
Abstract
Background
Myeloid-derived suppressor cells (MDSCs) are immature myeloid cells with immunosuppressive functions sub-classified into monocytic and polymorphonuclear MDSCs (M-MDSCs and PMN-MDSCs). Clinical studies reported increased levels of MDSCs that were associated with poor outcome in sepsis patients. Since sepsis patients exhibit signs of inflammation and immunosuppression, MDSCs may provide benefit by dampening deleterious inflammation in some patients. To test this hypothesis, we measured MDSCs in critically ill sepsis patients with pneumonia and multi-organ dysfunctions and a high likelihood of death.
Methods
This was a prospective multicenter observational cohort study performed in eight ICUs in Athens and Thessaloniki, Greece, enrolling critically ill patients with pneumonia and sepsis with multi-organ dysfunctions. A flow cytometry approach using blood collected at study inclusion in tubes containing lyophilized antibodies combined to unsupervised clustering was developed to quantify M-MDSCs and PMN-MDSCs.
Results
Forty-eight patients were included, of whom 34 died within 90 days. At study inclusion, M-MDSCs and PMN-MDSCs were increased in sepsis patients when compared to healthy subjects (3.07% vs 0.96% and 22% vs 2.1% of leukocytes, respectively; p < 10–4). Increased PMN-MDSCs were associated with secondary infections (p = 0.024) and new sepsis episodes (p = 0.036). M-MDSCs were more abundant in survivors than in patients who died within 28 days (p = 0.028). Stratification of patients according to M-MDSC levels revealed that high levels of M-MDSC were associated with reduced 90-day mortality (high vs low M-MDSCs: 47% vs 84% mortality, p = 0.003, hazard ratio [HR] = 3.2, 95% CI 1.4–7.2). Combining high M-MDSC levels with low Acute Physiology and Chronic Health Evaluation (APACHE) II score improved patient stratification (M-MDSCshigh/APACHE IIlow vs M-MDSCslow/APACHE IIlow: 20% vs 80% 90-day mortality, p = 0.0096, HR = 7.2, 95% CI 1.6–32). In multivariate analyses high M-MDSCs remained correlated with improved survival in patients with low APACHE II score (p = 0.05, HR = 5.26, 95% CI 1.0–27.8).
Conclusion
This is the first study to associate high levels of M-MDSCs with improved survival in sepsis patients.
Similar content being viewed by others


Introduction
Sepsis is defined as a dysregulated host response to an infection resulting in life-threatening organ dysfunction [1]. The prevalence of sepsis is increasing, and recent estimations suggest that sepsis affects about 48.9 million people and is responsible of 11.0 million sepsis-related deaths per year, representing 19.7% of all deaths worldwide [2]. Sepsis survivors frequently develop functional and cognitive impairments and worsening of chronic health conditions. Almost half of patients surviving sepsis are re-hospitalized within a year [3,4,5].
Exuberant proinflammatory responses during the early phase of sepsis, illustrated by the so-called “cytokine storm”, are implicated in tissue damage, organ dysfunctions and early mortality. A concomitant compensatory anti-inflammatory response participating to inflammation resolution and tissue repair promotes immunosuppression that can persist for extended periods of time. Immunosuppression includes features such as apoptosis-mediated depletion of dendritic cells, T cells and B cells, decreased expression of proinflammatory cytokines, increased expression of anti-inflammatory cytokines and inhibitory checkpoint molecules, and reduced expression of antigen-presenting molecules and costimulatory molecules by immune cells [5,6,7,8,9,10,11]. Persistent immunosuppression favors the development of secondary infections accounting for late mortality and morbidity. Hence, immunomodulatory therapies in sepsis should target inflammation or immunosuppression depending on patient’s status. Theragnostic approaches are promising for monitoring immune status and selecting the most appropriate host-directed immunotherapy to be implemented in a personalized manner [7, 10, 12, 13].
Myeloid-derived suppressor cells (MDSCs) are immature-like myeloid cells characterized by their immunosuppressive impact on innate and adaptive immune responses [14, 15]. Generally, MDSCs are subdivided into monocytic and polymorphonuclear MDSCs (M-MDSCs and PMN-MDSCs), yet additional subtypes have been proposed among which early stage and eosinophilic MDSCs [14,15,16,17]. MDSCs are rare in blood at homeostasis but expand when inflammatory and danger signals stimulate hematopoiesis. MDSCs may also be generated through the conversion of monocytes and neutrophils into pathologically activated MDSCs [14]. MDSCs have been primarily studied in the field of cancer, a condition in which these cells are enriched in tumor environment and impair anti-tumor immunity. MDSCs can rise in the blood of cancer patients to become one of the main leukocyte subtypes [18, 19]. Clinical trials targeting MDSCs are running to counterbalance tumor-associated immunosuppression in cancer patients [20, 21].
In the field of infection and sepsis, clinical studies have shown an association between high levels of PMN-MDSCs and/or M-MDSCs in the blood and development of nosocomial infections, morbidity and/or mortality [22,23,24,25,26,27,28,29,30,31]. These observations led to the proposal that MDSCs sustain immunosuppression, and could be targeted to reverse immunosuppression in septic patients. However, clinical studies included a limited number of patients with mixed infection etiologies and medical care. Moreover, MDSCs may have different impacts depending on disease progression [7, 26, 32]. Here, we conducted a prospective clinical study in patients with sepsis due to pneumonia, multi-organ failure and high likelihood of poor outcome to characterize MDSCs in severely ill sepsis patients.
