
Abstract
We show that Polycomb Repressive Complex-2 (PRC2) components EED and EZH2 maintain neural identity in cerebellar granule neuron progenitors (CGNPs) and SHH-driven medulloblastoma, a cancer of CGNPs. Proliferating CGNPs and medulloblastoma cells inherit neural fate commitment through epigenetic mechanisms. The PRC2 is an epigenetic regulator that has been proposed as a therapeutic target in medulloblastoma. To define PRC2 function in cerebellar development and medulloblastoma, we conditionally deleted PRC2 components Eed or Ezh2 in CGNPs and analyzed medulloblastomas induced in Eed-deleted and Ezh2-deleted CGNPs by expressing SmoM2, an oncogenic allele of Smo. Eed deletion destabilized the PRC2, depleting EED and EZH2 proteins, while Ezh2 deletion did not deplete EED. Eed-deleted cerebella were hypoplastic, with reduced proliferation, increased apoptosis, and inappropriate muscle-like differentiation. Ezh2-deleted cerebella showed similar, milder phenotypes, with fewer muscle-like cells and without reduced growth. Eed-deleted and Ezh2-deleted medulloblastomas both demonstrated myoid differentiation and progressed more rapidly than PRC2-intact controls. The PRC2 thus maintains neural commitment in CGNPs and medulloblastoma, but is not required for SHH medulloblastoma progression. Our data define a role for the PRC2 in preventing inappropriate, non-neural fates during postnatal neurogenesis, and caution that targeting the PRC2 in SHH medulloblastoma may not produce durable therapeutic effects.
Similar content being viewed by others


Introduction
During brain development, epigenetic inheritance specifies cell identities, directing progenitor cell differentiation along trajectories determined by their lineage. Thus, rhombic lip progenitors give rise to progeny with specific neural fates, including cerebellar granule neurons (CGNs) [1] and unipolar brush cell neurons (UBCs) [2]. Lineage tracing by scRNA-seq shows that CGNPs differentiate into neurons and not into other types of cells [3]. However, hyperactivation of SHH signaling can transform CGNPs, resulting in medulloblastoma [4,5,6,7], a primitive neuro-ectodermal tumor that is the most common malignant pediatric brain tumor. While most medulloblastoma cells adhere to a neural fate trajectory [8, 9], a small fraction of tumor cells differentiates as glia [3, 10]. In giving rise to glia, SHH medulloblastoma cells demonstrate increased pluripotency beyond the neural fate trajectory of CGNPs but maintain commitment to typically neuro-ectodermal fates. Epigenetic mechanisms thus maintain neuroectodermal lineage commitment through generations of proliferating progenitors during brain development and through generations of proliferating tumor cells in medulloblastoma.
PRC2 is a chromatin regulatory complex that has been shown to positively or negatively regulate neural progenitor proliferation in different contexts. The core subunits of the PRC2 include EZH1/2, EED, SUZ12, and RbAp46/48. By regulating trimethylation of H3K27 (H3K27me3), PRC2 suppresses CDKN2A, thus increasing neural progenitor proliferation in the hippocampus [11]. However, PRC2 also inhibits SHH-induced transcriptional regulation by depositing H3K27me3 at bivalent chromatin sites in the promoter regions of SHH pathway target genes [12]. As SHH signaling drives CGNP proliferation [13, 14] and medulloblastoma tumorigenesis [7, 15,16,17,18], PRC2-mediated repression of SHH target genes suggests the potential to inhibit proliferation in SHH-driven cells. In addition to affecting proliferation, PRC2 regulates hippocampal progenitor differentiation [11], consistent with of its role in fate commitment in diverse developmental contexts, from drosophila larva [19] to mammalian embryonic stem cells [20, 21]. PRC2 may similarly contribute to cerebellar development by regulating CGNP proliferation and differentiation.
