
Abstract
Results of our ab initio calculations of 〈111〉-oriented GaP, GaAs, GaSb, InP, InAs and InSb nanowires with the zinc-blende structure indicate morphology to crucially affect their electronic properties. For these nanowires, where {011} facets characterize their hexagonal cross section, the formation of small {112} facets between the adjacent {011} ones provides a more stable structure and removes surface states from the gap region even without hydrogen passivation. Our new structural model also predicts a crossover between the indirect and direct band gap in GaP, GaAs and GaSb nanowires when increasing diameters starting from 4 nm, while InP, InAs and InSb nanowires display the direct band gap at diameters of 1.5 nm and larger. Analysis of charge distribution between atoms suggests that {011} facets are positively charged even though a (011) surface of these materials is considered to be non-polar.
Similar content being viewed by others

1 Background
In small diameter III–V nanowires (NWs), where the surface-to-volume ratio is rather large, one can expect surface effects in addition to effects caused by quantum confinement to govern properties of these NWs. It is also important to trace a link between morphology and different properties of such NWs in order to open a way to their integration in various applications [1,2]. Even though in most cases III–V compounds have the zinc-blende structure, in the case of NWs both the zinc-blende and wurtzite structures are observed [2] because of larger density of surface dangling bonds for the zinc-blende structure as confirmed by first principles calculations [3-10]. However, high quality NWs in the zinc-blende structure mainly oriented along the 〈111〉 directions can be grown by different methods involving the vapor-liquid-solid and vapor-solid growth mechanisms [1,2,11]. It is found that their cross section is hexagonal with {011} or {112} facets [12-20], while a triangular-like morphology with {112} facets can also occur [15-17,21]. Theoretical predictions on stability of zinc-blende GaP, GaAs, InP and InAs NWs showed that they were close in total energy independently of morphology [3-10]. Moreover, investigations of electronic properties of bare GaAs, InP and InAs NWs, which had only {011} facets, indicated metallic properties because the Fermi level crossed some bands originated from states of surface atoms at edges [3,5,10,22]. However, passivation of dangling bonds by different chemical species was widely used to investigate a band-gap variation with NW diameter [7,8,22-27], while bare zinc-blende NWs with {112} facets and any bare wurtzite NW turned out to be semiconductors without passivation [3,5,9,10].
In this paper by means of ab initio calculations we show that the appearance of small {112} facets acting as edges between the adjacent {011} facets in zinc-blende GaP, GaAs, GaSb, InP, InAs and InSb NWs provides lowering in total energy and eliminates bands in the gap region leading to semiconducting properties.
2 Methods
The structural optimization and band structure calculations of zinc-blende III–V NWs have been performed by the total energy projector-augmented wave method (code VASP) [28]. Exchange and correlation potentials were included using the local density approximation of Ceperly and Alder [29]. We considered 〈111〉-oriented NWs with hexagonal cross sections involving {011} facets, while periodic boundary conditions were applied along the NW axis with the unit cell parameter (a ∥). To provide a negligible interaction between neighboring NWs at least 9 Å of vacuum were introduced. The further increasing in the vacuum thickness did not noticeably affect the total energy. All atoms in NWs were allowed to relax. We set the energy cutoff at 250 eV. The grid of 1 ×1×6 Monkhorst-Park points was found to be sufficient to assure total energy convergence with respect to number of k-points. Atomic relaxation was stopped when forces on the atoms were smaller than 0.04 eV/Å. The optimization of a ∥ was done by gradually increasing/decreasing its value along with the relaxation of the atomic positions till the equilibrium was reached. The bulk lattice parameters (a bulk ) summarized in Table 1 are very close to the experimental values. The charge transfers have been analyzed by utilizing the Bader method [30]. Since we did not perform quasiparticle calculations within the GW-approximation, the estimated gaps in our cases were smaller that experimental values for both bulk and NWs.
