
Abstract
Lewy body (LB) pathology commonly occurs in individuals with Alzheimer’s disease (AD) pathology. However, it remains unclear which genetic risk factors underlie AD pathology, LB pathology, or AD-LB co-pathology. Notably, whether APOE-ε4 affects risk of LB pathology independently from AD pathology is controversial. We adapted criteria from the literature to classify 4,985 subjects from the National Alzheimer’s Coordinating Center (NACC) and the Rush University Medical Center as AD-LB co-pathology (AD+LB+), sole AD pathology (AD+LB–), sole LB pathology (AD–LB+), or no pathology (AD–LB–). We performed a meta-analysis of a genome-wide association study (GWAS) per subpopulation (NACC/Rush) for each disease phenotype compared to the control group (AD–LB–), and compared the AD+LB+ to AD+LB– groups. APOE-ε4 was significantly associated with risk of AD+LB– and AD+LB+ compared to AD–LB–. However, APOE-ε4 was not associated with risk of AD–LB+ compared to AD–LB– or risk of AD+LB+ compared to AD+LB–. Associations at the BIN1 locus exhibited qualitatively similar results. These results suggest that APOE-ε4 is a risk factor for AD pathology, but not for LB pathology when decoupled from AD pathology. The same holds for BIN1 risk variants. These findings, in the largest AD-LB neuropathology GWAS to date, distinguish the genetic risk factors for sole and dual AD-LB pathology phenotypes. Our GWAS meta-analysis summary statistics, derived from phenotypes based on postmortem pathologic evaluation, may provide more accurate disease-specific polygenic risk scores compared to GWAS based on clinical diagnoses, which are likely confounded by undetected dual pathology and clinical misdiagnoses of dementia type.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) pathology has been the focus of many studies, but Lewy body (LB) pathology has received less attention. In individuals with AD, LB pathology frequently co-occurs with AD pathology, while LB pathology alone or accompanied by limited AD pathology is characteristic of Parkinson’s disease (PD) and LB dementia [29, 48, 52, 53]. Genetic variants associated with AD pathology, LB pathology, and/or AD-LB co-pathology remain to be distinguished. Characterizing each set of risk factors and any potential overlap will help clarify the etiology of AD and LB pathology. AD pathology is found in 19–67% of older individuals at autopsy, depending on the population, the individual’s age, and the diagnostic criteria used [43]. LB pathology is observed in 6–39% of older individuals, but in 60% of individuals with AD pathology [29, 33, 43]. Positive classification for LB pathology requires, at a minimum, the presence of α-synuclein-bearing Lewy bodies in the brainstem, with further extension of LB pathology into the limbic system associated with the clinical diagnosis of dementia [37]. Positive classification for AD pathology requires the presence of tau neurofibrillary tangles (NFTs) in the limbic system along with amyloid-β core neuritic plaques in the cortex [8, 38]. Lewy bodies and NFTs spread to the cortex late in the progression of either pathology [8, 37]. Braak staging is the primary scheme used to classify NFT progression, while the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) scoring is the primary scheme used to classify neuritic plaque density. AD has traditionally been defined pathologically as Braak stage IV or higher, and at least moderate CERAD [17], although some studies have used less stringent criteria (Braak stage III or higher, and at least sparse CERAD) [27]. Thal phasing of amyloid-β non-neuritic plaques [50], based on another staging devised by Braak [9], has more recently been used as a third essential metric to classify AD [11]. Of the five Thal phases, only the last two, in which plaques are present in the brainstem and cerebellum, are specific to dementia patients [17].
Clinical diagnoses of AD and LB dementia are challenging and error-prone in comparison to the gold standard of a pathologic diagnosis [19]. However, most study participants have only been clinically diagnosed due to the scarcity of postmortem pathologically confirmed data. In a study of 919 autopsied individuals comparing clinical diagnosis of AD to pathological diagnosis, the diagnosis of clinically probable AD had an 83% positive predictive value (PPV) for pathological criteria of Braak state III or higher and moderate/high CERAD [1]. This study was conducted before AD biomarkers like spinal fluid amyloid and tau levels, or amyloid and tau PET scans, were more commonly used, so the PPV of the clinical diagnosis is now likely higher than 83%; however, it remains imperfect. The PPV for a clinical diagnosis of probable LB dementia against the pathologic diagnosis is also around 80% [21, 44, 49]. In general, it has been difficult to clinically distinguish between AD without Lewy bodies, AD with Lewy bodies, and LB dementia [25]. Additionally, individuals who may have advanced pathology, but mild symptoms are frequently misdiagnosed clinically or missing from clinical datasets altogether because they do not seek medical attention.
Motor function and neuropsychiatric and cognitive symptoms have been suggested as diagnostic clues of AD pathology, LB pathology, or co-pathology [14, 41, 47]. Moreover, developing AD pathology biomarkers such as assays of amyloid-β, tau, and phosphorylated tau levels in the cerebrospinal fluid or blood plasma has been valuable in closing the gap between diagnosis during life and pathologic AD diagnosis [6, 31]. LB pathology biomarkers, including promising assays of α-synuclein aggregates in the cerebrospinal fluid, are similarly improving the diagnosis of LB dementia [35, 42, 46]. Still, because LB biomarkers have been developed more recently, most existing genetic datasets consist of only clinically diagnosed subjects. Ultimately, as a histological and molecular endophenotype, pathologic diagnosis offers the most reliable insights into the genetic drivers of disease.
