parsimony informative sites. Additionally, some genes had multiple introns and exons which may produce non-independent gene trees; in these cases, we selected the intron or exon with the most parsimony informative sites as a single representative. There were no CNEE cluster alignments with 10 or more parsimony informative sites, so we did not use CNEEs for coalescent species tree analysis with gene tree topologies. We estimated gene trees for these individual loci using RAxML v8.2.10 (Stamatakis 2014) with the GTRCAT model. We ran an ASTRAL analysis for sets of the intron trees, exon trees, and UCE trees. Quartet support for ASTRAL species trees was evaluated using local posterior probabilities (Sayyari and Mirarab 2016). We also ran a combined ASTRAL analysis on a set of trees that included all UCE trees, and for each gene, either the intron or exon that had the most parsimony informative sites.
The length of genomic regions that share the same evolutionary history (c-genes, as defined by Doyle 1997) is unclear. Intralocus recombination may be sufficiently common to render c-genes extremely short (Springer and Gatesy 2016), but other studies have implicitly assumed that entire transcription units are equivalent to c-genes (Song et al. 2012; Jarvis et al. 2014). C-gene lengths are likely to be sensitive to a number of factors, including the recombination rate and the species tree topology and branch lengths. Therefore, we took a conservative approach and sampled the most informative exon or intron from each gene to estimate gene trees for the ASTRAL analysis. Although there may be some intralocus recombination within the genus Gallus, using only a single exon or intron per gene should limit the number of cases where this is a problem. This conservative approach comes with a cost because it either excludes additional gene trees that could have been included (if c-genes are short) or it excludes sites that could increase the accuracy of gene tree estimation (if c-genes are long). However, it represents a straightforward way to avoid strong assumptions regarding the length of c-genes.
To complement the ASTRAL analysis of gene trees, we also used ASTRAL to analyze perfect transversions. We defined perfect transversions as parsimony informative sites with exactly two states and no gaps in the alignment column that differ by a transversion. We treated these sites as defining a single bipartition in a gene tree, so we generated partially resolved gene trees based on each perfect transversion site (rather than on a gene region, as above) and used those trees as input for ASTRAL. The ASTRAL analysis of perfect transversions has two desirable properties: (1) it permits the inclusion of all sequences in the analysis (since it focuses on sites, not on regions with more than 10 parsimony informative sites); and (2) it should be a consistent estimator for the species tree even if c-genes are very short (i.e., even if they are shorter than 100 bp). As described above, we used local posterior probabilities to examine support in this analysis.
Deviation from base composition stationarity (i.e., cases where taxa differ in base composition) has been suggested to mislead estimates of phylogeny (e.g. Conant and Lewis 2001; Betancur et al. 2013), and specifically estimates from avian exons (Jarvis et al. 2014). Therefore, we examined variation in base composition for all concatenated datasets using PAUP* 4.0a166 (Swofford 2019). Briefly, we deleted the Bambusicola outgroup, excluded parsimony uninformative sites, and used the “statefreq” command to obtain base composition data. Then we calculated the composition along three axes: (1) the strong–weak axis (the proportion of G and C); (2) the purine–pyrimidine axis (the proportion of A and G); and (3) the amino–keto axis (the proportion of A and C) for each species. We calculated the range (max‒min) for each pair of nucleotides (GC, AG, or AC) among the four species to determine whether species exhibited much difference in base composition.
Gene tree comparisons
We examined the fit of gene trees to the expectation given the multispecies coalescent in two ways. First, we calculated the expected spectrum of gene trees as described by Rosenberg (2002) using coalescent branch length estimates from ASTRAL. Code for this calculation is available from GitHub (see “asymtree” in the data availability section below). Second, we performed all possible single taxon deletions and examined the degree of asymmetry in the minority topologies (which are expected to be equiprobable if discordance among gene trees only reflects the multispecies coalescent; Pamilo and Nei 1988). Differences from the expectation given the multispecies coalescent were examined using the χ2 test. We also tested whether there was a difference among the spectra of gene tree topologies for trees generated using introns, exons, and UCEs. We did this because some previous analyses have shown that different data types can yield distinct topologies (Jarvis et al. 2014; Chen et al. 2017; Reddy et al. 2017). For the latter test we examined whether exons, introns, and UCEs were significantly different from expectation given homogeneity rather than testing for deviation from the multispecies coalescent (i.e., instead of a simple test using 15 counts of all rooted four taxon gene trees we used a 15 × 3 count matrix of all rooted four taxon trees generated using the introns, exons, and UCEs).
The simplest interpretation of asymmetries in the gene tree spectrum for quartets (which we observed for two pairs of taxa: G. gallus‒G. varius and G. gallus‒G. sonneratii; see below in “Results”) is introgression. This introgression could be relatively ancient, or it could reflect very recent introgression due to humans moving domesticated chickens into sympatry with other Gallus species or to hybridization among captive populations of various species. The individuals sequenced for this study were chosen because they showed substantial differences among all species at 15 nuclear loci and their mitogenomes (Kimball and Braun 2014), so they cannot be simple (F1) hybrids or backcrosses. However, it remains possible that they are relatively recent (e.g., fourth generation) backcrosses or that recent anthropogenic gene flow has been relatively high. We examined asymmetries in quartets for evidence of recent introgression; this would predict that gene trees with clades showing up in excess of the expectation from incomplete lineage sorting alone would also have an excess of very short (essentially zero length) branches for the relevant taxa. That is because alleles from one species that have very recently introgressed into the other will lead to identical alleles in the two species. Although population variation and/or mutation may result in one or two differences between species for these alleles, they will exhibit extremely short branches. To test the very recent introgression hypothesis, we conducted two analyses in which intronic gene trees were divided into two groups (based on observed asymmetries; see Results): (1) trees with a G. gallus‒G. varius clade and those without; and (2) trees with a G. gallus‒G. sonneratii clade and those without. We focused on intronic gene trees because introns generally accumulate substitutions at a higher rate than the other marker types. The distribution of terminal branch lengths for G. varius and G. sonneratii was then compared for the two groups of trees to determine whether there was an excess of very short terminal branches in the gene trees with the relevant clade.