3 Results and discussion
3.1 3.1 Morphology and stability
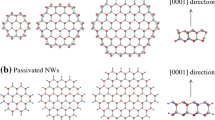
Possible cross sections of 〈111〉-oriented zinc-blende III–V NWs can be viewed as bounded {011} facets in the hexagonal shape (Figure 1, the top panel). Morphology without small {112} facets was used in all previous calculations [3-10,22-27], where three corner V and three corner III atoms at different edges had three and one dangling bands, respectively. However, the appearance of small {112} facets acting as edges between adjacent {011} facets by removing a pair of corner atoms was shown to be preferable in total energy for silicon NWs with 〈111〉 axes since it was possible to eliminate surface atoms with two dangling bonds by forming pentagon-like structures with dimers [31]. Contrary to silicon NWs, where all six {112} facets are equivalent, in the case of III–V NWs a half of {112} facets displays V–V dimers and no V atom with three dangling bonds, while another half has III–III dimers with virtually no dangling bonds. The V–V dimers are clearly seen in Figure 1, whereas atoms forming the III–III dimers show sizable inward relaxation leading to a slight incline of {011} facets towards {112} facets with V–V dimers. Dimer lengths are comparable to the corresponding III–V interatomic distances (Table 1) indicating a well-resolved correlation with the dimer composition (for example, d P−P is almost the same for GaP and InP NWs, while d G a−G a is very close for GaP, GaAs and GaSb NWs). The main structural features indicated in the cross sections (Figure 1, the top panel) are essentially the same for GaP, GaAs, GaSb, InP, InAs and InSb NWs regardless of diameter (in our case from 1.5 to 4.0 nm). Dimer lengths (Table 1) do not change with respect to NW diameter. We have also found a ∥ of all considered NWs to be slightly smaller than the initial value corresponding to the bulk case and it gets closer to the bulk value when diameter of a NW increases.

The top panel: cross sections of 〈111〉-oriented III–V NWs without (left) and with (right) small {112} facets and diameters of about 2.6 nm (the case of GaAs is shown) after full structural optimization. All facets are indicated. Pink and yellow spheres stand for III and V atoms, respectively. The bottom panel: the dependence of the total energy of GaAs NWs per the Ga-As formula unit on the number of formula units in the unit cell. The number of formula units is proportional to NW diameter. Zero at the energy scale corresponds to the energy of the GaAs formula unit in the bulk.
Now it is obvious that the formation of small {112} facets in III–V NWs decreases density of dangling bonds with respect to NWs without {112} facets because some V atoms at edges with multiple dangling bonds are eliminated. This issue, in turn, provides a clear lowering in total energy for all considered here NWs as can be seen in Figure 1, the bottom panel, for GaAs NWs.
We have estimated charge distribution between atoms in the III–V bulks and NWs. For the bulks of phosphides and arsenides the III atoms donate and the V atoms accept of about 0.53 – 0.58 e − (Table 1), while for the antimonide bulks the charge transfer is less (0.12 – 0.20 e −). In the case of NWs of phosphides and arsenides the effective atomic charges are essentially the same as for the corresponding bulks, while main differences can only be spotted for atoms at surface and especially for atoms forming dimers (Table 1) and back-bonds to the dimer atoms. For antimonide NWs a larger span of charge distribution than in the corresponding bulks (by 0.2 e −) is found.
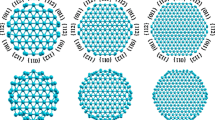
Since for 〈111〉-oriented III–V NWs any III–V atomic pair located between double layers is parallel to the NW axis, consists of the first neighbor atoms and does not coincide with any other pair within the unit cell in the plane of the cross section, we have summed up individual atomic charges for each III–V pair providing information about charge distribution projected to the NW cross section (Figure 2). We have assumed a III–V pair to be positively or negatively charged if the total charge per pair is respectively less or larger 8 e − (any III or V atom has three or five valence electrons, respectively). It is evident that {011} facets are positively charged, while {112} facets attract a negative charge. To neutralize the charge accumulated on the surface of NWs, the subsurface region gains a negative charge, while the inner core region has a mixture of positively and negatively charged III–V pairs (Figure 2). Such character of charge distribution is independent of NW composition and diameter. Even though the (011) surface is believed to be non-polar, in the case of considered III–V NWs the {011} facets get a positive charge, which is rather small: 0.04 – 0.06 e − per III–V pair except for some corner pairs with 0.14 – 0.20 e −. Thus, for a III–V NW with diameter of 4 nm (Figure 2) a positive charge of about 1 e − can be accumulated on each {011} facet if a NW length is equal to a double unit cell along the NW axis. The observed surface charge accumulation can affect parameters and performance of field effect transistors on III–V NWs [32].