Previous research has produced contrasting and somewhat ambiguous findings on the genetic risk loci for AD and LB pathology. This could be because most studies include only clinically assessed subjects or have relatively few pathologically assessed subjects. Importantly, most prior studies on AD and LB pathology, even with pathologic confirmation, do not stratify subjects into distinct groups for sole AD pathology (AD+LB–), LB pathology (AD–LB+), co-pathology (AD+LB+), and neither pathology (AD–LB–), making the results difficult to accurately interpret. It is well known that the ε4 allele of the Apolipoprotein E (APOE) gene is the strongest common genetic risk factor for AD [4, 30]. However, various studies have reported that APOE-ε4 is also associated with increased risk of sole LB pathology (AD–LB+) [18, 51], LB dementia [2, 5, 13, 24, 47], or increased risk of AD-LB co-pathology (AD+LB+) in AD individuals [14]. Walker and Richardson (2023) found that APOE-ε4 was associated with AD, LB, or limbic-predominant age-related TDP-43 encephalopathy pathology as well as with the presence of multiple of these pathologies [53]. This suggests that APOE-ε4 could be associated with AD–LB+ pathology.
How APOE-ε4 affects the severity of LB pathology has also been investigated. Studies reported that α-synuclein pathology mouse models expressing APOE-ε4 develop more extensive inclusions [16, 22]. In humans, LB pathology was found to be more severe among APOE-ε4 carriers independent of AD pathology severity [23], as well as among APOE-ε4-carrying AD–LB+ subjects relative to non-carriers [18, 22, 56]. However, when Kaivola et al. [27] categorized pathologically confirmed cases from the cohort in [13] by not only LB but also AD pathology status, APOE-ε4 was not associated with risk of AD–LB+ pathology [27]. Robinson et al. [45] found that APOE-ε4 was associated with cortical LB co-pathology (cortical LB pathology accompanied by an amyloidopathy, tauopathy, or TDP-43 proteinopathy) compared to sole LB pathology; however, APOE-ε4 was not associated with AD-LB co-pathology compared to sole AD pathology [45]. Furthermore, Dickson et al. [18] found that APOE-ε4 was not associated with more severe LB pathology in individuals with moderate or high AD pathology [18]. It, therefore, remains unclear whether APOE-ε4 in fact increases risk of AD–LB+ pathology.
Importantly, beyond APOE-ε4, there may be other pathology-specific genetic risk loci yet to be identified. Along this line, it is relevant to note that removing individuals that are not pathologically evaluated from study cohorts has been shown to reduce noise in genome-wide association studies (GWAS) and to improve polygenic risk score analyses of AD [15, 19, 20]. These observations emphasize the need for novel GWAS of AD/LB pathology to better characterize the genetic architecture of these complex dementias.
To this end, we assembled a preliminary cohort of 5,254 individuals with genetic data and autopsy-confirmed AD and LB pathology status, the largest such cohort to date. We adapted criteria from the literature to categorize these individuals as AD+LB+, AD+LB–, AD–LB+, or AD–LB–, yielding 1,072 AD+LB+, 2,492 AD+LB–, 158 AD–LB+, and 1,263 AD–LB– individuals in our study cohort (total N = 4,985). We compared each disease category to controls by performing separate GWAS meta-analyses. We also compared AD+LB+ pathology to AD+LB– pathology in another analysis.
Materials and methods
Study cohort
We analyzed data from individuals from the National Alzheimer’s Coordinating Center (NACC) and Rush University Medical Center databases who were evaluated postmortem for both AD and LB pathology. We excluded NACC individuals who were classified as having Lewy bodies in the olfactory bulb or in an “unspecified” region (individuals for whom the NACCLEWY parameter was equal to 4). We also excluded individuals missing sex or age-at-death information. In total, our preliminary cohort comprised 5,254 individuals before classification according to AD and LB pathology status. This cohort was distinct from that analyzed in [13], the largest genetic study of LB dementia (which included subjects without pathology verification), and [27], the largest previous genetic study of subjects categorized by both AD and LB pathology status.
Pathological criteria
We classified individuals as having both AD and LB pathology (AD+LB+), AD pathology only (AD+LB–), LB pathology only (AD–LB+), or neither pathology (AD–LB–) (Fig. 1). Individuals who could not be classified using our criteria were excluded (Fig. 1c). In sensitivity analyses, we applied the pathology criteria from [51] and [27] to our preliminary cohort (Fig. 1a–b). Criteria were set as follows for LB pathology.
-
LB+ pathology were individuals with Lewy bodies spread to the limbic system or cortex, as in [27] and [51].
-
LB– pathology were individuals with no Lewy bodies or Lewy bodies limited to the brainstem, as in [27], but not in [51], which excluded individuals with brainstem-limited Lewy bodies.
-
Some gray zones, representing rare subcategories with unclassified individuals, are defined based on Braak stage and CERAD score below.

Criteria for AD pathology were less straightforward because of borderline subcategories and possible confounding with other pathologies. In agreement with [27] and [51] (Fig. 1),
-
AD+ pathology included individuals with Braak stage IV or higher and CERAD score moderate or frequent.
-
AD– pathology included individuals with Braak stage II or lower, or Braak stage III/IV and CERAD score absent.