As in brain development, prior studies have shown divergent roles for the PRC2 in cancer, positively or negatively regulating tumor growth in different types of tumors [64, 65]. SHH medulloblastomas upregulate EZH2, the catalytic subunit of the PRC2, suggesting a growth-promoting function [12], and EZH2 inhibition has been proposed as a medulloblastoma therapy [22]. A more complex relationship, however, is suggested by the finding that pharmacologically increasing H3K27me3 levels in cultured medulloblastoma cells decreases tumor cell viability [12]. Moreover, prior studies show different effects on tumor growth in murine medulloblastoma models with widespread or mosaic Eed deletion [23]. The role of EZH2 in catalyzing inhibitory H3K27me3 marks and its non-canonical PRC2-independent functions has raised questions about the therapeutic potential of EZH2 inhibition [24,25,26].
To determine how PRC2 components affect cerebellar development, we deleted Eed conditionally in the CGNP-specific Atoh1. We then compared CGNP proliferation, apoptosis, differentiation, and gene expression in the resulting Eed-deleted mice, Eed-intact controls, and to mice with Ezh2-deleted CGNPs. To determine PRC2 function in SHH medulloblastoma, we bred Eed-deleted and Ezh2-deleted mouse lines with mice genetically engineered to develop SHH medulloblastoma from CGNPs. Our data show that CGNPs and medulloblastoma cells require PRC2 to maintain neural fate commitment, and that EED is specifically required for cerebellar growth, but neither EED nor EZH2 are required for medulloblastoma progression.
Results
EED is required for proper cerebellar growth
CGNPs showed robust EED and EZH2 expression during postnatal neurogenesis, with initially low but detectable H3K27 trimethylation that increased as CGNPs differentiated (Fig. 1A). To reduce variation in developmental age when making comparison between genotypes, we used Eedf/f littermates of EedcKO mice to represent normal development. These mice did not inherit Cre and showed intact EED expression. During the period of CGNP proliferation from postnatal day 1 (P1) through P15, cells of the CGNP lineage segregate spatially according to developmental state, with undifferentiated and early differentiating CGNPs in the external granule cell layer (EGL) and differentiated, post-mitotic CGNs in the internal granule cell layer (IGL). At P7, CGNPs throughout the EGL expressed EED and EZH2, and H3K27me3 was detectable in the EGL and IGL (Fig. 1A). H3K27me3 increased in the CGNs of the IGL by P21. Expression of PRC2 components thus continued throughout CGNP development and H3K27me3 correlated with neuronal maturation, suggesting a role for the PRC2 in the differentiation process.

PRC2 function is developmentally regulated in the postnatal cerebellum and is required for normal development. A–C Representative sagittal sections of A control, B EedcKO, and C Ezh2cKO cerebella at the indicated ages. H&E-stained sections show the distribution of cells in the EGL and IGL. IHC shows distribution of EED, EZH2, and H3K27me3, with DAPI counterstain. Red arrowheads highlight Purkinje cells, identified by their typical large nuclei. White arrow indicates a residual EGL population in P21 Eed knockouts. D Western blot for H3K27me3 and H3K27Ac in replicate samples of indicated genotypes, quantified below, using two-tailed Student’s t-tests. E Western blot for H3K4me3 and H2AK119ub in replicate samples of indicated genotypes, quantified below, using two-tailed Student’s t-tests. F IHC for pRB and cC3 in representative sections, with quantitative analysis using two-tailed Student’s t-tests. *, **, and *** denote p < 0.05, p < 0.01 and p < 0.001 respectively, relative to controls
In light these data, we analyzed Eed and Ezh2 function in CGNP using conditional genetic deletion. Prior studies showed that conditional Ezh2 deletion in the dorsal neural tube at embryonic day E12.5 results in cerebellar hypoplasia, with decreased numbers of both Purkinje cells and CGNPs [27]. However, the loss of Purkinje neurons may indirectly alter CGNPs, which depend on SHH released by Purkinje cells in order to proliferate [28]. To disrupt PRC2 genes specifically in CGNPs, we used conditional deletion by expressing Cre from the Atoh1 (aka Math1) promoter, which in the cerebellum is CGNP specific [29]. We interbred Math1-Cre transgenic mice that express Cre recombinase in the CGNP population with mice harboring conditional alleles of either Eed (Eedfl/fl) or Ezh2 (Ezh2fl/fl), to generate Math1-Cre/Eedf/f (EedcKO) and Math1-Cre/Ezh2f/f (Ezh2cKO) mice.