Methods
Study design and setting
This study was a prospective multicenter observational study performed in 8 ICUs in Athens and Thessaloniki, Greece. This was part of the INCLASS study (benefit of clarithromycin in patients with severe infections through modulation of the immune system study; registered at ClinicalTrials.gov, reference NCT03345992). The study was conducted in compliance with the declaration of Helsinki, and was approved by the central Ethics committee (52086/2017) and the National organization for Medicines-EOF (51239/01-06-2017) in Athens, Greece. Eighteen healthy volunteers serving as controls were recruited at Lausanne University Hospital. Exclusion criteria for healthy volunteers were prior diagnosis of sepsis or SARS-CoV-2 infection, acute or chronic viral hepatitis, autoimmune disease, immunodeficiency and use of immunomodulatory drugs. The study was approved by the Commission cantonale d'éthique de la recherche sur l'être humain, Canton de Vaud, Switzerland (CER-VD, Lausanne, Switzerland). Written informed consent was obtained from study participants or legal representatives prior to enrollment.
Patients
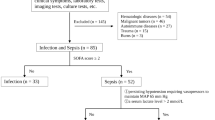
Between December 2017 and February 2019, 48 adult patients with pneumonia were prospectively recruited from eight hospitals in Athens and Thessaloniki, Greece (Table 1). Inclusion criteria were a Sequential Organ Failure Assessment (SOFA) score ≥ 7, including respiratory failure (PiO2/FiO2 < 200), and any other organ system failure with SOFA score of ≥ 3. Most patients were on the ICU when included (n = 28), with a median time of 5 days (interquartile range: 2–7 days). Exclusion criteria were pregnancy, corticosteroid intake, macrolide treatment, allergy to macrolides, neutropenia (< 1000/mm3), HIV infection (with CD4+ T cells < 200/mm3), neoplasm or transplantation. Ethylenediamine tetraacetic acid-anticoagulated blood samples were collected at study inclusion and 5 and 10 days later. Patients were followed up for 28 days, recording all-cause mortality and incidence of secondary infections and new sepsis episodes. The definitions of ventilator-associated pneumonia (VAP), hospital-acquired pneumonia (HAP), healthcare-associated pneumonia (HCAP), secondary infections and new sepsis episodes used in the INCLASS study (available on clinicaltrials.gov) are listed in Additional file 1: Table S1. A late assessment of mortality at 90 days was performed. Mortality was coded as follows: (1) sepsis-related mortality/multi-organ failure (due to progression of the initial septic episode), (2) mortality due to secondary sepsis/infection, and (3) mortality due to other causes. Lactate and CRP levels were measured in routine laboratories of hospitals. Interleukin-6 (IL-6) and ferritin were quantified by enzyme-linked immunosorbent assay (IL-6: Invitrogen, Carlsbad, CA, lower limit of detection: 10 pg/mL; ferritin: ORGENTEC Diagnostika GmbH, Mainz, Germany, lower limit of detection: 75 ng/mL).
Flow cytometry analysis
We established a procedure to limit variability resulting from sample handling/labeling and analysis. To that end, we used a targeted flow cytometry approach using blood samples collected in tubes containing a mixture of lyophilized fluorescently labeled antibodies targeting MDCSs developed in collaboration with DURAClone (Beckman Coulter, Brea, CA). These tubes contained antibodies (clone/fluorochrome) directed against human CD3 (UCHT1/APC-AF700), CD11b (Bear1/PE-Cy7), CD14 (RMO52/APC-AF750), CD15 (80H5/Pacific Blue), CD16 (3G8/ECD), CD19 (J3-119/APC-AF700), CD33 (D3HL60.251/APC), CD45 (J33/Krome Orange), CD56 (NKH-1/APC-AF700), CD124 (G077F6/PE) and human leukocyte antigen (HLA)-DR (Immu-357/FITC). CD3, CD19, CD56 labeled with the same fluorochrome were used to filter lineage-positive leukocytes. One hundred microliter of blood were added to the tubes (all from the same batch) and gently pipetted up and down 10 times. After 20 min, 900 μL of 1 × BD FACS™ lysing solution (BD Biosciences, San Jose, CA) were added. Samples were vortexed and frozen at − 80 °C until all samples were acquired. Samples were thawed, washed with cell stain medium (PBS containing 0.5% BSA and 0.02% sodium azide) and acquired in a single day using an Attune NxT Flow Cytometer (Thermo Fisher Scientific, Waltham, MA, USA). Debris, doublets and CD45-negative cells were excluded from analysis by manual gating using FlowJo™ (v10.6.2, Ashland, OR) (Additional file 2: Fig. S1). We then applied FlowSOM for unsupervised clustering using the biexponential transformed and normalized expression levels of cell surface markers CD3/CD19/CD56 (lineage), CD11b, CD14, CD15, CD16, CD33, CD45, CD124 and HLA-DR and relative side scatter area (SSC-A). Metaclusters were set on 30 populations, merged into 8 populations based on biological knowledge and marker expression, as represented in tSNE and heatmap plots. M-MDSCs corresponded to CD11b+ CD14+ CD15−/low CD16− CD33+ HLA-DR−/low cells, while PMN-MDSCs corresponded to CD11b+ CD14− CD15+ CD16+ CD33− HLA-DR− cells.