Other subcategories were largely classified differently between [27] and [51], and we settled on the following criteria. First, we defined three gray zones of unclassified individuals corresponding to rare pathologic profiles (N = 269 total).
-
Individuals who had Braak stage V/VI and CERAD score absent, regardless of LB category, were not classified for the reason of likely having a rare tauopathy distinct from AD [40].
-
Similarly, individuals with absent or brainstem Lewy bodies (LB–) who had Braak stage V/VI and CERAD score sparse were not classified.
-
Braak stage III and CERAD score frequent in LB– individuals were insufficient for classification as AD+, but too high for a confident classification as AD–. Ultimately, our goal was to obtain a clean control (AD–LB–) group.
Second, certain borderline subcategories were classified differently (AD+ or AD–) depending on LB category.
-
Individuals with absent or brainstem Lewy bodies (LB–) were AD– for Braak stage III and CERAD score sparse or moderate; or Braak stage IV and CERAD score sparse.
-
Individuals with limbic or cortical Lewy bodies (LB+) were AD+ for Braak stage III or higher and CERAD score sparse; or Braak stage III and CERAD score moderate or frequent.
Using our criteria (Fig. 1c), we categorized our preliminary cohort into 1,072 AD+LB+, 2,495 AD+LB–, 158 AD–LB+, and 1,263 AD–LB– individuals; these 4,985 individuals formed our study cohort (Table 1). Overall, the set of four phenotypes was better represented in our study than in previous studies (Additional file 1: Table S1), which stratified subjects less completely or had fewer individuals in total (Tsuang et al.) (Table 2).
Genome-wide analysis
We performed a meta-analysis of separate GWAS in the NACC and Rush subsets of our cohort for each of AD+LB+, AD+LB–, and AD–LB+ pathology compared to AD–LB– pathology, as well as for AD+LB+ versus AD+LB– pathology. We used PLINK 2.0 for logistic regression and included sex, age-at-death, and the top ten principal components accounting for genetic ancestry as covariates [11]. We removed duplicates and first-degree relatives within and between genomic datasets using KING [36]. In each pair of relatives, the relative with younger age at death was preferentially kept or the oldest control in the absence of pathology cases. We filtered out genetic variants that had a minor allele frequency below 0.01, departed from Hardy–Weinberg equilibrium with a significance below P < 10−5, or had a missingness rate above 20%. We imputed data on the TOPMed reference panel as described in [32] and considered variants with R2 > 0.8. We meta-analyzed the separate NACC and Rush GWAS using the inverse variance weighted method in METAL [54]. Manhattan plots from summary statistics were produced using the R package CMplot [55]. The significance threshold was set at P < 5 × 10−8, the standard threshold for genome-wide significance. We estimated the association of APOE-ε4 with risk of AD+LB+, AD+LB–, and AD–LB+ pathology relative to AD–LB– pathology, and the association with risk of AD+LB+ pathology relative to AD+LB– pathology, in terms of odds ratio (OR). We also estimated the association of APOE-ε2. We compared our estimates to those in the literature [2, 10, 13, 14, 18, 27, 45, 47, 51] and when relevant we computed measures of linkage disequilibrium between variants in European ancestry populations using LDlink [34]. We examined loci besides APOE that led to genome-wide significant signals. We explored lead variant annotation at significant loci using gnomAD [28]. Finally, we surveyed AD and PD risk loci reported in large clinical case–control GWAS [3, 12, 39] and report the ones associated with pathology at the nominal significance level (P < 0.05) in our study.
Results
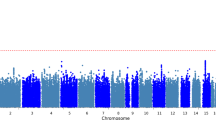
We observed that APOE-ε4 (rs429358) was associated with risk of AD+LB+ pathology versus AD–LB– pathology (OR = 4.24, 95% CI = 3.52–5.10, P = 1.5 × 10−52) and risk of AD+LB– pathology versus AD–LB– pathology (OR = 4.22, 95% CI = 3.60–4.96, P = 1.4 × 10−69) (Fig. 2a–b; Table 4). We did not observe an association of APOE-ε4 with the risk of AD–LB+ pathology versus AD–LB– pathology (OR = 0.93, 95% CI = 0.60–1.43, P = 0.73) or risk of AD+LB+ pathology versus AD+LB– pathology (OR = 1.01, 95% CI = 0.90–1.13, P = 0.83) (Fig. 2c–d; Table 4). Another gene locus that yielded significant associations was BIN1. Like APOE-ε4, we observed that rs4663105 on the BIN1 locus was associated with risk of AD+LB– pathology compared to AD–LB– pathology (OR = 1.40, 95% CI = 1.26–1.56, P = 6.5 × 10−10) and risk of AD+LB+ pathology compared to AD–LB– pathology (OR = 1.53, 95% CI = 1.35–1.75, P = 1.4 × 10−10) (Fig. 2a–b; Table 5). rs4663105 was not observed to be associated with risk of AD–LB+ pathology versus AD–LB– pathology (OR = 1.10, 95% CI = 0.85–1.41, P = 0.48) or risk of AD+LB+ pathology versus AD+LB– pathology (OR = 1.13, 95% CI = 1.02–1.25, P = 0.019) at the genome-wide significance level (Fig. 2c–d; Table 5). When using pathological criteria from Tsuang et