Eed deletion resulted in marked depletion of the CGNP lineage, with markedly less populous EGL in 5/5 P7 EedcKO cerebella compared to controls (Fig. 1B). All EedcKO mice subjected to immunohistochemistry (n = 3) showed loss of both EED protein and EZH2 protein throughout the EGL (Fig. 1B), consistent with reports that EED loss destabilizes EZH2 [30]. H3K27me3 expression was absent in the EedcKO CGNPs, consistent with PRC2 disruption (Fig. 1B). In contrast, Purkinje cells showed robust EED, EZH2, and H3K27me3; however, these cells, which typically localize at the outer margin of the CGNs, were scattered throughout the depopulated IGL and remained identifiable by their typical morphology with large nuclei and cell bodies (Fig. 1B, red arrows). At P21, 5/5 EedcKO cerebella were clearly hypoplastic, with no clear IGL, and with inappropriately persistent, small populations of H3K27me3 + cells in the EGL (Fig. 1B; white arrow). Consistent with cerebellar impairment, EedcKO mice showed tremor and ataxia and frequently fell while walking. EED was thus required for PRC2 stability, demonstrated by loss of EZH2 protein in EedcKO CGNPs, for H3K27 tri-methylation in CGNPs, and for cerebellar growth and function.
Ezh2 deletion in contrast did not cause neurologic abnormalities or overt cerebellar hypoplasia (Fig. 1C). P7 Ezh2cKO CGNPs showed intact EED protein expression despite widespread absence of EZH2 (Fig. 1C). H3K27me3 was clearly reduced in EedcKO CGNPs and CGNs compared to controls (Fig. 1C). Thus, while H3K27me3 was altered in both EedcKO and Ezh2cKO mice, Eed deletion more effectively disrupted the PRC2 complex, as demonstrated by loss of EZH2 in EedcKO cerebella, and more profoundly altered cerebellar growth.
We compared chromatin marks in EedcKO and Ezh2cKO cerebellar lysates, using controls mice that did not inherit the Cre transgene. Western blot analysis showed that both Eed and Ezh2 deletions disrupted PRC2 function and we did not identify differences in chromatin marks. Both deletions reduced H3K27me3 as expected (Fig. 1D). However, residual H3K27me3 from cerebellar cells outside the Atoh1 lineage that were not subject to conditional deletion produce detectable signal that may have obscured differences. We noted a trend toward increased H3K27 acetylation in both EedcKO and Ezh2cKO (Fig. 1D) that was not statistically significant but would be consistent with the previously observed increased H3K27Ac in H3K27me3-depleted embryonic stem cells [31, 32]. EedcKO CGNPs did not show differences in H2AK119 monoubiquitination, indicating that PRC1 function was not altered, and did not show differences in H3K4me3 (Fig. 1E). Within the resolution of our western blot assays, therefore, deletion of Eed or Ezh2 resulted in similar chromatin changes, limited to H3K27 modification.
Eed deletion decreases progenitor proliferation and increases apoptosis
We investigated developmental processes that mediate growth failure in EedcKO cerebella. Eed deletion was previously shown to decrease proliferation and increased apoptosis in hippocampal progenitors [11]. As both decreased CGNP proliferation and increased CGNP apoptosis can cause cerebellar hypoplasia [13, 33,34,35], we quantified proliferative CGNPs, identified by expression of phosphorylated-RB (pRB) and apoptotic CGNPs, identified by expression of cleaved Caspase-3 (cC3). We used as controls littermates of EedcKO mice that had at least one intact Eed allele, including Eedf/f mice and Math1-Cre/Eedf/+ mice. Similar to hippocampal progenitors, EedcKO CGNPs showed decreased proliferation and increased apoptosis (Fig. 1F), implicating both processes in the cerebellar hypoplasia of EedcKO mice.