Statistical analysis
Baseline patient characteristics were compared using Chi-square exact test, Mann–Whitney U test, and Kruskal–Wallis test as appropriate. The comparison between cell populations and clinical data were evaluated using the Mann–Whitney U test and correlation studies performed using Spearman’s rank-order correlation. M-MDSCs ≤ 4.3% and > 4.3% of leukocytes were considered as low and high percentages, respectively. The cutoff value was based on highest tertile of % M-MDSCs in sepsis patients. APACHE II scores ≤ 20 and > 20 (cutoff values based on median) were considered as low and high. Statistical differences between survival and event curves were assessed with the log-rank test. The contribution of MDSCs to mortality was analyzed using the Cox proportional hazard model. Statistics and figure design were performed using R v.3.6.0 (R-Foundation for Statistical Computing, Vienna, Austria). p values < 0.05 were considered to be statistically significant.
Results
Clinical data
We included 48 patients with sepsis due to pneumonia (33 [69%] with ventilator-associated/hospital-acquired pneumonia and 15 [31%] with healthcare-associated pneumonia). Fourteen patients (29.2%) survived, 23 patients (47.9%) died within 28 days and 34 patients (70.8%) died within 90 days (Table 1). The median age of 90-day survivors was significantly lower than the median age of 90-day non-survivors (median and interquartile range [IQR]): 57 [47–74] vs 75 [67–86] years; p = 0.0014). At study inclusion, 90-day survivors and non-survivors had similar SOFA scores, leukocyte counts, and C-reactive protein (CRP), IL-6 and ferritin levels. Survivors had lower APACHE II scores (16 [14–21] vs 23 [18–27]; p = 0.009), Charlson comorbidity indexes (4 [1–5] vs 6 [5–9]; p = 0.002) and lactate levels (1.4 [0.85–2.2] vs 2.8 [1.6–2.8] mmol/L; p = 0.02). Survivors had longer length of hospital stays (36 [24–48] vs 14 [8–28] days; p = 0.003) (Table 1).
Differential expression of M-MDSCs and PMN-MDSCs in sepsis patients
We used a targeted flow cytometry approach combined to unsupervised automatic clustering to identify M-MDSCs and PMN-MDSCs in whole blood (see “Methods” and Fig. 1). M-MDSCs and PMN-MDSCs represented 0.96% [0.46–1.5] and 2.1% [0.7–3.1] of leukocytes in healthy individuals (n = 18), respectively. Considering all sepsis patients analyzed at study inclusion, M-MDSCs and PMN-MDSCs represented 3.1% [2.04–4.85] and 22% [7.9–43.0] of leukocytes (p < 10–4 vs healthy individuals) (Additional file 1: Table S2). The percentages and absolute counts of M-MDSCs and PMN-MDSCs remained elevated and were not statistically significantly altered over a 10-day follow-up period (Additional file 2: Fig. S2). Therefore, subsequent analyses were performed using the levels of M-MDSCs and PMN-MDSCs measured at study inclusion.

Identification of MDSCs by flow cytometry and unsupervised clustering. Blood was collected in tubes containing lyophilized fluorescently labeled antibodies targeting MDCSs and analyzed as described in “Methods”. t-SNE plots of leukocyte populations (left) and relative side scatter area (SSC-A) and expression levels of surface markers (right). Lin+: lineage (i.e., CD3, CD19 or CD56) positive; DCs: dendritic cells
The percentages of M-MDSCs at study inclusion showed a trend towards lower levels in 90-day non-survivors when compared to survivors (2.9% [1.8–4.1] vs 4.6% [2.6–6.5]; p = 0.052) (Table 1). The percentages of M-MDSCs did not correlate with the percentages of PMN-MDSCs (Spearman’s correlation coefficient [ρ] = − 0.003, p = 0.98) (Fig. 2A). M-MDSCs inversely correlated with lactate levels (ρ = − 0.43, p = 0.002), IL-6 levels, (ρ = − 0.29, p = 0.045), and ferritin levels (ρ = − 0.32, p = 0.028) (Fig. 2A), while PMN-MDSCs inversely correlated with CRP levels (ρ = − 0.39, p = 0.047). M-MDSCs but not PMN-MDSCs inversely correlated with the age of sepsis patients, while no such correlation was observed in the group of healthy controls for both M-MDSCs and PMN-MDSCs (Fig. 2B and Additional file 2: Fig. S3).