al. [51] (Fig. 1a), effect estimates for APOE-ε4 differed considerably from those reported in the original study, particularly so for the effect on AD–LB+ vs. AD–LB– (Table 4). On the contrary, there was fair agreement when using pathological criteria from Kaivola et al. [27] (Fig. 1b; Table 4). APOE-ε2 showed similar results to APOE-ε4, except with the opposite direction of effect in the GWAS where APOE-ε4 exhibited an association (Additional file 1: Table S3). Overall, we observed an enrichment among the 79 variants listed in the clinical AD GWAS (Bellenguez et al. [3]) and tested in our analyses; we observed an enrichment of nominally significant associations with concordant direction of effect: 20.3% variants (16/79) in the AD+LB− vs. AD−LB− contrast and 24.1% (19/79) in the AD+LB+ vs. AD−LB− (with the chance level being at 2.5%). In contrast, we did not observe a significant enrichment for the 76 variants identified in the PD clinical GWAS Chang et al. [12] and Nalls et al. [39]: 2.6% (2/76) in the AD+LB+ vs. AD−LB− contrast, 3.9% (3/76) in the AD−LB+ vs. AD−LB− contrast, and 2.6% (2/76) in the AD+LB+ vs. AD+LB− contrast. Among known AD risk loci besides BIN1 and APOE reported by Bellenguez et al. [3], ADAM17 (rs72777026), COX7C (rs62374257), HLA (rs6605556), TREM2 (rs143332484), HS3ST5 (rs785129), SEC61G (rs76928645), CLU (rs11787077), ECHDC3 (rs7912495), TPCN1 (rs6489896), FERMT2 (rs17125924), DOC2A (rs1140239), PRDM7 (rs56407236), ABI3 (rs616338), ABCA7 (rs12151021), and SIGLEC11 (rs9304690) were concordant and nominally associated with AD+LB+ versus AD–LB–; and CR1 (rs679515), ADAM17 (rs72777026), INPP5D (rs10933431), CLNK/HS3ST1 (rs6846529), ANKH (rs112403360), COX7C (rs62374257), HLA (rs6605556), TREM2 (rs143332484), ZCWPW1/NYAP1 (rs7384878), PTK2B (rs73223431), CLU (rs11787077), ECHDC3 (rs7912495), PICALM (rs3851179), SORL1 (rs11218343), FERMT2 (rs17125924), APH1B (rs117618017), MAF (rs450674), and ABCA7 (rs12151021) were concordant and nominally associated with AD+LB– pathology versus AD–LB– (Table 6; Additional file 1: Table S5). Among known PD risk loci reported by Chang et al. [12] and Nalls et al. [39], SCN3A (rs353116) and HLA-DRB6/HLA-DQA1 (rs9275326) were concordant and nominally associated with AD+LB+ versus AD–LB–; TMEM175/DGKQ (rs34311866), FAM200B/CD38 (rs11724635), and SNCA (rs356182) were concordant and nominally associated with AD–LB+ versus AD–LB–; and GBA (rs35749011) and TMEM175/DGKQ (rs34311866) were concordant and nominally associated with AD+LB+ versus AD+LB– (Table 6; Additional file 1: Table S5). Notably, the TPCN1 locus, reported to be associated with LB dementia by Kaivola et al. [26], was associated with AD+LB+ pathology and AD+LB– pathology versus AD–LB– pathology below or near the nominal significance level, but not so with AD–LB+ pathology versus AD–LB– pathology or AD+LB+ pathology versus AD+LB– pathology.

Manhattan plots of genetic association with pathology contrasts. a. Association with AD+LB+ pathology versus AD–LB– pathology. b. Association with AD+LB– pathology versus AD–LB– pathology. c. Association with AD–LB+ pathology versus AD–LB– pathology. d. Association with AD+LB+ pathology versus AD+LB– pathology. Variants at two novel loci exhibited genome-wide significant associations in the AD–LB+ versus AD–LB– analysis (rs112017605 on both an intron of AC024598.1 and an intron of AC067752.1 on chromosome 10 and rs116691607 on an intron of BLMH on chromosome 17) (c; Additional file 1: Table S2), but we do not discuss these candidates in the main text because neither was flanked by a set of nearby variants in linkage disequilibrium, raising concern that these could be spurious signals
Discussion
Our study emphasizes that APOE-ε4 is associated with the risk of both AD+LB– pathology (OR = 4.22, P = 1.4 × 10−69) and AD+LB+ co-pathology (OR = 4.24, P = 1.5 × 10−52) compared to AD–LB– pathology. These OR estimates were lower than the estimates in Tsuang et al. for APOE-ε4-associated risk of AD+LB– pathology (OR = 12.6, P = 2.1 × 10−28) and AD+LB+ co-pathology (OR = 9.9, P = 1.2 × 10−24) (Table 4) [51]. Tsuang et al. appear to have overestimated the true effect size of APOE-ε4 in their positive pathology subjects, possibly due to the smaller size of their cohort [51] (N = 640 versus N = 4,985 in the current study) (Table 3). Our estimate of APOE-ε4-associated risk for AD+LB+ pathology is in line with the estimate in Kaivola et al., the next largest study of pathologically assessed individuals (OR = 4.25, P = 1.29 × 10−32) [27]. Our data substantiate that APOE-ε4 is a driver of AD pathology. Notably, we estimated a similar effect size of APOE-ε4 on the risk of AD+LB– pathology (OR = 4.22) and on the risk of AD+LB+ co-pathology (OR = 4.24) versus AD–LB– pathology, suggesting that APOE-ε4 does not have a specific effect on the development of LB pathology in individuals with AD pathology. Consistent with this finding, we did not observe APOE-ε4 to be associated with the risk of AD+LB+ co-pathology compared to sole AD+LB– pathology (OR = 1.01, P = 0.83).