Projection of charge distribution on the plane of the cross section for III–V NWs with small {112} facets and largest considered diameter of about 4 nm.
3.2 3.2 Morphology and band structure
All band structures of GaP, GaAs, GaSb, InP, InAs and InSb NWs without {112} facets and with different diameters indicate metallic nature because the Fermi level crosses several bands (Figure 3, top panel). These bands originate mainly from p-states of III and V atoms with dangling bonds located at the edges between adjacent {011} facets. The same issue was observed in previous theoretical calculations [3,5,10,22]. In addition to the fact that III–V NWs with small {112} facets are predicted to be more stable (Figure 1), these NWs are semiconductors because the Fermi level is in the band gap (Figure 3, the bottom panel). Moreover, GaP and GaAs NWs have a competitive character of the gap: the lowest in energy conduction band has two minima in the Γ and Z points, which are almost equal in energy, while the maximum of the valence band is in the Γ point. The GaSb NW is characterized by the indirect band gap since the minimum of the conduction band is in the Z point and the maximum of the valence band can be found in the Γ point. Besides a band-gap reduction due to quantum confinement effect attenuation when increasing diameter of GaP, GaAs and GaSb NWs, we have revealed a crossover to the direct band gap. Thus, at diameters of about 4 nm these NWs show the first direct transition in the Γ point (Figure 3, the bottom panel) because the second minimum of the conduction band in the Z point goes up in energy. At the same time, InP, InAs and InSb NWs independently of diameter have the direct band gap displaying the first direct transition in the Γ point. Dispersion of the top valence band and bottom conduction band of hydrogenated GaP [24], GaSb [23], InP [8,26] and InAs [23] NWs is different from that of the bare NWs presented in Figure 3 because hydrogen passivation leads to the well-resolved maximum of the valence band and minimum of the conduction band (both in the Γ point), which are characterized by s- and p-states of atoms belonging to the inner part of NWs. Moreover, hydrogenated GaP [24] and GaSb [23] NWs always have the direct band gap even at diameters less than 4 nm, while a crossover from direct transition to the indirect one is previously predicted [24] for GaP NWs with diameters starting from 4 nm and larger.

The top panel: band structures of III–V NWs without {112} facets and diameters of about 2.6 nm, which cross section is shown in Figure 1 . Zero at the energy scale corresponds to the Fermi level indicated by the red dashed lines. The bottom panel: band structures of III–V NWs with {112} facets and diameters of about 2.6 nm, which cross section is shown in Figure 1. For GaP, GaAs and GaSb NWs with diameter of 4 nm the top valence band and the bottom conduction band are presented by the blue dashed lines. The bands are aligned to the top of the valence band in the Γ point. Zero at the energy scale corresponds to the top of the valence band.
We have also revealed that independently of diameter phosphide NWs have both the maximum of the valence band and the minimum of the conduction band to be mainly defined by p-states of III and V surface atoms, while for arsenide NWs valuable contributions to the top of the valence band also come from the inner part atoms and eventually for antimonide NWs p-states of almost all atoms can be found in the extrema points, as can be clearly seen in Figure 4. For the sake of comparison in the cases of GaP, GaAs and GaSb NWs such an analysis has been performed for both conduction band minima in the Γ and Z points indicating that in the Z point less atoms contribute their states with respect to the Γ point for GaP and GaAs. Contrary to hydrogenated III–V NWs [8,23,24,26], bare III–V NWs are characterized by lack of s-states in the band extrema points that can compromise optical properties.