A Correlation plot matrix of M-MDSCs, PMN-MDSCs, CRP, leukocytes, lactate, IL-6 and ferritin levels. *M-MDSCs inversely correlated with lactate (ρ = − 0.43, p = 0.002), IL-6, (ρ = − 0.29, p = 0.045), and ferritin (ρ = − 0.32, p = 0.028), and PMN-MDSCs inversely correlated with CRP (ρ = − 0.39, p = 0.047). B Scatterplots of M-MDSCs (% of leukocytes) and age in healthy controls (left) and sepsis patients (right)
Twenty-six (66.7%) patients developed a secondary infection, among which 23 (47%) were associated with a new sepsis episode (see definitions in Additional file 1: Table S1). Patients who developed or not a secondary infection were similar in age and gender, had similar APACHE II and SOFA scores at admission, and comparable 90-day mortality rates. However, patients who developed a secondary infection stayed 3.6- to 4.1-fold longer in hospital (33 [27–43] vs 8 [5–12] days; p < 0.0001) and ICUs (29 [20–36] vs 8 [5–10] days; p < 0.0001) and had lower CRP levels (75 [27–144] vs 164 [130–194] mg/L; p = 0.003) than patients who did not develop a secondary infection (Table 2). Moreover, patients who developed a secondary infection presented higher levels of PMN-MDSCs than patients that did not develop a secondary infection (31% [13–46] vs 11% [7–26]; p = 0.03) and new sepsis episode (33% [14–45] vs 11% [7–26]; p = 0.04) (Fig. 3A). Besides, patients whose mortality was related to secondary sepsis/infection expressed 3.7-fold higher levels of PMN-MDSCs than patients whose mortality was related to the primary sepsis event (36% [26–49] vs 9.8% [7.3–14.5] of leukocytes, p = 0.0021) (Additional file 2: Fig. S4). No difference in M-MDSCs was observed for all these parameters. PMN-MDSCs and M-MDSCs were similarly represented in patients with documented Gram-negative (n = 23) and Gram-positive (n = 8) infections.

A PMN-MDSCs in relation with secondary infection and new sepsis episode. B M-MDSCs and PMN-MDCs in survivors (n = 14), early deaths (≤ 28 days, n = 23) and late deaths (> 28 days, n = 12). Boxplots show median, upper and lower quartiles. Whiskers show 5 to 95 percentiles. Each dot represents an individual sample
Survivors and early and late deaths (i.e., ≤ 28 and > 28 days) expressed similar levels of PMN-MDSCs (Fig. 3B). In contrast, survivors expressed 1.64-fold more M-MDSCs than early deaths (4.6% [2.6–6.7] vs 2.8% [1.5–3.6], p = 0.028) and, albeit not significant, 1.55-fold more M-MDSCs than late deaths (3.0% [2.3–4.7], p = 0.19) (Fig. 3B). Subsequently, we stratified patients according to the expression of M-MDSCs (low and high levels: ≤ 4.3% and > 4.3%) and the APACHE II score (low and high: ≤ 20 and > 20) (see “Methods”).
Ninety-day mortality was decreased in patients with high levels of M-MDSCs (high vs low MDSCs: 47% vs 84% mortality, p = 0.007, hazard ratio [HR] = 3.2, 95% confidence interval [95% CI] 1.4–7.2) (Table 2 and Fig. 4A), while hospital stay was increased (high vs low MDSCs: 33 [12–44] vs 16 [8–28] days; p = 0.04) (Table 2). Patients with high levels of M-MDSCs showed reduced leukocytes counts, lactate levels and ferritin levels at admission (p = 0.04, 0.01 and 0.03, respectively) (Table 2).

Kaplan–Meier 90-day survival curves based on low and high levels of M-MDSCs (≤ 4.3% and > 4.3% of leukocytes) (A) and on the combination of low and high levels of MDSCs and low and high APACHE II scores (≤ 20 and > 20) (B). Statistical differences were assessed using the log-rank test
Combining M-MDSCs (low and high levels) and APACHE II score (low and high) in analyses increased patient stratification. The 90-day mortality rate was 20% in patients with high M-MDSCs and low APACHE II score, while it was 71–88% in the three other groups (overall comparison: p = 0.0062; M-MDSCshigh/APACHE IIlow vs M-MDSCslow/APACHE IIlow: 20% vs 80%, p = 0.0096, HR = 7.2, 95% CI 1.6–32) (Fig. 4B). In multivariate analyses including baseline factors associated with mortality (age, APACHE II score, Charlson comorbidity index), high M-MDSCs remained associated with improved survival in patients with low APACHE II score (p = 0.05, HR = 5.26, 95% CI 1.0–27.8) (Table 3).
Discussion
To our knowledge, this is the first study reporting that high expression levels of M-MDSCs are associated with improved outcome of sepsis patients with pneumonia.
M-MDSCs and PMN-MDSCs remained stably elevated during 10 days of follow-up. These data corroborate the persistence of MDSCs for 14 to 28 days in sepsis and ICU surgical patients, and of M-MDSCs and PMN-MDSCs for 8 days in sepsis patients [23, 27,28,29]. M-MDSCs and PMN-MDSCs were similarly expressed in patients with Gram-negative and Gram-positive infections, while few studies reported the preferential expansion of M-MDSCs and PMN-MDSCs in Gram-negative infections and Gram-positive infections, respectively [22, 25]. Thus, while chronical elevation of MDSCs may be an attractive biomarker for sepsis [9, 26, 32], additional studies will be required to outline whether the expansion of specific subpopulations of MDSCs in sepsis results from different kinds of infections. We did not detect a correlation between the levels of M-MDSCs and PMN-MDSCs. This might suggest that, under pathological conditions, these populations result from different hematopoietic drivers [33].
Experimental investigations and all clinical studies to date suggested that MDSCs are detrimental during sepsis [7, 22,23,24,25,26,27,28,29,30,31]. High levels of MDSCs at admission correlated with early mortality of surgical septic shock patients [23], and high levels of M-MDSCs on days 6–8 correlated with mortality and secondary infections in septic shock patients [27]. Persistent expansion of MDSCs might be implicated in the establishment of persistent inflammation, immunosuppression and catabolism syndrome (PICS) observed in a subset of chronic critically ill patients (CCI) who experienced sepsis [34]. In our cohort, high levels of PMN-MDSCs were not associated with mortality, but were associated with the occurrence of secondary infections and new sepsis episodes. It is possible that either an association with mortality was missed because of sample size, or that the absence of association was genuine. In line with the second option, a recent study failed to detect an association between mortality and the expansion of PMN-MDSCs in blood sampled from sepsis patients at ICU admission and 3 days later [35].