Eed deletion produces discrete patterns of transcriptomic change
To resolve the effects of Eed-deletion in individual cells, we subjected Eed-deleted and control CGNPs to scRNA-seq analysis. We harvested cerebella from 3 replicate P7 EedcKO mice and subjected them to Drop-seq bead-based scRNA-seq preparation [66]. We then sequenced the resulting bar-coded libraries and identified cells by bead-specific bar codes. After QC and filtering, we included 2377 cells for analysis. We compared these cells to previously sequenced data from 6631 cells from 5 replicate P7 WT cerebella. To adjust for differences in sequencing depth in EedcKO and WT cells, we down-sampled the WT cells to 46.5% of their original depth to achieve similar sequencing depth between conditions, consistent with best practices [36].
We subjected scRNA-seq data from EedcKO and WT cells to principal component analysis (PCA) and Louvain clustering, as in our prior studies [3, 37, 38], to identify 20 clusters with distinctive gene expression, numbered from 0, most populous, to 19, least populous (Additional file 1: Fig. S1). We then determined cluster-specific gene expression profiles by comparing the expression of each detected gene in cells within the cluster versus all cells outside the cluster (Additional file 2: Data 1). In Clusters 4 and 6, we noted that cluster-specific genes were expressed by discrete subpopulations, suggesting that further sub-clustering of these clusters would be informative. Re-iterative clustering split Cluster 4 into 4_0 and 4_1 and Cluster 6 into 6_0 and 6_1, which showed discrete, cluster-specific patterns of gene expression (Additional file 2: Data 1). We mapped these 22 clusters by color code on the UMAP projection to visualize the different populations (Fig. 2A).

scRNA-seq shows differences in the composition of EedcKO cerebella, including a population of myoid cells not found in normal brain. A UMAP projection of cells from EedcKO and control cerebella, grouped by transcriptomic similarities into color-coded clusters. B Bubble Plot shows the magnitude and frequency of the expression of indicated cell type markers in each cluster. The genes and clusters are ordered according to the hierarchical clustering analysis, with groupings indicated along the top and right margins of the Bubble Plot. C UMAP projection of cells from EedcKO and control cerebella, disaggregated by genotype, with clusters indicated by the same color code as in (A). D Comparison of each normalized cluster population in EedcKO and control cerebella. Dots represent values for individual replicates, bars indicate the means and whiskers indicate the SEM. *** indicates p < 0.0001,** indicates p < 0.001, * indicates p < 0.05; Dirichlet regression was used to compare normalized cluster populations. E Bar graphs showing the mean proportional population of each cluster in EedcKO and control cerebella, with the subsets of proliferating CGNPs and differentiating CGNPs and CGNs also shown separately for clarity. In B and D, CGNP and CGN cluster numbers are presented in red for clarity
The differential gene expression patterns identified the cell-type of each cluster and demonstrated diverse types of cells that were expected in cerebellar tissue (Fig. 2A,B; Table 1). We identified Clusters 0, 1, 2, 3, 4_1, 5, 6_1, 10, 14, 16, and 17 as CGNP lineage cells in a spectrum of differentiation states, from cycling CGNPs to differentiated CGNs, based on expression of SHH target genes including Sfrp1, neural progenitor-related genes including CGNP transcription factor Barhl1 and axonal pathfinding receptor Cntn2, and neuronal genes including the transcription factor Rbfox3. Different markers identified diverse types of non-neural cells, including astrocytes (Cluster 7), oligodendrocytes (Cluster 9), vascular fibroblasts (Cluster 11), myeloid cells (Cluster 12), endothelial cells (Cluster 13), pericytes (Cluster 18), and ependymal cells (Cluster 19). Markers also identified neural populations that were outside the Atoh lineage but expected in the cerebellum, including gabaergic neural progenitors (Cluster 6_0), identified by expression of Ascl1 and Pax3, gabaergic interneurons (Cluster 4_0) identified by Pax2 and Gad1, and Purkinje cells identified by Gad1 and Calb1 [3, 39]. In contrast to all other clusters, which demonstrated markers expected in the brain, Cluster 15 expressed the muscle cell marker Myog, suggesting myoid differentiation.