This finding contradicts the results presented by Chung et al. (2015), where APOE-ε4 was found to be associated with AD+LB+ co-pathology when compared to sole AD+LB– pathology (P = 0.03) [14]. However, their AD+LB+ group was five times smaller than ours (N = 215 versus N = 1,072), and the AD+LB– group was eight times smaller (N = 316 versus N = 2,492). Our result is consistent with the finding in Robinson et al. [45] that APOE-ε4 is not associated with the co-occurrence of AD pathology with other pathologies (OR = 0.71, P = 0.64 for intermediate AD pathology; and OR = 0.93, P = 0.83 for high AD pathology). Our result is also consistent with the finding in Dickson et al. [18] that APOE-ε4 is not associated with higher Lewy body count in individuals with moderate AD pathology (P ≥ 0.30 for all regions) or high AD pathology (P ≥ 0.069 for all regions) [18]. Taken together, APOE-ε4 appears similarly prevalent in AD pathology cases with or without LB pathology.
Furthermore, we did not find APOE-ε4 to be associated with risk for sole LB pathology (AD–LB+) pathology (OR = 0.93, P = 0.73) compared to no pathology (AD–LB–). This is in contradiction with [51] where APOE-ε4 was strongly associated with risk for AD–LB+ pathology (OR = 6.1, P = 1.3 × 10−10). This discrepancy could be because Tsuang et al. used a more stringent definition of AD pathology (Fig. 1a), leaving open the possibility that individuals whom we would have classified as AD+LB+ with our criteria were instead classified as AD–LB+. Indeed, when we categorized our initial pathologically evaluated cohort using the exact same criteria as in [51], testing the association of APOE-ε4 with risk of AD–LB+ pathology yielded a modestly higher OR and nominal significance (OR = 1.46, P = 5.5 × 10−3) (Table 4). The main difference between our criteria and those of Tsuang et al. is that we lower the threshold for AD pathology to Braak stage III NFTs plus sparse neuritic plaques in LB+ individuals, suggesting that LB pathology commonly occurs in APOE-ε4 carriers with potential early-stage AD but not in firmly non-AD APOE-ε4 carriers. Knowing the breakdown of the APOE-ε4-positive subjects in [51] by Braak stage and CERAD score would establish further support for this interpretation (Tables 5, 6).
Dickson et al. also found an association of APOE-ε4 with the risk of diffuse LB pathology and low AD pathology (OR = 3.46, P = 0.001) while classifying individuals with Braak stage III NFTs and Thal phase 0, 1, or 2 amyloid-β non-neuritic plaques as having low AD pathology [18]. In this AD–LB+ group, the median Braak stage was III and the median Thal phase was 1. We would have classified this subset as AD+LB+. A Thal phase of 1 tends to correspond to a CERAD score of sparse neuritic plaques or higher [7]. Therefore, many individuals in this AD–LB+ group in [18] had a Braak stage of III and at least sparse neuritic plaques; this subset was likely the source of the elevated frequency of APOE-ε4 in the group. We would have classified this subset as AD+LB+ instead. It should be noted that this AD–LB+ group in Dickson et al. was smaller than ours (N = 54 versus N = 158) and the controls were not pathologically confirmed. Another cause of the discrepancy between our result that APOE-ε4 was not associated with the risk of AD–LB+ pathology and Tsuang et al.’s finding that APOE-ε4 increased risk for LB pathology may have been that the pathologically confirmed AD–LB– group in [51] was more than four times smaller than ours (N = 269 versus N = 1,263). Remarkably, the frequency of APOE-ε4 was 31.9% in the AD–LB+ group [51], which was far higher than in our AD–LB+ group (8.9%). Our result was consistent with the finding in Dickson et al. that APOE-ε4 was not associated with the risk of the AD–LB+ phenotype of transitional (limbic) LB pathology and low AD pathology (OR = 0.73, P = 0.31) [18].
In a larger study than [51] of pathologically confirmed LB dementia cases and mixed pathologic and clinical controls, Chia et al. found that APOE-ε4 was associated with risk of LB dementia: OR = 2.45 with P = 4.65 × 10−63 for rs769449, which is in linkage disequilibrium with APOE-ε4 with R2 = 0.766 [13]. However, this could have been because Chia et al. did not categorize individuals by AD pathology status, and many AD+LB+ individuals were inevitably included in the LB+ case group. When Kaivola et al. studied the cohort in Chia et al. using more precise pathological categorization, APOE-ε4 was not observed to have a significant effect on risk for AD–LB+ pathology (OR = 0.75, P = 0.31) [27]. Associations of APOE-ε4 with risk of LB pathology (OR = 1.63, P = 2.8 × 10−11) and LB dementia (OR = 2.71, P = 7.1 × 10−35; OR = 2.40, P = 1.05 × 10−48; and OR = 2.94, P = 6.6 × 10−20) reported in Beecham et al., Bras et al., Guerreiro et al., and Sabir et al. [2, 10, 24, 47], respectively, could similarly have been because these studies did not exclude AD+LB+ individuals from their LB+ case groups. Our finding was also consistent with the result in Robinson et al. that APOE-ε4 was associated with the co-occurrence of cortical LB pathology with other pathologies, including AD pathology, versus sole cortical LB pathology (OR = 9.32, P = 0.003) [45]. The latter result would imply that APOE-ε4 was rarer in the AD–LB+ individuals in [45] than in LB+ individuals with advanced LB pathology. Presumably, because 80% of LB+ individuals also had AD pathology, the prevalence of APOE-ε4 in LB+ individuals was most likely unrelated to the presence of LB pathology.