We also evaluated the presence of historical introgression by analyzing our ML gene tress with PhyloNetworks (Solís-Lemus et al. 2017). PhyloNetworks uses distributions of quartets to evaluate the pseudolikelihood of a phylogenetic network under a coalescent model. Similar to our analyses of gene tree spectra, an excess of quartets that conflict with the species tree is interpreted as introgression. We ran PhyloNetworks for 20 iterations while allowing for zero, one, or two reticulations, and plotted profiles of pseudolikelihoods for each number of reticulations to determine the appropriate network topology with slope heuristics (Solís-Lemus and Ané 2016).
Results
Assembly and annotation of three novel Gallus genomes
Assembly quality and depth of coverage of the three new Gallus genomes (Additional file 2: Table S1) were similar to those we obtained for Bambusicola thoracicus (Tiley et al. 2018). The de novo assemblies recovered most of the genome based on comparisons to G. gallus. Although our genome assemblies are highly fragmented due to a lack of long-range information for scaffolding (Additional file 2: Table S1), such genomes can provide valuable information for phylogenetic studies (e.g., Jarvis et al. 2014) and for analyses of molecular evolution (e.g., Tiley et al. 2018). Indeed, our recovery of 7513 one-to-one orthologous clusters, which yielded 40,621 one-to-one intron and 40,859 one-to-one exon alignments, emphasizes the value of relatively low coverage genomes for phylogenomic analyses.
Analysis of repeat content further emphasized the completeness of our Gallus genome assemblies. All new Gallus genomes contained about 9% transposable element content with the majority of repeats annotated as chicken repeat 1 elements (CR1s), consistent with other bird genomes (Table 1). This estimated repeat content is only slightly lower than the repeat content of the reference G. gallus genome, suggesting little information was lost due to our use of a single library of short insert length.
Phylogenomic analysis of the genus Gallus
The ML tree generated using the complete alignment of over 32 megabase pairs (Mbp) of orthologous sequences from the four Gallus species and the Bambusicola outgroup had 100% bootstrap support for both internal nodes (Fig. 3). The topology of the Gallus species tree obtained using ASTRAL to summarize 3406 gene trees was identical to the estimate of Gallus phylogeny generated by analysis of concatenated data (Fig. 3); the local posterior probabilities for both branches in the ASTRAL tree were maximal (1.0). Finally, the topology of the ASTRAL tree generated using 35,920 “perfect transversions” extracted from the complete alignment was identical to trees generated by ML concatenation and the ASTRAL analysis of 3406 gene trees, and they also had local posterior probabilities of 1.0, although the coalescent branch lengths estimated using perfect transversions were slightly shorter than those estimated using gene trees (Fig. 3).

Whole genome phylogeny for Gallus. ML and the ASTRAL species trees were topologically identical. ML bootstrap support for both nodes was 100%, while both ASTRAL (estimated from gene trees or from perfect transversions) had local posterior probabilities of 1.0. Estimated branch lengths for the internal branches (A and B) are presented; the units for branch lengths are substitutions per site for ML and coalescent units for ASTRAL
Of the 15 possible rooted topologies for the four species within Gallus, the distribution of gene trees with each topology appears to generally fit the expectation given the multispecies coalescent (Fig. 4). Although it is possible for a rooted four-taxon species tree to be in the “anomaly zone” (where the most common gene tree does not match the species tree), our estimates of coalescent branch lengths indicated that the most common gene tree for Gallus should match the species tree (Fig. 4 and Additional file 3). Almost 36% of gene trees were topologically identical to the estimated species tree (i.e., speciodendric; Rosenberg 2002). This is slightly fewer than expected given the ASTRAL branch lengths estimated using gene trees as input (Fig. 4) and slightly more than expected given the ASTRAL branch lengths generated using perfect transversion data (Additional file 3). Although the observed spectrum of gene tree topologies appeared to have a fairly good fit to the multispecies coalescent based on a simple visual assessment, a perfect fit to expectation was strongly rejected (χ2 = 341.74; df = 14; p = 2.2 × 10‒64). Specifically, many topologies expected to be relatively common, including the speciodendric topology, were slightly less common than expected given the multispecies coalescent. Instead, there was a modest excess of non-speciodendric gene trees that included a G. gallus‒G. varius clade (topologies 1, 14, and 15) as well as a slight excess of gene trees with topology 10 (Fig. 4).

The observed spectrum of Gallus gene tree topologies is similar to expectation given the multispecies coalescent. Expected numbers of gene trees with each topology were calculated assuming the topology and branch lengths that were estimated by ASTRAL when gene trees were used as input. The topology for each gene tree is presented as a rooted newick string using a single letter (first letter of the species name) to indicate each Gallus species. The speciodendric gene tree is shaded in pink
The impact of data type on phylogenomic analyses of Gallus
Independent concatenated ML analyses of four data types (CNEEs, exons, introns, and UCEs) all resulted in a topology identical to the combined topology (Fig. 3), although support differed among the data types (Table 2). Bootstrap support was not a simple function of the number of sites in each subset of the data; support was actually lowest for the exon dataset and maximal for the UCE dataset. In addition to expected differences in overall evolutionary rate (the treelength for CNEEs was only 1/5 that of the combined dataset), data type also had an impact on relative branch lengths. Specifically, the internal branches make up a much smaller proportion of the total treelength for the CNEE tree than they do for the other trees (Table 2).