We disaggregated the UMAP to make separate projections from EedcKO and WT mice, demonstrating that each genotype distributed differently across the clusters (Fig. 2C). To compare the cluster populations in EedcKO and WT samples statistically, since each replicate contributed different numbers of cells to the analysis, we normalized the population of cells in each cluster from each replicate to the total number of cells from that replicate. These proportional populations were inter-related by the normalization to the whole, and therefore could not be analyzed by individual t-tests, which assume independence. We therefore used Dirichelet regression analysis to compare the proportional cluster populations [40].
As suggested by the disaggregated UMAP (Fig. 2C), Dirichelet regression analysis showed that the populations of specific clusters were significantly different in EedcKO cerebella compared to controls (Fig. 2D). We found significant differences in the populations of Atoh1-lineage cell types that were subject to conditional deletion, and also of clusters outside the Atoh1 lineage. Within the Atoh1 lineage, CGNP/CGN Clusters 0, 1, 2, 3, 10, and 16 were more populous in controls, while CGNP/CGN Clusters 4_1, 5, 6_1, and 8 were more populous in the EedcKO (Fig. 2D). Outside the Atoh1 lineage, Purkinje cells, astrocytes, and vascular fibroblasts were increased in controls and pericytes were increased in the EedcKO. These cell types were not subject to Eed deletion, and differences in their populations reflect non-cell autonomous effects. Additionally, the myoid Cluster 15 was present only in the EedcKO cerebella (Fig. 2C,D). While proliferative Clusters 5 and 6_1 were relatively increased in EedcKO mice, proliferative Clusters 0, 3, and 10 were decreased, resulting in a net decrease in proliferative populations (Fig. 2E), consistent with the decrease in pRB + CGNPs.
Eed deletion permits divergence from the neural fate of CGNPs
The Myog-expressing Cluster 15 showed multiple genes typical of muscle cells, including troponins, Myl1, Cav3, and Smyd1 (Fig. 3A). We did not observe this myoid transcriptomic pattern in any cells from control mice. Cluster 15 cells also up-regulated Cdkn1a and Cdkn1c (Fig. 3A,B), which are known to be suppressed by the PRC2 in other types of cells [41,42,43,44]. Based on the up-regulation of PRC2-suppressed genes, we infer that Cluster 15 cells were within the Atoh1 lineage, and that conditional Eed deletion caused these Atoh1-lineage cells to diverge from the expected CGNP trajectory.

Expression of PRC2 targets in EedcKO and Ezh2cKO cerebella. A Bubble Plot shows the magnitude and frequency of the expression of indicated genes in each cluster, and feature plot of Cdkn1a + cells (red), color-coded over the UMAP from (2C). B Feature plots of Hoxa9+, Hoxa7+, and Cdkn2a+ cells (red), color-coded over the UMAP from (2C). C and D Representative immunofluorescence in sagittal sections of WT, EedcKO, and Ezh2cKO cerebella, showing expression of C CDKN2A or D MYOG and SMYD1, with DAPI counterstain. The CDKN2A + and MYOG + fractions in cerebellar sections from 3 replicate mice of indicated genotypes are graphed on the right. EedcKO, and Ezh2cKO replicates were compared to controls using one-tailed Student’s t-tests and to each other using two-tailed Student’s t-tests. *, **, and *** denote p < 0.05, p < 0.01 and p < 0.001 respectively
Eed deletion permits expression of PRC2-inhibited CDK inhibitors and Hox genes
While only Cluster 15 cells showed Cdkn1a and Cdkn1c, the broad set of Atoh1-lineage cells, including CGNPs, CGNs, and Cluster 15 cells expressed other genes typically suppressed by the PRC2, including Cdkn2a, Hoxa9, and Hoxa7 [44,45,46,47] (Fig. 3B). Indeed, these markers differentiated the CGNP/CGN clusters enriched in EedcKO cerebella (Clusters 4_1, 5, 6_1, and 8) from the CGNP/CGN clusters enriched in WT samples (Clusters 0, 1, 2, 3, 10, and 16). The absence of Cdkn2a, Hoxa9, and Hoxa7 in WT cells (Fig. 3B) indicates that each of these genes are normally suppressed by the PRC2 in CGNP-lineage cells.