The balance of evidence thus suggests that APOE-ε4 does not affect risk for AD–LB+ pathology when strictly defined to exclude possible early-stage AD. Subjects with LB pathology and no AD pathology have been rare, and more are needed to substantiate this conclusion. This concept does not oppose the observations in Dickson et al. and Zhao et al. [18, 56] that APOE-ε4 was associated with higher LB counts in AD–LB+ subjects, as neither analysis compared cases to controls without pathology. Similarly compatible is the observation in Goldberg et al. [23] that APOE-ε4 was associated with further propagated Lewy bodies; although Goldberg et al. adjusted for AD pathology level, their analysis did not specifically compare LB+ to LB– subjects. It is conceivable that APOE-ε4 worsens LB pathology but does not influence its actual emergence in individuals without AD pathology. Overall, our data suggest APOE-ε4 is most likely not involved in the emergence of LB pathology in the absence or presence of AD pathology. This interpretation is further supported by the lack of an effect of APOE-ε4 in the largest GWAS of clinically defined PD (OR = 1.02, P = 0.49) [39].
A second gene locus that yielded significant associations was BIN1. As for APOE-ε4, the BIN1 lead variant was associated with the risk of sole AD (AD+LB–) and AD+LB+ co-pathology, but not sole LB (AD–LB+) when compared to no pathology (AD–LB–). BIN1 was also not associated with the risk of AD+LB+ co-pathology when compared to sole AD pathology (AD+LB–) pathology. These results further corroborate that BIN1 is also a driver of AD pathology. In the largest previous GWAS of LB pathology, Chia et al. [13] found that BIN1 is a risk locus for pathologically confirmed LB dementia (OR = 1.25, P = 4.16 × 10−9 for rs6733839, in linkage disequilibrium with rs4663105 with R2 = 0.8968). However, when gathering individuals, Chia et al. did not select against AD pathology, which was presumably far more prevalent in the LB+ case group than in the control group. Given that we do not observe an association of rs4663105 with risk for AD–LB+ pathology, the BIN1 association reported in [13] may have been driven by the AD+LB+ subgroup within the LB+ group. However, our findings are limited by the size of our AD–LB+ group (N = 158); the lower statistical power of the AD–LB+ pathology versus AD–LB– pathology GWAS was likely also the reason that known synucleinopathy risk loci like GBA and SNCA did not yield genome-wide significant associations in this analysis. Altogether, the current balance of evidence suggests that variants on the BIN1 locus behave like APOE-ε4: pathogenic BIN1 variants increase the overall risk of LB pathology simply by increasing the risk of AD pathology (which is frequently accompanied by LB pathology), but they do not affect the risk of AD–LB+ pathology or the risk of co-pathology (AD+LB+) among AD+ individuals. It is worth mentioning that the effect of BIN1 on the risk of AD pathology may be lifestyle-dependent, as we did not observe any association of BIN1 with pathology in the subset of Rush individuals alone (Additional file 1: Fig. S2; Additional file 1: Table S4). The monastic life of these subjects likely militates against disease.
Future studies should continue the effort of determining the risk loci for AD pathology, LB pathology, or AD-LB co-pathology using pathologically well-categorized and clinically unbiased cohorts. It may be worth focusing on comparing AD+LB+ to AD+LB– groups to identify LB pathology risk loci because the sample size of either phenotype is larger than AD–LB+. Further study of the AD+LB+ versus AD+LB– and AD–LB+ versus AD–LB– contrasts may also reveal possible differences between genetic risk factors underlying LB pathology in the presence or absence of AD pathology; we propose a hypothetical genetic model in Additional file 1: Fig. S3.
Conclusion
In conclusion, our set of GWAS meta-analyses indicates that while APOE-ε4 is a risk factor for AD pathology and increases risk of AD-LB co-pathology, it is not a risk factor for LB pathology independent of AD pathology or along with AD pathology. This is also true of variants on the BIN1 locus; therefore, neither APOE-ε4 nor BIN1 variants appear to play a specific mechanistic role in the emergence of LB pathology. We provide GWAS meta-analysis summary statistics that will enable more reliable, pathologically precise polygenic risk score calculations for AD, LB dementia, and related disorders. Ultimately, we shed light on the genetic bases of AD and LB pathology, which will be useful for further unraveling the etiology of these debilitating pathologies and developing accurate and effective interventions.