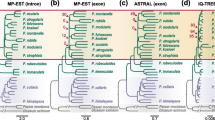
Differences were also evident among gene tree distributions for exons, introns, and UCEs (Fig. 5). We were able to reject the null hypothesis of homogeneity for the three data types (χ2 = 60.239; df = 28; p = 0.00038); the primary difference among the spectra appeared to be the large excess of trees with the G. sonneratii–G. gallus clade for the exons (topology 10; Additional file 3). However, one limitation of the use of gene trees for this analysis is that many regions did not have enough informative sites to provide a reliable estimate of the gene tree topology (we only considered regions with at least ten parsimony informative sites). Our analyses of perfect transversions allowed us to include CNEEs (in addition to exons, introns, and UCEs); doing so revealed additional differences among data types. ASTRAL branch lengths were generally longer for the analyses of gene trees than they were for perfect transversions (Table 3), which we expected based on the analyses of all data types (Fig. 3). However, analysis of the CNEE data resulted in longer coalescent branch length estimates than the other data types, and the estimate of the length of the branch uniting G. gallus, G. sonneratii, and G. lafayettii was actually longer than all of the branch length estimates based on gene trees. Despite the observed differences among data types, all ASTRAL analyses resulted in the same species tree topology, and both branches had maximal (local posterior probability of 1.0) support (Fig. 3 and Additional file 4: Treefile 1).

Observed spectra of gene tree topologies for each data type. The gene trees spectra are expressed as proportions for the three data types that have a sufficient number of gene trees. The order of the topologies for the individual gene trees presented is identical to Fig. 4
The observation that analyses of all data partitions resulted in the same topology suggests that factors like deviation from base composition stationarity have not affected our estimates of Gallus phylogeny. However, shifts in base composition over time, which have been observed in avian phylogenomic studies focused on longer time scales (Jarvis et al. 2014; Braun et al. 2019), could explain some of the inconsistent results obtained in previous studies of Gallus phylogeny (Fig. 2). We did observe some variation across the four Gallus species in the base composition of parsimony informative sites for the different data types (Table 4). The greatest variation was typically along the GC-AT axis, though CNEEs had slightly more variation along the AC-GT axis. Exons exhibited substantially more variation than the non-coding data partitions (exhibiting nearly 2‒3 × more variation in base composition than the other data types), consistent with the observation that exons exhibit more variation in GC-content than introns when all birds are examined (Reddy et al. 2017). The mean GC-content for informative sites was close to 50% for all data types (Table 4). However, the GC-content was higher than this average for G. gallus and G. varius and lower for G. sonneratii and G. lafayettii for most data types. The largest degree of variation was in GC-content for three of the four data types and the largest effect was for exons. The exceptional data type was CNEEs, where variation in the proportion of AC (i.e., variation on the amino-keto axis) was slightly higher than variation in the proportion of GC. However, the small number of informative sites in CNEEs (Table 2) is expected to result in a high sampling variance, so this is unlikely to reflect genuinely higher variance on the amino-keto axis. The dominant pattern is the relatively high GC-content variant along with the more than 2.5-fold greater variation in exons.
Asymmetries in Gallus gene tree spectra suggest ancient introgression
Examining the patterns evident in the full spectrum of the 15 possible gene trees is challenging, but single taxon deletions can simplify the expectations. When only four taxa are considered, the multispecies coalescent makes two predictions: (1) the majority gene tree topology will be congruent with the species tree; and (2) the two minority gene tree topologies will be equiprobable (Pamilo and Nei 1988). Deviations from this expectation can be driven by asymmetric patterns of gene flow (e.g., introgression) or by systematic biases in gene tree estimation that favor one of the two minority topologies. We found strong asymmetries in four of the five possible taxon deletions (Table 5). More specifically, there was a large excess of gene trees that had a G. gallus‒G. varius clade (evident when either G. lafayettii or G. sonneratii was deleted) and a somewhat more modest excess of gene trees with a G. gallus‒G. sonneratii clade (evident when G. varius or Bambusicola was deleted). In contrast to these asymmetries, deletion of G. gallus resulted in nearly equal minority topologies (Table 5).
The same asymmetries were evident in the perfect transversion data (Additional file 5: Table S2). The counts of transversions that support each topology were of course much higher than the number of gene trees, but the magnitude of the differences for the minority topologies was very similar. The perfect transversion data further emphasized the near equality of two minority topologies when after deletion of G. gallus; the modest excess of gene tree topologies with a G. varius‒G. sonneratii clade relative to a G. varius‒G. lafayettii clade (Table 4) is reversed in the perfect transversion data (2781 transversions support G. varius + G. sonneratii and 2813 support G. varius + G. lafayettii). The simplest interpretation of these data is the existence of gene flow after the radiation of the four species that involves two specific pairs of species: (1) G. gallus and G. varius, and (2) G. gallus and G. sonneratii. If introgression had occurred very recently (i.e., since the domestication of chickens), we would expect zero (or very few) substitutions since the time of introgression. This would lead to virtually identical sequences for G. gallus and the other taxon involved in the introgression, which would result in an excess of very short terminal branches for these pairs of taxa. We looked for the excess of very short terminal branch lengths in the intronic gene trees that could reflect introgression, but we did not observe a difference in the distribution of terminal branch lengths (Additional file 6: Fig. S1), suggesting much of the introgression was ancient.
Our analyses with phylogenetic networks also supported introgression between G. gallus and G. varius. We observed an improvement in the pseudolikelihood score when allowing for one reticulation (Additional file 7: Fig. S2). The best network allowing for one reticulation showed that about 15% of gene trees are introgressed between G. gallus and G. varius, which is consistent with our analyses of gene tree spectra (Additional file 8: Fig. S3).