Ezh2 deletion, like Eed deletion, permits myoid differentiation
We used immunohistochemistry (IHC) to determine if specific transcripts from PRC2-target genes were translated in EedcKO cerebella, and to probe Ezh2cKO cerebella for similar patterns of expression. Cells expressing CDKN2A, MYOG, and SMYD1 were absent in controls, including WT cerebella and cerebella from Eedf/f and Eedf/+ littermates of EedcKO mice. As the frequency of CDKN2A + , MYOG + , and SMYD1 + cells in mutant cerebella could not be lower than in control cerebella, we used one-tailed statistical tests to compare mutant to control replicates, and two-tailed statistical tests for comparison between EedcKO and Ezh2cKO genotypes.
EedcKO cerebella showed CDKN2A + CGNPs throughout the EGL; the fraction of CDKN2A + cells was significantly greater than in WT or Ezh2cKO cerebella (Fig. 3C). Ezh2cKO cerebella showed significantly more CDKN2A + CGNPs than controls, but fewer than EedcKO cerebella. Both PRC2 component mutations thus resulted in up-regulation of CDKN2A, with EedcKO showing a higher fraction of affected cells.
Both EedckO and Ezh2ckO cerebella showed cells expressing the muscle cell transcription factor MYOG, while no cells expressed MYOG control cerebella, EedcKO cerebella (Fig. 3D). MYOG + cells were significantly more numerous in EedckO than Ezh2cKO cerebella (Fig. 3D). EedckO cerebella also showed cells expressing the muscle cell chromatin regulator SMYD1, which was absent in Ezh2ckO and control cerebella (Fig. 3D). Pairwise comparison of SMYD1 expression in EedckO cerebella (present in 3/3) versus Ezh2ckO (absent in 3/3) or controls (absent in 3/3) using the Barnard’s exact test showed p values of 0.03. Transcripts that were not found in control CGNPs, including Cdkn2a and Myog, were thus translated into proteins in both EedckO and Ezh2ckO cerebella, with SMYD1 expression in EedcKO cerebella demonstrating an additional myoid gene ectopically expressed.
Increased activation of the intrinsic apoptotic pathway in Eed cKO CGNPs
To probe the mechanisms of increased cell death in EedcKO CGNPs, we analyzed p53 function and the expression of genes regulating apoptosis. CGNPs are highly sensitive to p53-mediated activation of the intrinsic apoptotic pathway [48, 49] and also to direct activation the intrinsic apoptotic pathway triggered by changes in apoptotic regulators [35, 50]. Cdkn1a expression in Cluster 15 suggested that Eed deletion might activate p53-dependent transcription, potentially mediating the observed increased apoptosis. To determine if other apoptotic regulators were altered in EedcKO CGNPs, we compared the expression of pro-apoptotic BH3-only genes in the CGNP and CGN clusters of EedcKO and control mice (Fig. 4A).