Availability of data and materials
Genome-wide association summary statistics of our pathologically confirmed case-control analyses have been deposited to the European Bioinformatics Institute GWAS Catalog under accession no. GCST90276157, GCST90276158, GCST90276159, GCST90276160. https://www.ebi.ac.uk/gwas/studies/GCST90276157
References
Beach TG, Monsell SE, Phillips LE, Kukull W (2012) Accuracy of the clinical diagnosis of Alzheimer disease at national institute on aging Alzheimer disease centers, 2005–2010. J Neuropathol Exp Neurol 71:266–273. https://doi.org/10.1097/NEN.0b013e31824b211b
Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ et al (2014) Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. https://doi.org/10.1371/journal.pgen.1004606
Bellenguez C, Küçükali F, Jansen IE et al (2022) New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54:412–436. https://doi.org/10.1038/s41588-022-01024-z
Belloy ME, Napolioni V, Greicius MD (2019) A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron 101:820–838. https://doi.org/10.1016/j.neuron.2019.01.056
Berge G, Sandro SB, Ronge A et al (2014) Apolipoprotein E ε2 genotype delays onset of dementia with Lewy bodies in a Norwegian cohort. J Neurol Neurosurg Psychiatry 85:1227–1231. https://doi.org/10.1136/jnnp-2013-307228
Blennow K, Zetterberg H (2018) Biomarkers for Alzheimer’s disease: current status and prospects for the future. J Intern Med 284:643–663. https://doi.org/10.1111/joim.12816
Boluda S, Toledo JB, Irwin DJ et al (2014) A comparison of Aβ amyloid pathology staging systems and correlation with clinical diagnosis. Acta Neuropathol 128:543–550. https://doi.org/10.1007/s00401-014-1308-9
Braak H, Alafuzoff A, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. https://doi.org/10.1007/s00401-006-0127-z
Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18:351–357. https://doi.org/10.1016/s0197-4580(97)00056-0
Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O et al (2014) Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet 23:6139–6146. https://doi.org/10.1093/hmg/ddu334
Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ et al (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. https://doi.org/10.1186/s13742-015-0047-8
Chang D, Nalls M, Hallgrímsdóttir I et al (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49:1511–1516. https://doi.org/10.1038/ng.3955
Chia R, Sabir MS, Bandres-Ciga S, Saez-Atienzar S, Reynolds RH et al (2021) Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet 53:294–303. https://doi.org/10.1038/s41588-021-00785-3
Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC et al (2015) Clinical features of Alzheimer disease with and without Lewy bodies. JAMA Neurol 72:789–796. https://doi.org/10.1001/jamaneurol.2015.0606
Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M et al (2010) Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet 19:3295–3301. https://doi.org/10.1093/hmg/ddq221
Davis AA et al (2020) APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aay3069
DeTure MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegeneration. https://doi.org/10.1186/s13024-019-0333-5
Dickson DW, Heckman MG, Murray ME, Soto AI, Walton RL et al (2018) APOE ε4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 91:1182–1195
Escott-Price V, Hardy J (2022) Genome-wide association studies for Alzheimer’s disease: bigger is not always better. Brain Commun 4:125. https://doi.org/10.1093/braincomms/fcac125
Escott-Price V, Myers AJ, Huentelman M, Hardy J (2017) Polygenic risk score analysis of pathologically confirmed Alzheimer disease. Ann Neurol 82:311–314. https://doi.org/10.1002/ana.24999
Ferman TJ, Boeve BF, Smith GE, Lin SC, Silber MH et al (2011) Inclusion of RBD improves the diagnostic classification of dementia with Lewy bodies. Neurology 77:875–882. https://doi.org/10.1212/WNL.0b013e31822c9148
Fyfe I (2020) APOE*ε4 promotes synucleinopathy. Nat Rev Neurol. https://doi.org/10.1038/s41582-020-0335-5
Goldberg TE, Huey ED, Devanand DP (2020) Association of APOE e2 genotype with Alzheimer’s and non-Alzheimer’s neurodegenerative pathologies. Nat Commun. https://doi.org/10.1038/s41467-020-18198-x
Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T et al (2018) Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol 17:64–74. https://doi.org/10.1016/s1474-4422(17)30400-3
Hohl U, Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J (2000) Diagnostic accuracy of dementia with Lewy bodies. Arch Neurol 57:347–351. https://doi.org/10.1001/archneur.57.3.347
Kaivola K et al (2023) Genome-wide structural variant analysis identifies risk loci for non-Alzheimer’s dementias. Cell Genom. https://doi.org/10.1016/j.xgen.2023.100316
Kaivola K, Shah Z, Chia R, International LBD Genomics Consortium, Scholz S (2022) Genetic evaluation of dementia with Lewy bodies implicates distinct disease subgroups. Brain 145:1757–1762. https://doi.org/10.1093/brain/awab402
Karczewski KJ, Francioli LC, Tiao G et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Kotzbauer PT, Trojanowski JQ, Lee VMY (2001) Lewy body pathology in Alzheimer’s disease. J Mol Neurosci 17:225–232. https://doi.org/10.1385/JMN:17:2:225
Koutsodendris N, Nelson MR, Rao A, Huang Y (2022) Apolipoprotein E and Alzheimer’s disease: findings, hypotheses, and potential mechanisms. Annu Rev Path 17:73–99. https://doi.org/10.1146/annurev-pathmechdis-030421-112756
Lantero Rodriguez J, Karikari TK, Suárez-Calvet M et al (2020) Plasma p-tau181 accurately predicts Alzheimer’s disease pathology at least 8 years prior to post-mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol 140:267–278. https://doi.org/10.1007/s00401-020-02195-x
Le Guen Y, Belloy ME, Napolioni V et al (2021) A novel age-informed approach for genetic association analysis in Alzheimer’s disease. Alzheimer’s Res Therapy. https://doi.org/10.1186/s13195-021-00808-5