Discussion
A strongly supported Gallus species tree combined with introgression
Phylogenomic analyses of the genus Gallus using thousands of loci of four different data types produced a strongly supported topology (Fig. 3) that was congruent with published UCE studies (Hosner et al. 2016, 2017). For the individuals included in our study, there was evidence of ancient, but not recent, introgression, suggesting that these genomes are good representatives of their respective species. The well-resolved phylogeny and availability of genomes will allow future studies to better understand patterns of change within this group Additional file 8: Fig. S3.
Sampling variance and errors in phylogenetic estimation
The strong support in this study raises the question of why previous studies (prior to the UCE studies; Fig. 2) have failed to find consensus, or even to recover the topology we found in all of our analyses. There are two main reasons why phylogenetic analyses might fail to recover the true topology: sampling error and systematic error. Our results suggest the failure of most previous analyses to recover the topology found in this study is likely to reflect sampling error.
There are two types of sampling error that may have been important; the first reflects why phylogenetic studies can benefit from sampling large numbers of loci and the importance of the multispecies coalescent. Given the branch lengths estimated by ASTRAL, we expect 29.1% to 36.5% of gene trees (based on the perfect transversion and gene tree analysis, respectively) to be speciodendric (i.e., match the species tree). The largest number of gene trees sampled prior to the Hosner et al. studies was 15 (Kimball and Braun 2014). The expectation is that ~ 4‒5 of those trees would match the species tree, with the probability of not sampling any speciodendric gene trees being relatively low (p = 0.0011 for the higher probability and p = 0.0058 for the lower probability, binomial test). However, these probabilities assume the gene trees are fully resolved. Examination of unpublished gene trees from Kimball and Braun (2014) revealed that six (of 15) trees were unresolved (i.e., they exhibited a polytomy within Gallus for the ML tree), and three of the unresolved gene trees were compatible with the species tree (Additional file 9: Treefile 2). Observing three speciodendric gene trees given the Kimball and Braun (2014) sample size is not unexpected (0.144 < p < 0.324, binomial test), assuming all three of those the unresolved trees were actually speciodendric trees.
The lack of resolution observed in the Kimball and Braun (2014) gene trees, where 40% (6 of 15) gene trees were unresolved, emphasizes the second type of sampling variance: sampling insufficient information in each gene tree to resolve the gene tree. Sampling more sites within loci can provide better resolution (more likely to match the true underlying tree), emphasizing that the short segments that are often used to estimate gene trees can be problematic (Patel et al. 2013; Gatesy and Springer 2014; Mirarab et al. 2014). For this study, we limited the impact of uninformative gene regions by limiting our gene tree analyses to loci with at least 10 informative sites, in contrast to prior analyses where the number of informative sites given the four Gallus species and the Bambusicola outgroup was not considered. These results emphasize the fact that phylogenomics, particularly when it reflects the use of data extracted from whole genomes (where longer loci can be obtained), can provide a way to overcome both types of sampling variance (Gee 2003).
In contrast to sampling error, which one can be overcome by adding loci and by sampling more informative loci, systematic error reflects cases where adding data (either loci, sites, or both) actually increases support for an incorrect tree. One type of systematic error is “localized biases”, where specific loci within phylogenomic datasets exhibit strong (and potentially misleading) signals (e.g., Kimball et al. 2013; Brown and Thomson 2016; Shen et al. 2017). This is the only type of systematic error that could explain the failure of previous Gallus phylogenies to recover the topology found in this dataset, assuming previous studies sampled one or more misleading loci. However, previous studies varied in the loci included, making this explanation unlikely. Taken as a whole, our results suggest that the simplest explanation for the failure of previous studies to recover the topology found in this study is simply the limited number of loci and sites sampled in those studies.
Introgression
Given that hybridization can occur among all of the Gallus species, it is certainly possible that introgression has affected genomes in this genus. As a first step to understand the possible role of introgression, we looked for asymmetries in gene trees. Analyses of gene trees and perfect transversions suggested the existence of some gene flow after speciation, with the gene flow involving two specific pairs of taxa: G. gallus + G. varius and G. gallus + G. sonneratii. The distribution of terminal branch lengths in G. varius and G. sonneratii (Additional file 6: Fig. S1) indicates that, if the asymmetries reflect introgression, the vast majority of the introgression occurred prior to domestication. We cannot exclude the possibility of very limited amounts of post-domestication gene flow driven by hybridization of Domestic Chickens with other species (or hybridization among captive populations of these species) followed by extensive backcrossing. Likewise, we cannot exclude contamination of the reference G. gallus genome assembly (ICGSC 2004) with some alleles from other species. This could reflect genes introgressed during domestication that subsequently moved into wild-type G. gallus (given that wild-type G. gallus populations are known to have some in recent contamination from domesticated chicken; Peterson and Brisbin 1998), but it is extremely unlikely that this recent introgression explains the observed asymmetric patterns we observed in our gene trees. Instead, it is likely that changes in ranges during the history of Gallus may have led to periods of sympatry where hybridization may have occurred. The introgression between G. gallus and G. varius was also supported by the phylogenetic network analyses (Additional file 8: Fig. S3). Considering that the excess of quartet frequencies from our gene tree spectra analyses supported introgression between G. gallus and G. sonneratii to a lesser extent, and that we may not be able to identify multiple reticulations involving the same edge, it is not surprising that both events were not supported by the network analyses.
Data type effects are evident in the Gallus radiation
One interesting aspect of genome evolution revealed early in phylogenomic era is the extensive incongruence observed for individual gene trees. Far from “ending incongruence”, as the discussions of early phylogenomic papers asserted (Gee 2003), phylogenomics has instead heralded a new “beginning of incongruence” (Jeffroy et al. 2006). While processes such as deep coalescence were recognized prior to the phylogenomic era (e.g., Pamilo and Nei 1988; Maddison 1997), phylogenomics has also revealed sources of bias that affect subsets of the genome. While we did not identify any data type effects on the topology within Gallus, as has been noted previously (Braun et al. 2019), we observed differences among the data type partitions that may explain why exploring distinct data partitions can be important.