Increased spontaneous, p53-indepenedent CGNP apoptosis contributes to growth failure in Eed-deleted cerebella. A Heat map of BH3-only gene expression in each CGNP/CGN cluster. Hierarchical clustering based on BH3-only genes grouped together the CGNP clusters that were enriched in EedcKO mice (red box), and identified a set of apoptosis regulators (blue box) that was up-regulated in this group. B Heat map of BH3-only gene expression in the combined set of CGNP/CGN clusters, comparing EedcKO and control genotypes, with the blue box highlighting the same apoptosis regulators as in (A). C Representative H&E-stained sagittal sections of P21 WT, EedcKO, Eed/Trp53dKO, and Eed/Bax/BaktKO mice. * indicates regions of IGL. Arrow indicates regions of persistent or absent EGL
Hierarchical clustering based on BH3-only gene expression distinguished the 3 proliferative clusters enriched in EedcKO cerebella, Clusters 4_1, 5, and 6_1, which showed increased expression of Pmaip1 (aka Noxa), Bad, Bcl2l11 (aka Bim), and Hrk (Fig. 4A). Similarly, comparing the combined set of all cells in CGNP and CGN clusters, EedcKO cells showed a distinctive pattern of BH3-only gene expression, with increased Bbc3 (aka Puma), Pmaip1 (aka Noxa), Bad, Bcl2l11 (aka Bim), and Hrk (Fig. 4B). These data suggested that Eed deletion may increase apoptosis by direct activation of the intrinsic apoptotic pathway.
To determine whether p53 signaling or the intrinsic apoptotic pathway contributed to cerebellar hypoplasia in EedcKO mice, we combined Eed deletion with deletion of either Trp53 or both Bax and Bak. We bred EedcKO mice with Trp53fl/fl mice to generate Math1-Cre/Eedfl/fl/Trp53fl/fl (Eed/Trp53dKO) mice and bred EedcKO mice with Baxfl/fl/Bak−/− mice to generate Math1-Cre/Eedfl/fl/Baxfl/fl/Bak−/− (Eed/Bax/BaktKO) mice. Eed/Trp53dKO mice showed cerebellar hypoplasia similar to EedcKO mice (Fig. 4C). In contrast, cerebella in Eed/Bax/BaktKO mice were markedly less abnormal, with relatively increased CGNs within the IGL and more appropriate layering of Purkinje cells between the IGL and molecular layers (Fig. 4C). Bax/Bak co-deletion did not fully rescue the effects of Eed knockout, as the IGL remained less densely populated than WT controls and the molecular layer contained ectopic cells (Fig. 4C). The absence of rescue in Eed/Trp53dKO mice indicates that growth failure in the EedcKO cerebella was p53-independent. The partial rescue by co-deletion of Bax and Bak, however, demonstrates that p53-independent activation of the intrinsic apoptosis pathway contributed to cerebellar hypoplasia in EedcKO mice. Bax/Bak co-deletion also increased the myoid population (Additional file 1: Fig. S2), indicating that myoid cells were typically removed from EedcKO cerebella by apoptosis.
PRC2 function is not required for SHH medulloblastoma tumorigenesis
Mutations that disrupt cerebellar growth may identify genes required for growth of medulloblastoma [51] and the PRC2 has been proposed as a target for medulloblastoma therapy. Therefore, to determine whether medulloblastomas depend on Eed and the PRC2, we bred EedcKO and Ezh2cKO mice with SmoM2 mice [52]. SmoM2 mice harbor a Cre-conditional transgene with an oncogenic allele of the SHH receptor component Smo. Mice that inherit both Math1-Cre and SmoM2 develop medulloblastoma with 100% penetrance and without treatment die of tumor progression by P50 [6]. We have shown that these tumors recapitulate the gene expression patterns and cellular diversity of SHH medulloblastomas resected from patients [3, 38, 53]. By interbreeding EedcKO and Ezh2cKO mice with SmoM2 mice, we generated pups with the genotypes Math1-Cre/Eedfl/fl/SmoM2 (M-Smo/EedcKO) and Math1-Cre/Ezh2fl/fl/SmoM2 (M-Smo/Ezh2cKO). To generate control mice with SHH medulloblastomas with intact PRC2, we bred Math1-Cre and SmoM2 mice to generate Math1-Cre/SmoM2 (M-Smo) controls. We then compared medulloblastomas in M-Smo/EedcKO, M-Smo/Ezh2cKO, and M-Smo mice.