Leverenz JB, Fishel MA, Peskind ER et al (2006) Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol 63:370–376. https://doi.org/10.1001/archneur.63.3.370
Machiela MJ, Chanock SJ (2015) LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31:3555–3557. https://doi.org/10.1093/bioinformatics/btv402
Majbour N, Aasly J, Abdi I, Ghanem S, Erskine D, van de Berg W, El-Agnaf O (2022) Disease-associated α-synuclein aggregates as biomarkers of Parkinson disease clinical stage. Neurology. https://doi.org/10.1212/WNL.0000000000201199
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM (2010) Robust relationship inference in genome-wide association studies. Bioinformatics 26:2867–2873. https://doi.org/10.1093/bioinformatics/btq559
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J et al (2017) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB consortium. Neurology 89:88–100. https://doi.org/10.1212/WNL.0000000000004058
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ et al (1991) The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486. https://doi.org/10.1212/wnl.41.4.479
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S et al (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18:1091–1102
Nelson PT, Alazufoff I, Bigio EH, Bouras C, Braak H et al (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362–381. https://doi.org/10.1097/nen.0b013e31825018f7
Pillai JA, Bena J, Bonner-Jackson A, Leverenz JB (2021) Impact of APOE ε4 genotype on initial cognitive symptoms differs for Alzheimer’s and Lewy body neuropathology. Alzheimer’s Res Therapy. https://doi.org/10.1186/s13195-021-00771-1
Poggiolini I, Gupta V, Lawton M, Lee S, El-Turabi A et al (2022) Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain 145:584–595. https://doi.org/10.1093/brain/awab431
Rahimi J, Kovacs GG (2014) Prevalence of mixed pathologies in the aging brain. Alzheimer’s Res Therapy. https://doi.org/10.1186/s13195-014-0082-1
Rizzo G, Arcuti S, Copetti M, Alessandria M, Savica R et al (2018) Accuracy of clinical diagnosis of dementia with Lewy bodies: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 89:358–366. https://doi.org/10.1136/jnnp-2017-316844
Robinson JL, Lee EB, Xie SX, Rennert L, Suh E et al (2018) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141:2181–2193. https://doi.org/10.1093/brain/awy146
Russo MJ, Orru CD, Concha-Marambio L et al (2021) High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-021-01282-8
Sabir MS, Blauwendraat C, Ahmed S, Serrano GE, Beach TG et al (2019) Assessment of APOE in atypical parkinsonism syndromes. Neurobiol Dis 127:142–146
Savica R, Beach TG, Hentz JG et al (2019) Lewy body pathology in Alzheimer’s disease: a clinicopathological prospective study. Acta Neurol Scand 139:76–81. https://doi.org/10.1111/ane.13028
Skogseth R, Hortobágyi T, Soennesyn H, Chwiszczuk L, Ffytche D et al (2017) Accuracy of clinical diagnosis of dementia with Lewy bodies versus neuropathology. J Alzheimer’s Dis 59:1139–1152. https://doi.org/10.3233/jad-170274
Thal DR, Rub U, Orantes M, Braak H (2002) Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. https://doi.org/10.1212/WNL.58.12.1791
Tsuang D, Leverenz JB, Lopez OL et al (2013) APOE ϵ4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 70:223–228. https://doi.org/10.1001/jamaneurol.2013.600
Twohig D, Nielsen HM (2019) α-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener. https://doi.org/10.1186/s13024-019-0320-x
Walker JM, Richardson TE (2023) Cognitive resistance to and resilience against multiple comorbid neurodegenerative pathologies and the impact of APOE status. J Neuropathol Exp Pathol 82:110–119. https://doi.org/10.1093/jnen/nlac115
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191. https://doi.org/10.1093/bioinformatics/btq340
Yin L, Zhang H, Tang Z, Xu J, Yin D et al (2021) rMVP: a memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom Proteomics Bioinform 19:619–628. https://doi.org/10.1016/j.gpb.2020.10.007
Zhao N et al (2020) APOE4 exacerbates alpha-synuclein pathology and related toxicity independent of amyloid. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aay1809
Acknowledgements
This research is based on data collected by the National Alzheimer’s Coordinating Center (NACC), the Rush University Medical Center, and genetic data distributed by the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS).
Funding
This work was supported by the National Institute on Aging grants 1R01-AG060747 (MDG) and K99AG075238 (MEB), the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie Actions grant 890650 (YLG), and the Alzheimer’s Association grant AARF-20–683984 (MEB).
Author information
Authors and Affiliations
Contributions
Concept and design: YLG, MDG. Data analysis: ST and YLG. Drafting of the manuscript: ST, YLG, NK, MDG. Critical review of the manuscript: ST, YLG, NK, MEB, and MDG. Funding acquisition: YLG, MEB, and MDG.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Participants or their caregivers provided written informed consent in the original studies. The current study protocol was granted an exemption by the Stanford University institutional review board because the analyses were carried out on deidentified data; therefore, additional informed consent was not required.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary Materials
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Talyansky, S., Le Guen, Y., Kasireddy, N. et al. APOE-ε4 and BIN1 increase risk of Alzheimer’s disease pathology but not specifically of Lewy body pathology. acta neuropathol commun 11, 149 (2023). https://doi.org/10.1186/s40478-023-01626-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-023-01626-6