One explanation for data type effects in birds appears to be the greater GC-content variation across species in coding exons relative to non-coding regions (Jarvis et al. 2014; Reddy et al. 2017). Perhaps surprisingly given the recent radiation of Gallus species (and thus limited time to accumulate differences), we observed greater GC-content variation for exons than for non-coding regions in this study. This observation is consistent with existence of a bias toward AT→GC substitutions documented in G. gallus (Webster et al. 2006). These biases are generally thought to reflect GC-biased gene conversion (Mugal et al. 2015; Bolívar et al. 2019). However, our results modify earlier observations of increased GC-content in parts of the chicken genome by providing evidence for when these shifts might have occurred. Specifically, there appear to be biases toward GC substitutions in G. gallus and G. varius (suggesting the bias toward GC is the ancestral state for the genus Gallus given our estimate of the species tree) along with a bias toward AT in G. lafayettii and G. sonneratii. It seems likely that the lower bootstrap support for the species tree in the concatenated tree for exons (Table 2) reflects, at least in part, these biases in the substitution process. However, the shift in base composition for introns is very modest and therefore unlikely to be an explanation for the topologies differences observed in previous studies.
A second data type effect is the shorter internal branches observed for CNEEs. There is a strong overlap between CNEEs and UCEs as classes of genetic elements (although we excluded overlapping CNEEs and UCEs in this study). Our CNEEs were identified by homology to the Edwards et al. (2017) CNEE set, which were trimmed to a conserved region (and so averaged ~ 500 bp), whereas we identified UCEs by homology to Jarvis et al. (2014) who included a short, conserved core (~ 100 bp), and then up to 1000 bp of flanking sequence (so most were > 2000 bp). Population genomic studies in humans indicate that UCE/CNEE core regions are “ultraselected” for conservation rather than representing mutational cold spots (Katzman et al. 2007); it seems likely that these regions are also subject to strong purifying selection in birds. This strong purifying selection is expected to result in local depression of the effective population size due to the Hill and Robertson (1966) effect. If Hill-Robertson effects due to strong purifying selection on CNEEs explain their conservation, they would be expected to have longer coalescent branch lengths in an ASTRAL analysis. This is exactly what we observed (Table 3). The fact that estimates of coalescent branch lengths obtained for the CNEE data were longer than those obtained using other data types is especially surprising since we were only able to obtain CNEE coalescent branch lengths using perfect transversion data. For the other three data types estimates of coalescent branch length obtained using prefect transversions were shorter than those obtained using gene tree topologies. This suggests that coalescent branch lengths are genuinely longer for CNEEs. Mendes and Hahn (2017) suggested that internal branches are overestimated in concatenated analyses due to the site patterns that appear to be homoplastic given the assumption that all genomic regions have the same underlying tree topology. Hill–Robertson effects would lead to greater agreement among gene trees that are also congruent with the species tree, reducing this overestimation.
A corollary of the hypothesis that CNEEs have a locally reduced effective population size is that the amount of intralocus recombination is sufficient to reduce the impact of Hill–Robertson effects in our UCE data. This brings up the possibility that intralocus recombination could distort our estimates of the species tree (Springer and Gatesy 2016; but see Xu and Yang 2016). However, the congruence between estimates of the species tree generated using ASTRAL on gene trees and those generated using perfect transversions indicates that intralocus recombination cannot be distorting our topology because the transversion data treats each perfect transversion site as a partially resolved gene tree. Scoring biallelic markers as partially resolved is expected to yield accurate estimates of coalescent branch lengths as long as the markers are truly homoplasy-free (Springer and Gatesy 2019). However, our observation that perfect transversion branch lengths are shorter than those estimated using gene trees (Table 2) suggests that the transversion data are unlikely to be homoplasy-free. Linkage among the perfect transversions is also likely to inflate the local posterior probabilities calculated by ASTRAL, but this is not especially problematic given the high support observed in the analysis of gene trees. We view the use of gene trees and transversions as complementary. The ASTRAL tree generated using gene trees is expected to be accurate if there is little intralocus recombination, but the ASTRAL transversion tree is expected to be more accurate if intralocus recombination is very common. Since both ASTRAL species trees have identical topologies, similar branch lengths, and maximal support values, we are confident in our inference.
One data type that we did not examine was mitochondrial data. Multiple mitochondrial trees for the genus Gallus have been published, and they also show conflicting resolutions for the relationship within Gallus (Fig. 2). Since the mitochondrial genome is maternally inherited, it will have a lower effective population size, and therefore a higher probability of congruence with the species tree (Moore 1995). Yet none of the mitochondrial topologies for relationships among Gallus (Fig. 2) match the species tree we estimated, even though several of those trees were estimated from mitochondrial data collected from the same individuals used for the genomes in this study. There are many situations that can lead to differences between mitochondrial and nuclear trees, including introgression and incomplete lineage sorting, as well as analytical artefacts such as insufficient taxon sampling and inadequate model fit (e.g., Meiklejohn et al. 2014; Tamashiro et al. 2019). Our results suggest introgression is unlikely, given the limited evidence for gene flow. However, simple gene tree–species tree discordance is possible. The mitochondrial genome does not recombine in birds (Berlin and Ellegren 2001), so it ultimately represents a single gene tree. However, the simplest hypothesis is topological error. That hypothesis can explain why the many published analyses of the mitochondrial data have presented different topologies (Fig. 2). The observation that even studies using complete mitogenomes have resulted in alternative topologies within Gallus (Fig. 2) further corroborates the hypothesis that estimates of mitochondrial gene trees may simply be incorrect. Analyses of mitochondrial data can be challenging (Braun and Kimball 2002), and models of sequence evolution used in phylogenetic analyses often have a relatively poor fit to the data (e.g., Meiklejohn et al. 2014; Tamashiro et al. 2019). Overall, we believe that the observation that analyses of mitochondrial data fail to converge on a single topology provides evidence that estimates of the Gallus mitochondrial trees are not correct, and error provides a good explanation for the observed discordance with the species tree.