Cross sections of 〈111〉-oriented III–V NWs with diameters of about 2.6 nm where III (blue spheres) and V (green spheres) atoms, whose states characterize the top of the valence band and the bottom of the conduction band, are highlighted. The rest of the III and V atoms are pink and yellow spheres, respectively.
4 Conclusions
We have suggested a new structural model of III–V NWs with the zinc-blende structure and {011} facets bounded in the hexagonal shape, which involves small {112} facets between the adjacent {011} ones acting as edges. In this case it is possible to reduce number of dangling bonds at the surface because of the formation of III–III and V–V dimers and to provide a sizable lowering in total energy. We have also predicted {011} and {112} facets to be positively and negatively charged, respectively, whereas essential charge distribution can be spotted for surface atoms located near edges. Moreover, such III–V NWs are semiconductors since there is no band associated with surface states to be crossed by the Fermi level and no hydrogen passivation is necessary to investigate semiconducting properties of these NWs. Moreover, previously published and reported here changes in direct/indirect character of the gap and band dispersion near the gap region in III–V NWs with different morphology and with or without surface passivation can be viewed as a valuable tool for band-gap engineering in order to tune electronic, optical and transport properties of such NWs targeting specific applications.
References
W Lu, CM Lieber, Semiconductor nanowires. J. Phys. D: Appl. Phys. 39, 387 (2006).
S Barth, F Hernandez-Ramirez, JD Holmes, A Romano-Rodriguez, Synthesis and applications of one-dimensional semiconductors. Prog. Mater. Sci. 55, 563 (2010).
M Galicka, M Bukala, R Buczko, P Kacman, Modelling the structure of GaAs and InAs nanowires. J. Phys.: Condens. Matter. 20, 454226 (2008).
T Yamashita, T Akiyama, K Nakamura, T Ito, Effects of facet orientation on relative stability between zinc blende and wurtzite structures in group III–V nanowires. Jpn. J. Appl. Phys. 49, 055003 (2010).
M Rosini, R Magri, Surface effects on the atomic and electronic structure of unpassivated GaAs nanowires. ACS Nano. 4, 2010.
R Leitsmann, F Bechstedt, Surface influence on stability and structure of hexagon-shaped III–V semiconductor nanorods. J. Appl. Phys. 102, 2007.
M Dionizio Moreira, P Vanazuela, RH Miwa, InP and InAs nanowires hetero- and homojunctions: energetic stability and electronic properties. Nanotechnology. 21, 285204 (2010).
TM Schmidt, RH Miwa, P Vanazuela, A Fazzio, Stability and electronic confinement of free-standing InP nanowires: Ab initio calculations. Phys. Rev. B. 72, 193404 (2005).
T Akiyama, K Nakamura, T Ito, Structural stability and electronic structures of InP nanowires: Role of surface dangling bonds on nanowire facets. Phys. Rev. B. 73, 235308 (2006).
S Cahangirov, S Ciraci, First-principles study of GaAs nanowires. Phys. Rev. B. 79, 2009.
P Mohan, R Bag, S Singh, A Kumar, R Tyagi, Mechanism of self-assembled growth of ordered GaAs nanowire arrays by metalorganic vapor phase epitaxy on GaAs vicinal substrates. Nanotechnology. 23, 025601 (2012).
J Johansson, LS Karlsson, CPT Svensson, T Martensson, BA Wacaser, K Deppert, L Samuelson, W Seifert, The structure of <111>B oriented GaP nanowires. J. Cryst. Growth. 298, 635 (2007).
M Jeppsson, KA Dick, JB Wagner, P Caroff, K Deppert, L Samuelson, LE Wernersson, GaAs/GaSb nanowire heterostructures grown by MOVPE. J. Cryst. Growth. 310, 4115 (2008).
M Björk, H Schmid, CM Breslin, L Gignac, H Riel, InAs nanowire growth on oxide-masked <111> silicon. J. Cryst. Growth. 344, 31 (2012).
MA Verheijen, RE Algra, MT Borgström, G Immink, E Sourty, WJP van Enckevort, E Vlieg, EPAM Bakkers, Three–dimensional morphology of GaP–GaAs nanowires revealed by transmission electron microscopy tomography. Nano Lett. 7, 3051 (2007).