Somehow astonishing, no human study ever reported a positive role of MDSCs during sepsis. Yet, previous studies included patients with diverse infection etiologies and medical care (ED, medical/surgical ICUs), while we enrolled ICU patients with severe ventilator, hospital, and healthcare-associated pneumosepsis. We cannot exclude that a beneficial role of M-MDSCs is restricted to this kind of patients. In the same line of idea, patients with sepsis caused by CAP displayed a specific blood gene expression signature on ICU admission [36]. Moreover, sepsis patients with pneumonia were differentially distributed into Mars1–4 endotypes than sepsis patients with peritonitis [37]. Therefore, the site of infection and/or surgical intervention may affect blood gene expression profile, reflecting different cellular fates.
One could imagine that MDSCs play a dual role during sepsis. Although hypothetical, this supposition is founded on several facts. First, MDSCs are phagocytic cells, which can help fighting infections through ingestion and killing of microorganisms. Second, MDSCs can dampen systemic or local inflammation induced by molecular patterns of pathogen or endogenous origin, the latter being released upon stress or during tissue injury. Third, MDSCs harvested from septic mice protected recipient mice from acute lethal infections including cecal ligation and puncture-induced sepsis and Pseudomonas pneumonia [38,39,40,41]. Remarkably, we observed that high levels of M-MDSCs were associated with reduced 90-day mortality in sepsis patients with multi-organ failure and high likelihood of mortality. Increased M-MDSCs might drive beneficial effects through dampening inflammation-induced organ dysfunction in severely ill sepsis patients. Of importance, high levels of M-MDSCs were not beneficial in patients with a high APACHE II score at study enrollment. Albeit speculative, a possible explanation is that these patients were on a trajectory more or less invariably conducting to death, and that the influence of M-MDSCs in those conditions was negligible.
As observed in other conditions, PMN-MDSCs were more abundant than M-MDSCs in sepsis patients (and healthy controls). It has been shown that M-MDSCs are more potent immunosuppressive cells than PMN-MDSCs on a per cell basis [26]. This may explain, at least in part, how a minor subpopulation of MDSCs may have a significant impact. Supporting the concept of MDSCs as inflammatory brakes, M-MDSCs negatively correlated with lactate, IL-6 and ferritin levels, and PMN-MDSCs with CRP levels. M-MDSCs and PMN-MDSCs mediate immunosuppressive functions through different mechanisms involving, for example the expression of IL-10, transforming growth factor β, nitric oxide and programmed death-ligand 1 (PDL1) by M-MDSCs and the expression of arginase 1 by PMN-MDSCs. PMN-MDSCs may also preferentially use reactive oxygen species, peroxynitrite and possibly prostaglandin E2 to drive immunosuppression [14, 42,43,44,45,46].
Several factors may explain discrepancies reported in the literature such as the causative agent and site of infection, the inflammatory status which impacts on myelopoiesis and the generation of MDSCs, the timing of blood sampling and downstream treatment, or the immunophenotyping of MDSCs [48]. Indeed, both mouse and human studies revealed that MDSCs evolve after sepsis onset, acquiring superior suppressive functions over time [29, 47]. A pitfall of (early) studies on MDSCs is the lack of harmonization of sample handling, and eventually the lack of discrimination of MDSC subpopulations. For instance, MDSCs are better detected in whole blood than in PBMCs [49]. PMN-MDSCs but not M-MDSCs are sensitive to freezing/thawing of PBMCs, while M-MDSCs are more sensitive than PMN-MDSCs to delayed blood processing [50, 51]. Even though using a consensus protocol, the multicenter Mye-EUNITER MDSC Monitoring Initiative reported important center-related differences in PMN-MDSCs detection in the blood healthy donors [52]. To minimize analytical variations, we labeled whole blood immediately after drawing using DURAClone tubes, and used unsupervised clustering strategies to analyze flow cytometry data. However, as often inevitable in multicenter studies, samples were cryopreserved before analysis. Finally, we acknowledge that there is still no definite perfect phenotyping protocol of MDSCs. Unbiased transcriptomics and unsupervised flow and mass cytometry might help identifying new markers of MDSCs, such as LDL receptor 1 (LOX-1) expressed by PMN-MDSCs [53,54,55].
Our study has several limitations including the characterization of MDSCs by phenotypic and not functional analyses, and the absence of immunological correlates. Yet, several studies reported the immunosuppressive function of MDSCs based on their phenotype [22, 25, 56]. The sample size may have affected the detection of associations between MDSCs and sepsis parameters. We focused on patients with sepsis due to pneumonia, while the role of M-MDSCs may vary in different disease processes. Patients were aged, and our observations may not be verified in a younger population. Aging is a condition that might influence MDSCs [57]. However, contrary to expectations, we detected a negative correlation between age and M-MDSCs in sepsis patients. As recently argued, it might be difficult to differentiate an increase of MDSCs to due aging (as a consequence of inflammageing) from that due to disease-mediated expansion [58]. Finally, the risk of mortality in our study population was high (71% of patients died within 90 days). However, poor outcome likely represented a favorable condition to detect a positive role of MDSCs in sepsis.