Conclusions
Taken as a whole, our results emphasize the need to reevaluate prior estimates of avian phylogeny using phylogenomic data. The availability of whole genome data allowed us to recover a very strongly supported topology that was consistent across all analyses (Fig. 3) and matched previous studies based on sequence capture of UCEs. However, we were also able to significantly extend the previous UCE studies by providing evidence both for historical introgression and for modest (but detectable) data type effects. The observation that coding exons exhibit greater GC-content variation than non-coding data adds to the body of evidence showing that non-coding sequences are likely to represent the most useful source of information for avian phylogenomics. Overall, these phylogenomic analyses provide a rigorous framework for evolution of the genus Gallus and information that should be useful for phylogenomic studies in the future.
Availability of data and materials
New sequencing data, genome assemblies, and annotations have been deposited under NCBI BioProject accession PRJNA396570 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA396570). Alignments and tree files can be found in Zenodo (https://doi.org/10.5281/zenodo.3525340). Software used for the project is available from github (the profile HMM sequence extraction pipeline from https://github.com/aakanksha12/Extract_seq and the expected gene tree spectrum calculator from https://github.com/ebraun68/asymtree). All code is in Perl, platform independent, and available under free software licenses (GNU GPL 3.0 for Extract_seq and MIT for asymtree).
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Armstrong MH, Braun EL, Kimball RT. Phylogenetic utility of avian ovomucoid intron G: a comparison of nuclear and mitochondrial phylogenies in the Galliformes. Auk. 2001;118:799–804.
Berlin S, Ellegren H. Evolutionary genetics: clonal inheritance of avian mitochondrial DNA. Nature. 2001;413:37–8.
Betancur-R R, Li C, Munroe TA, Ballesteros JA, Ortí G. Addressing gene tree discordance and non-stationarity to resolve a multi-locus phylogeny of the flatfishes (Teleostei: Pleuronectiformes). Syst Biol. 2013;62:763–85.
Bolívar P, Guéguen L, Duret L, Ellegren H, Mugal CF. GC-biased gene conversion conceals the prediction of the nearly neutral theory in avian genomes. Genome Biol. 2019;20:5.
Braun EL, Kimball RT. Examining basal avian divergences with mitochondrial sequences: model complexity, taxon sampling and sequence length. Syst Biol. 2002;51:614–25.
Braun EL, Cracraft J, Houde P. Resolving the avian tree of life from top to bottom: the promise and potential boundaries of the phylogenomic era. In: Kraus RHS, editor. Avian genomics in ecology and evolution—from the lab into the wild. Cham: Springer; 2019. p. 151–210.
Brown JM, Thomson RC. Bayes factors unmask highly variable information content, bias, and extreme influence in phylogenomic analyses. Syst Biol. 2016;66:517–30.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST + : architecture and applications. BMC Bioinform. 2008;10:421.
Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–96.
Chen MY, Liang D, Zhang P. Phylogenomic resolution of the phylogeny of laurasiatherian mammals: exploring phylogenetic signals within coding and noncoding sequences. Genome Biol Evol. 2017;9:1998–2012.
Chojnowski J, Kimball RT, Braun EL. Introns outperform exons in analyses of basal avian phylogeny using clathrin heavy chain genes. Gene. 2008;410:89–96.
Conant GC, Lewis PO. Effects of nucleotide composition bias on the success of the parsimony criterion in phylogenetic inference. Mol Biol Evol. 2001;18:1024–33.
Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–9.
Doyle JJ. Trees within trees: genes and species, molecules and morphology. Syst Biol. 1997;46:537–53.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–5.
Edwards SV, Cloutier A, Baker AJ. Conserved nonexonic elements: a novel class of marker for phylogenetics. Syst Biol. 2017;66:1028–44.
Eriksson J, Larson G, Gunnarsson U, Bed’hom B, Tixier-Boichard M, Strömstedt L, et al. Identification of the Yellow Skin gene reveals a hybrid origin of the domestic chicken. PLoS Genet. 2008;4:e1000010.
Faircloth BC, McCormack JE, Crawford NG, Harvey MG, Brumfield RT, Glenn TC. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst Biol. 2012;61:717–26.
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–91.
Gatesy J, Springer MS. Phylogenetic analysis at deep timescales: unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol Phylogenet Evol. 2014;80:231–66.
Gee H. Evolution: ending incongruence. Nature. 2003;425:782.
Gill F, Donsker D. IOC world bird list (v9.2). 2019. https://doi.org/10.14344/ioc.ml.9.2.
Hill WG, Robertson A. The effect of linkage on limits to artificial selection. Genet Res. 1966;8:269–94.
Hosner PA, Faircloth BC, Glenn TC, Braun EL, Kimball RT. Avoiding missing data biases in assembling the landfowl tree of life (Aves: Galliformes). Mol Biol Evol. 2016;33:1110–25.
Hosner PA, Tobias JA, Braun EL, Kimball RT. How do seemingly non-vagile clades accomplish trans-marine dispersal? Trait and dispersal evolution in the landfowl. Proc Roy Soc Lond B. 2017;284:20170210.
Imsland F, Feng C, Boije H, Bed’Hom B, Fillon V, Dorshorst B, et al. The Rose-comb mutation in chickens constitutes a structural rearrangement causing both altered comb morphology and defective sperm motility. PLoS Genet. 2012;8:e1002775.
International Chicken Genome Sequencing Consortium (ICGSC). Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716.
Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346:1320–31.