RE Algra, MA Verheijen, LF Feiner, W GG.Immink, R Theissmann, WJP van Enckevort, E Vlieg, EPAM Bakkers, Paired twins and {112} morphology in GaP nanowires. Nano Lett. 10, 2349 (2010).
HJ Joyce, Q Gao, J Wong-Leung, Y Kim, HH Tan, C Jagadish, Tailoring GaAs, InAs, and InGaAs nanowires for optoelectronic device applications. IEEE J. Select Topics Quantum. Electr. 17, 766 (2011).
K Ikejiri, F Ishizaka, K Tomioka, T Fukui, Bidirectional growth of indium phosphide nanowires. Nano Lett. 12, 4770 (2012).
T Xu, KA Dick, S Plissard, TH Nguyen, Y Makoudi, M Berthe, JP Nys, X Wallart, B Grandidier, P Caroff, Faceting, composition and crystal phase evolution in III–V antimonide nanowire heterostructures revealed by combining microscopy techniques. Nanotechnology. 23, 095702 (2012).
J Bauer, H Paetzelt, V Gottschalch, G Wagner, GaAs nanowires grown by MOVPE. Phys. Status Solidi B. 247, 1294 (2010).
JH Kang, Q Gao, P Parkinson, HJ Joyce, HH Tan, Y Kim, Y Guo, H Xu, J Zou, C Jagadish, Precursor flow rate manipulation for the controlled fabrication of twin-free GaAs nanowires on silicon substrates. Nanotechnology. 23, 415702 (2012).
TM Schmidt, Hydrogen and oxygen on InP nanowire surfaces. Appl. Phys. Lett. 89, 123117 (2006).
WF Sun, MC Li, LC Zhao, Ab initio study of electronic structures of InAs and GaSb nanowires along various crystallographic orientations. Comp. Mater Sci. 50, 780 (2010).
CBE Santos, TM Schmidt, Direct band gap GaP nanowires predicted through first principles. J. Appl. Phys. 108, 103715 (2010).
M Dionizio Moreira, P Vanazuela, TM Schmidt, The effects of oxygen on the surface passivation of InP nanowires. Nanotechnology. 19, 065203 (2008).
D Li, Z Wang, F Gao, First-principles study of the electronic properties of wurtzite, zinc-blende, and twinned InP nanowires. Nanotechnology. 21, 505709 (2010).
CL dos Santos, P Piquini, Diameter dependence of mechanical, electronic, and structural properties of InAs and InP nanowires: A first-principles study. Phys. Rev. B. 81, 075408 (2010).
G Kresse, J Hafner, Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B. 49, 14251 (1994).
JP Perdew, A Zunger, Self-interaction correction to density functional approximation for many electron systems. Phys. Rev. B. 23, 5048 (1981).
W Tang, E Sanville, G Henkelman, A grid-based Bader analysis algorithm without lattice bias. J. Phys.: Condens Mater. 21, 084204 (2009).
DB Migas, VE Borisenko, The role of morphology in stability of Si nanowires. J. Appl. Phys. 105, 104316 (2009).
SA Dayeh, C Soci, PKL Yu, ET Yu, D Wang, Extraction of transport coefficients and their variability in the presence of surface states in semiconductor nanowires. Appl. Phys. Lett. 90, 162112 (2007).
Acknowledgements
This work has been supported by Belarusian Republican Foundation for Fundamental Research under the Grant No. F14U-001 and by the Singapore Ministry of Education (project reference MOE2013-T2-1-044). The authors thank Professor V. E. Borisenko for useful suggestions and comments on the results presented in the paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DBM and DAY have generated structural models of NWs and performed calculations. DAY and ABF dealt with figure preparation. DBM, ABF and CS performed the data analysis and wrote the paper. R. directed the project. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Migas, D.B., Filonov, A.B., Yatsyna, D.A. et al. Role of edge facets on stability and electronic properties of III–V nanowires. Nano Convergence 2, 14 (2015). https://doi.org/10.1186/s40580-015-0045-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40580-015-0045-7