Conclusions
This represents the first report of an association between high levels of M-MDSCs and improved outcome of patients with pneumosepsis. We believe that these observations should provide impetus for additional studies to appreciate the role of MDSCs in patients with severe sepsis and multi-organ failure, and for deciphering the mechanisms regulating the expansion and the activation of MDSCs in bacterial sepsis. Such investigations will be required to assess whether MDSCs are prognostic and/or theragnostic biomarkers in sepsis.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author on reasonable request. Restrictions apply to the availability of data associated with the INCLASS study, which is not finalized (contact Prof E.J. Giamarellos-Bourboulis).
Abbreviations
- APACHE II:
-
Acute Physiology and Chronic Health Evaluation II
- CRP:
-
C-reactive protein
- HAP:
-
Hospital-acquired pneumonia
- HCAP:
-
Healthcare-associated pneumonia
- HR:
-
Hazard ratio
- IL-6:
-
Interleukin-6
- IQR:
-
Interquartile range
- MDSCs:
-
Myeloid-derived suppressor cells
- PMN-MDSCs:
-
Polymorphonuclear-MDSCs
- M-MDSCs:
-
Monocytic myeloid-derived suppressor cells
- SOFA:
-
Sequential Organ Failure Assessment
- VAP:
-
Ventilator-associated pneumonia
- 95% CI:
-
95% Confidence interval
References
Singer M, Deutschman CS, Seymour CW et al (2016) The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315:801–810. https://doi.org/10.1001/jama.2016.0287
Rudd KE, Johnson SC, Agesa KM et al (2020) Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395:200–211. https://doi.org/10.1016/S0140-6736(19)32989-7
Prescott HC, Angus DC (2018) Enhancing recovery from sepsis: a review. JAMA 319:62–75. https://doi.org/10.1001/jama.2017.17687
Shankar-Hari M, Saha R, Wilson J et al (2020) Rate and risk factors for rehospitalisation in sepsis survivors: systematic review and meta-analysis. Intensive Care Med 46:619–636. https://doi.org/10.1007/s00134-019-05908-3
van der Poll T, Shankar-Hari M, Wiersinga WJ (2021) The immunology of sepsis. Immunity 54:2450–2464. https://doi.org/10.1016/j.immuni.2021.10.012
van der Poll T, van de Veerdonk FL, Scicluna BP et al (2017) The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol 17:407–420. https://doi.org/10.1038/nri.2017.36
Venet F, Monneret G (2018) Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol 14:121–137. https://doi.org/10.1038/nrneph.2017.165
Torres LK, Pickkers P, van der Poll T (2021) Sepsis-induced immunosuppression. Annu Rev Physiol. https://doi.org/10.1146/annurev-physiol-061121-040214
Venet F, Demaret J, Gossez M et al (2021) Myeloid cells in sepsis-acquired immunodeficiency. Ann N Y Acad Sci 1499:3–17. https://doi.org/10.1111/nyas.14333
Rubio I, Osuchowski MF, Shankar-Hari M et al (2019) Current gaps in sepsis immunology: new opportunities for translational research. Lancet Infect Dis 19:e422–e436. https://doi.org/10.1016/S1473-3099(19)30567-5
Hotchkiss RS, Moldawer LL, Opal SM et al (2016) Sepsis and septic shock. Nat Rev Dis Primers 2:16045. https://doi.org/10.1038/nrdp.2016.45
Stanski NL, Wong HR (2020) Prognostic and predictive enrichment in sepsis. Nat Rev Nephrol 16:20–31. https://doi.org/10.1038/s41581-019-0199-3
Peters van Ton AM, Kox M, Abdo WF et al (2018) Precision immunotherapy for sepsis. Front Immunol 9:1926. https://doi.org/10.3389/fimmu.2018.01926
Veglia F, Sanseviero E, Gabrilovich DI (2021) Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 21:485–498. https://doi.org/10.1038/s41577-020-00490-y
Talmadge JE, Gabrilovich DI (2013) History of myeloid-derived suppressor cells. Nat Rev Cancer 13:739–752. https://doi.org/10.1038/nrc3581
Goldmann O, Beineke A, Medina E (2017) Identification of a novel subset of myeloid-derived suppressor cells during chronic staphylococcal infection that resembles immature eosinophils. J Infect Dis 216:1444–1451. https://doi.org/10.1093/infdis/jix494
Bronte V, Brandau S, Chen SH et al (2016) Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 7:12150. https://doi.org/10.1038/ncomms12150
Gabrilovich DI (2017) Myeloid-derived suppressor cells. cancer. Immunol Res 5:3–8. https://doi.org/10.1158/2326-6066.CIR-16-0297
Ugel S, De Sanctis F, Mandruzzato S et al (2015) Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 125:3365–3376. https://doi.org/10.1172/JCI80006
Law AMK, Valdes-Mora F, Gallego-Ortega D (2020) Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. https://doi.org/10.3390/cells9030561
Grover A, Sanseviero E, Timosenko E et al (2021) Myeloid-derived suppressor cells: a propitious road to clinic. Cancer Discov 11:2693–2706. https://doi.org/10.1158/2159-8290.CD-21-0764
Janols H, Bergenfelz C, Allaoui R et al (2014) A high frequency of MDSCs in sepsis patients, with the granulocytic subtype dominating in gram-positive cases. J Leukoc Biol 96:685–693. https://doi.org/10.1189/jlb.5HI0214-074R