Jeffroy O, Brinkmann H, Delsuc F, Philippe H. Phylogenomics: the beginning of incongruence? Trends Genet. 2006;22:225–31.
Johnsgard PA. The pheasants of the world. 2nd ed. Oxford: Oxford University Press; 1999. p. 92–9.
Kan XZ, Li XF, Lei ZP, Chen L, Gao H, Yang ZY, et al. Estimation of divergence times for major lineages of galliform birds: evidence from complete mitochondrial genome sequences. Afric J Biotech. 2010a;9:3073–8.
Kan XZ, Yang JK, Li XF, Chen L, Lei ZP, Wang M, et al. Phylogeny of major lineages of Galliform birds (Aves: Galliformes) based on complete mitochondrial genomes. Genet Mol Res. 2010b;9:1625–33.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Katzman S, Kern AD, Bejerano G, Fewell G, Fulton L, Wilson RK, et al. Human genome ultraconserved elements are ultraselected. Science. 2007;317:915.
Kimball RT, Braun EL. A multigene phylogeny of Galliformes supports a single origin of erectile ability in non-feathered facial traits. J Avian Biol. 2008;39:438–45.
Kimball RT, Braun EL. Does more sequence data improve estimates of galliform phylogeny? Analyses of a rapid radiation using a complete data matrix. PeerJ. 2014;2:e361.
Kimball RT, Braun EL, Zwartjes P, Crowe TM, Ligon JD. A molecular phylogeny of the pheasants and partridges suggests that these lineages are not monophyletic. Mol Phylogenet Evol. 1999;11:38–54.
Kimball RT, Mary CM, Braun EL. A macroevolutionary perspective on multiple sexual traits in the Phasianidae (Galliformes). Int J Evol Biol. 2011;2011:423938.
Kimball RT, Wang N, Heimer-McGinn V, Ferguson C, Braun EL. Identifying localized biases in large datasets: a case study using the avian tree of life. Mol Phylogenet Evol. 2013;69:1021–32.
Lawal RA, Al-Atiyat RM, Aljumaah RS, Silva P, Mwacharo JM, Hanotte O. Whole-genome resequencing of red junglefowl and indigenous village chicken reveal new insights on the genome dynamics of the species. Front Genet. 2018;9:264.
Li D, Li Y, Li M, Che T, Tian S, Chen B, et al. Population genomics identifies patterns of genetic diversity and selection in chicken. BMC Genomics. 2019;20:263.
Li L, Stoeckert J Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
Ligon JD, Kimball RT, Merola-Zwartjes M. Mate choice in red junglefowl: the issues of multiple ornaments and fluctuating asymmetry. Anim Behav. 1998;55:41–50.
Maddison WP. Gene trees in species trees. Syst Biol. 1997;46:523–36.
Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioninformatics. 2011;27:764–70.
Marcais G, Yorke JA, Zimin A. QuorUM: an error corrector for Illumina reads. PLoS ONE. 2015;10:e0130821.
McCarthy EM. Handbook of avian hybrids of the world. New York: Oxford University Press; 2006.
Meiklejohn KA, Danielson MJ, Braun EL, Faircloth BC, Glenn TC, Kimball RT. Incongruence among different mitochondrial regions: a case study using complete mitogenomes. Mol Phylogenet Evol. 2014;78:314–23.
Meiklejohn KA, Faircloth BC, Glenn TC, Kimball RT, Braun EL. Analysis of a rapid evolutionary radiation using ultraconserved elements: evidence for a bias in some multispecies coalescent methods. Syst Biol. 2016;65:612–27.
Mendes FK, Hahn MW. Gene tree discordance causes apparent substitution rate variation. Syst Biol. 2017;65:711–21.
Mirarab S, Bayzid MS, Boussau B, Warnow T. Statistical binning improves species tree estimation in the presence of gene tree incongruence. Science. 2014;346:1250463.
Moore WS. Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution. 1995;49:718–26.
Mugal CF, Weber CC, Ellegren H. GC-biased gene conversion links the recombination landscape and demography to genomic base composition: gC-biased gene conversion drives genomic base composition across a wide range of species. BioEssays. 2015;37:1317–26.
Nishibori M, Shimogiri T, Hayashi T, Yasue H. Molecular evidence for hybridization of species in the genus Gallus except for Gallus varius. Anim Genet. 2005;36:367–75.
Pamilo P, Nei M. Relationships between gene trees and species trees. Mol Biol Evol. 1988;5:568–83.
Patel S, Kimball RT, Braun EL. Error in phylogenetic estimation for bushes in the tree of life. J Phylogenet Evol Biol. 2013;1:110.
Peterson AT, Brisbin IL. Genetic endangerment of wild Red Junglefowl Gallus gallus? Bird Conserv Int. 1998;8:387–94.
Reddy S, Kimball RT, Pandey A, Hosner PA, Braun MJ, Hackett SJ, et al. Why do phylogenomic data sets yield conflicting trees? data type influences the avian tree of life more than taxon sampling. Syst Biol. 2017;66:857–79.
Rosenberg NA. The probability of topological concordance of gene trees and species trees. Theor Pop Biol. 2002;61:225–47.
Saitou N, Nei M. The number of nucleotides required to determine the branching order of three species, with special reference to the human-chimpanzee-gorilla divergence. J Mol Evol. 1986;24:189–204.
Sayyari E, Mirarab S. Fast coalescent-based computation of local branch support from quartet frequencies. Mol Biol Evol. 2016;33:1654–68.
Shen XX, Hittinger CT, Rokas A. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat Ecol Evol. 2017;1:0126.
Shen YY, Dai K, Cao X, Murphy RW, Shen XJ, Zhang YP. The updated phylogenies of the Phasianidae based on combined data of nuclear and mitochondrial DNA. PLoS ONE. 2014;9:e95786.