Mathias B, Delmas AL, Ozrazgat-Baslanti T et al (2017) Human myeloid-derived suppressor cells are associated with chronic immune suppression after severe sepsis/septic shock. Ann Surg 265:827–834. https://doi.org/10.1097/SLA.0000000000001783
Patera AC, Drewry AM, Chang K et al (2016) Frontline science: defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol 100:1239–1254. https://doi.org/10.1189/jlb.4HI0616-255R
Uhel F, Azzaoui I, Gregoire M et al (2017) Early expansion of circulating granulocytic myeloid-derived suppressor cells predicts development of nosocomial infections in patients with sepsis. Am J Respir Crit Care Med 196:315–327. https://doi.org/10.1164/rccm.201606-1143OC
Schrijver IT, Theroude C, Roger T (2019) Myeloid-derived suppressor cells in sepsis. Front Immunol 10:327. https://doi.org/10.3389/fimmu.2019.00327
Waeckel L, Venet F, Gossez M et al (2020) Delayed persistence of elevated monocytic MDSC associates with deleterious outcomes in septic shock: a retrospective cohort study. Crit Care 24:132. https://doi.org/10.1186/s13054-020-02857-y
Coudereau R, Waeckel L, Cour M et al (2021) Emergence of immunosuppressive LOX-1+ PMN-MDSC in septic shock and severe COVID-19 patients with acute respiratory distress syndrome. J Leukoc Biol. https://doi.org/10.1002/JLB.4COVBCR0321-129R
Hollen MK, Stortz JA, Darden D et al (2019) Myeloid-derived suppressor cell function and epigenetic expression evolves over time after surgical sepsis. Crit Care 23:355. https://doi.org/10.1186/s13054-019-2628-x
Darcy CJ, Minigo G, Piera KA et al (2014) Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit Care 18:R163. https://doi.org/10.1186/cc14003
Guerin E, Orabona M, Raquil MA et al (2014) Circulating immature granulocytes with T-cell killing functions predict sepsis deterioration*. Crit Care Med 42:2007–2018. https://doi.org/10.1097/CCM.0000000000000344
Sayyadioskoie SR, Schwacha MG (2021) Myeloid-derived suppressor cells (MDSCs) and the immunoinflammatory response to injury (mini review). Shock 56:658–666. https://doi.org/10.1097/SHK.0000000000001795
Mastio J, Condamine T, Dominguez G et al (2019) Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J Exp Med 216:2150–2169. https://doi.org/10.1084/jem.20181952
Mira JC, Brakenridge SC, Moldawer LL et al (2017) Persistent inflammation, immunosuppression and catabolism syndrome. Crit Care Clin 33:245–258. https://doi.org/10.1016/j.ccc.2016.12.001
De Zuani M, Hortova-Kohoutkova M, Andrejcinova I et al (2021) Human myeloid-derived suppressor cell expansion during sepsis is revealed by unsupervised clustering of flow cytometric data. Eur J Immunol 51:1785–1791. https://doi.org/10.1002/eji.202049141
Scicluna BP, Klein Klouwenberg PM, van Vught LA et al (2015) A molecular biomarker to diagnose community-acquired pneumonia on intensive care unit admission. Am J Respir Crit Care Med 192:826–835. https://doi.org/10.1164/rccm.201502-0355OC
Scicluna BP, van Vught LA, Zwinderman AH et al (2017) Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med 5:816–826. https://doi.org/10.1016/S2213-2600(17)30294-1
Sander LE, Sackett SD, Dierssen U et al (2010) Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med 207:1453–1464. https://doi.org/10.1084/jem.20091474
Chang S, Kim YH, Kim YJ et al (2018) Taurodeoxycholate increases the number of myeloid-derived suppressor cells that ameliorate sepsis in mice. Front Immunol 9:1984. https://doi.org/10.3389/fimmu.2018.01984
Namkoong H, Ishii M, Fujii H et al (2018) Clarithromycin expands CD11b+Gr-1+ cells via the STAT3/Bv8 axis to ameliorate lethal endotoxic shock and post-influenza bacterial pneumonia. PLoS Pathog 14:e1006955. https://doi.org/10.1371/journal.ppat.1006955
Derive M, Bouazza Y, Alauzet C et al (2012) Myeloid-derived suppressor cells control microbial sepsis. Intensive Care Med 38:1040–1049. https://doi.org/10.1007/s00134-012-2574-4
Nagaraj S, Gupta K, Pisarev V et al (2007) Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 13:828–835. https://doi.org/10.1038/nm1609
Rastad JL, Green WR (2016) Myeloid-derived suppressor cells in murine AIDS inhibit B-cell responses in part via soluble mediators including reactive oxygen and nitrogen species, and TGF-beta. Virology 499:9–22. https://doi.org/10.1016/j.virol.2016.08.031
Ohl K, Tenbrock K (2018) Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front Immunol 9:2499. https://doi.org/10.3389/fimmu.2018.02499
Wang D, DuBois RN (2016) The role of prostaglandin E(2) in tumor-associated immunosuppression. Trends Mol Med 22:1–3. https://doi.org/10.1016/j.molmed.2015.11.003
Krishnamoorthy M, Gerhardt L, Maleki Vareki S (2021) Immunosuppressive effects of myeloid-derived suppressor cells in cancer and immunotherapy. Cells. https://doi.org/10.3390/cells10051170
Brudecki L, Ferguson DA, McCall CE et al (2012) Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun 80:2026–2034. https://doi.org/10.1128/IAI.00239-12
Gabrilovich DI (2014) Editorial: The intricacy of choice: can bacteria decide what type of myeloid cells to stimulate? J Leukoc Biol 96:671–674. https://doi.org/10.1189/jlb.4CE0514-271R