Shen YY, Liang L, Sun YB, Yue BS, Yang XJ, Murphy RW, et al. A mitogenomic perspective on the ancient, rapid radiation in the Galliformes with an emphasis on the Phasianidae. BMC Evol Biol. 2010;10:132.
Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0. https://www.repeatmasker.org. Accessed 8 Dec 2015.
Solís-Lemus C, Ané C. Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 2016;12:e1005896.
Solís-Lemus C, Bastide P, Ané C. PhyloNetworks: a package for phylogenetic networks. Mol Biol Evol. 2017;34:3292–8.
Song S, Liu L, Edwards SV, Wu S. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc Natl Acad Sci USA. 2012;109:14942–7.
Springer MS, Gatesy J. The gene tree delusion. Mol Phylogenet Evol. 2016;94:1–33.
Springer MS, Gatesy J. Retroposon insertions within a multispecies coalescent framework suggest that ratite phylogeny is not in the ‘Anomaly Zone’. bioRxiv. 2019. https://doi.org/10.1101/643296.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006;7:62.
Stein RW, Brown JW, Mooers AØ. A molecular genetic time scale demonstrates Cretaceous origins and multiple diversification rate shifts within the order Galliformes (Aves). Mol Phylogenet Evol. 2015;92:155–64.
Swofford DL. PAUP*: phylogenetic analysis using parsimony (*and other methods). Version 4.0a166. 2019.
Tamashiro RA, White ND, Braun MJ, Faircloth BC, Braun EL, Kimball RT. What are the roles of taxon sampling and model fit in tests of cyto-nuclear discordance using avian mitogenomic data? Mol Phylogenet Evol. 2019;130:132–42.
Tiley GP, Kimball RT, Braun EL, Burleigh JG. Comparison of the Chinese Bamboo Partridge and Red Junglefowl genome sequences highlights the importance of demography in genome evolution. BMC Genomics. 2018;19:336.
Wang N, Kimball RT, Braun EL, Liang B, Zhang Z. Assessing phylogenetic relationships among Galliformes: a multigene phylogeny with expanded taxon sampling in Phasianidae. PLoS ONE. 2013;8:e64312.
Wang N, Kimball RT, Braun EL, Liang B, Zhang Z. Ancestral range reconstruction of Galliformes: the effects of topology and taxon sampling. J Biogeogr. 2017;44:122–35.
Wang MS, Li Y, Peng MS, Zhong L, Wang ZJ, Li QY, et al. Genomic analyses reveal potential independent adaptation to high altitude in Tibetan chickens. Mol Biol Evol. 2015;32:1880–9.
Webster MT, Axelsson E, Ellegren H. Strong regional biases in nucleotide substitution in the chicken genome. Mol Biol Evol. 2006;23:1203–16.
Wheeler TJ, Eddy SR. nhmmer: DNA homology search with profile HMMs. Bioinformatics. 2013;29:2487–9.
Xi Z, Liu L, Davis CC. Genes with minimal phylogenetic information are problematic for coalescent analyses when gene tree estimation is biased. Mol Phylogenet Evol. 2015;92:63–71.
Xu B, Yang Z. Challenges in species tree estimation under the multispecies coalescent model. Genetics. 2016;204:1353–68.
Yang S, Shi Z, Ou X, Liu G. Whole-genome resequencing reveals genetic indels of feathered-leg traits in domestic chickens. J Genet. 2019;98:47.
Yi G, Qu L, Liu J, Yan Y, Xu G, Yang N. Genome-wide patterns of copy number variation in the diversified chicken genomes using next-generation sequencing. BMC Genom. 2014;15:962.
Zhang C, Rabiee M, Sayyari E, Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018;19:153.
Zimin AV, Marcais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. The MaSuRCA genome assembler. Bioinformatics. 2013;29:2669–77.
Acknowledgements
Not applicable.
Funding
Funding for this project was provided by the Florida International University Tropics Program and the Susan S. Levine Trust. RTK and ELB also received support from the United States National Science Foundation (DEB-1118823 and DEB-1655683). The funding bodies had no role in the design of the study or collection, analysis, or interpretation of data.
Author information
Authors and Affiliations
Contributions
All authors participated in the design of the experiments, data analysis and interpretation, and writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No permissions were required for this study as blood was collected by breeders and given to R. Kimball for previous research rather than for the purpose of this study.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Supplementary information
Additional file 1:
This file corresponds to Fig. 1 and its five parts, each of which is a photograph of a taxon included in the analyses we report. Credits (and licenses) for each photograph are indicated below.
Additional file 2: Table S1.
Comparison of Bambusicola assemblies and published genomes utilized in this study.
Additional file 3:
One sheet (Gene tree counts): The graph presents the expected counts of gene trees given the coalescent branch lengths in the ASTRAL trees (GT = ASTRAL tree based on gene trees; TV = ASTRAL tree based on perfect transversions); the other sheet (Data type info): The graph presents the expected frequencies of each gene tree topology given the GT ASTRAL tree and the observed frequencies of each gene tree after subdividing trees by their source (exon, intron, or UCE).
Additional file 4:
This file includes four sets of phylogenomic trees for the genus Gallus.
Additional file 5: Table S2.
This file comprises two sheets with information on the fit of gene trees or transversional changes to the three possible quartets produced by deleting a single taxon.
Additional file 6: Fig. S1.
The distribution of terminal branch lengths for selected Gallus species are independent of topology.
Additional file 7: Fig. S2.
Profile pseudolikelihood scores for networks with h reticulations.
Additional file 8: Fig. S3.
Best phylogenetic network with one reticulation.
Additional file 9:
Trees from Kimball and Braun (2014)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tiley, G.P., Pandey, A., Kimball, R.T. et al. Whole genome phylogeny of Gallus: introgression and data-type effects. Avian Res 11, 7 (2020). https://doi.org/10.1186/s40657-020-00194-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40657-020-00194-w