Apodaca MC, Wright AE, Riggins AM et al (2019) Characterization of a whole blood assay for quantifying myeloid-derived suppressor cells. J Immunother Cancer 7:230. https://doi.org/10.1186/s40425-019-0674-1
Grutzner E, Stirner R, Arenz L et al (2016) Kinetics of human myeloid-derived suppressor cells after blood draw. J Transl Med 14:2. https://doi.org/10.1186/s12967-015-0755-y
Kotsakis A, Harasymczuk M, Schilling B et al (2012) Myeloid-derived suppressor cell measurements in fresh and cryopreserved blood samples. J Immunol Methods 381:14–22. https://doi.org/10.1016/j.jim.2012.04.004
Cassetta L, Bruderek K, Skrzeczynska-Moncznik J et al (2020) Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J Immunother Cancer. https://doi.org/10.1136/jitc-2020-001223
Condamine T, Dominguez GA, Youn JI et al (2016) Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. https://doi.org/10.1126/sciimmunol.aaf8943
Darden DB, Bacher R, Brusko MA et al (2021) Single-cell RNA-seq of human myeloid-derived suppressor cells in late sepsis reveals multiple subsets with unique transcriptional responses: a pilot study. Shock 55:587–595. https://doi.org/10.1097/SHK.0000000000001671
Roussel M, Ferrell PB Jr, Greenplate AR et al (2017) Mass cytometry deep phenotyping of human mononuclear phagocytes and myeloid-derived suppressor cells from human blood and bone marrow. J Leukoc Biol 102:437–447. https://doi.org/10.1189/jlb.5MA1116-457R
Damuzzo V, Pinton L, Desantis G et al (2015) Complexity and challenges in defining myeloid-derived suppressor cells. Cytom B Clin Cytom 88:77–91. https://doi.org/10.1002/cyto.b.21206
Verschoor CP, Johnstone J, Millar J et al (2013) Blood CD33(+)HLA-DR(-) myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol 93:633–637. https://doi.org/10.1189/jlb.0912461
Pawelec G, Picard E, Bueno V et al (2021) MDSCs, ageing and inflammageing. Cell Immunol 362:104297. https://doi.org/10.1016/j.cellimm.2021.104297
Acknowledgements
The authors thank the patients, families and clinical staff who contributed to the study.
Funding
This work was supported by grants from the Swiss National Science Foundation (SNSF) Grant number 310030_173123 to TR and the Hellenic Institute for the Study of Sepsis to EJGB. EJGB, TC and TR were supported by the European Sepsis Academy Horizon 2020 Marie Skłodowska-Curie Action: Innovative Training Network (MSCA-ESA-ITN, grant number 676129). ITS and CT received a scholarship from the Société Académique Vaudoise (Lausanne, Switzerland).
Author information
Authors and Affiliations
Contributions
ITS, EK, EJGB, TC and TR conceptualized and designed the study. ITS, PB, AH and TR developed custom-made DURAClone tubes. EK and EJGB designed the clinical study and organized blood sampling. ITS and CT analyzed the samples. All the authors interpreted the data. ITS and TR wrote the manuscript. All the authors revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in compliance with the declaration of Helsinki, and was approved by the central Ethics committee (52086/2017) and the National organization for Medicines-EOF (51239/01-06-2017) in Athens, Greece, and by the Commission cantonale d'éthique de la recherche sur l'être humain, Canton de Vaud, Switzerland (CER-VD) in Lausanne, Switzerland. Written informed consent was obtained from study participants or legal representatives prior to enrollment.
Consent for publication
Not applicable.
Competing interests
The authors do not have any conflict of interest regarding this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Definitions used in the study. Table S2. MDSCs in healthy subjects and sepsis patients.
Additional file 2: Fig. S1.
Gating strategy to exclude doublets and non-hematopoietic (CD45-) cells. Fig. S2. M-MDSCs and PMN-MDSCs expressed in % of leukocytes and absolute counts in healthy controls, and in sepsis survivors and non-survivors analyzed at days 1 (study inclusion), 5 and 10. Boxplots show median, upper and lower quartiles. Whiskers show 5 to 95 percentiles. Each dot represents an individual sample. No significant differences were detected in longitudinal analyses. Fig. S3. Scatterplots of PMN-MDSCs and age in healthy controls (left) and sepsis patients (right). Fig. S4. MDSCs (in % of leukocytes) in relation with the cause of 90-day mortality (primary sepsis related mortality n = 13; due to secondary infection/sepsis n = 16; other causes n = 6). Boxplots show median, upper and lower quartiles. Whiskers show 5 to 95 percentiles. Each dot represents an individual sample.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schrijver, I.T., Karakike, E., Théroude, C. et al. High levels of monocytic myeloid-derived suppressor cells are associated with favorable outcome in patients with pneumonia and sepsis with multi-organ failure. ICMx 10, 5 (2022). https://doi.org/10.1186/s40635-022-00431-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40635-022-00431-0