M-Smo/EedcKO, M-Smo/Ezh2cKO, and M-Smo mice all developed medulloblastomas with 100% frequency by P10 (Fig. 5A). M-Smo tumors showed heterogeneous H3K27me3, with strongest H3K27me3 expression in the most differentiated elements, similar to WT cerebella (Fig. 5B). Deletion of either Eed or Ezh2 disrupted PRC2 function, as both M-Smo/EedcKO and M-Smo/Ezh2cKO showed minimal H3K27me3 in tumor cells, with residual H3K27me3 in interspersed stromal cells demonstrating the effectiveness of the staining technique (Fig. 5B). M-Smo/EedcKO and M-Smo/Ezh2cKO mice showed shorter survival times compared to controls, indicating that PRC2-mutant medulloblastomas progressed more quickly (Fig. 5C).

Loss of PCR2 function accelerated progression of SHH medulloblastomas. A Representative H&E-stained sagittal sections of medulloblastomas in M-Smo, M-Smo/EedcKO, and M-Smo/Ezh2cKO mice at postnatal day 12 (P12) and comparison of cross-sectional area of tumors in midline sections of replicate mice of each genotype, usiong two-tailed Student’s t-test. B Representative IF stains showing H3K27me3 in M-Smo, M-Smo/EedcKO, and MSmo/Ezh2cKO tumors, and H3K27me3 western blots of medulloblastomas from 3 replicate mice of each genotype. C Kaplan–Meier curves compare the survival times of M-Smo, M-Smo/EedcKO, and MSmo/Ezh2cKO mice, using the Log-Rank test. D IHC for cC3 in representative sections, with quantitative analysis of replicate samples, compared by two-tailed Student’s t-test. E IHC for NEUN in representative sections, with western blot of replicate samples quantified on the right, compared by two-tailed Student’s t-test. F–J Flow cytometry analysis of dissociated tumors of indicated age and genotype, showing F, H pRB + fractions and G, I cell cycle distribution of pRB + cells, and J EdU MFI, compared by two-tailed Student’s t-test. *, **, and *** denote p < 0.05, p < 0.01 and p < 0.001 respectively, relative to controls
We compared apoptosis and terminal differentiation in M-Smo/EedcKO, M-Smo/Ezh2cKO, and M-Smo tumors. Eed-deleted and Ezh2-deleted medulloblastomas showed less apoptosis, demonstrated smaller fractions of cC3 + cells compared to control tumors (Fig. 5D), in contrast to the increased cell death that we noted in EedcKO cerebella. Medulloblastomas in M-Smo/EedcKO and M-Smo/Ezh2cKO mice also showed reduced neuronal differentiation, demonstrated by less abundance of neuronal marker NEUN (Fig. 5E). PRC2 disruption therefore decreased both apoptosis and terminal differentiation in SHH medulloblastoma.
Transient growth suppression in M-Smo/Eed cKO medulloblastomas
While M-Smo/EedcKO tumors progressed more rapidly than M-Smo control tumors, we noted consistently smaller cross-sectional area in midline sections of M-Smo/EedcKO tumors at P12 compared to either M-Smo/Ezh2cKO or control tumors (Fig. 5A), suggesting that Eed deletion might produce an initial growth suppression, followed by more rapid growth. To analyze tumor growth dynamics, we compared RB phosphorylation and cell cycle progression in M-Smo/EedcKO and M-Smo tumors. We injected EdU into 3–5 replicate mice of each genotype at either P12 or P18, then harvested tumors 1 h after EdU injection and quantified pRB and EdU uptake by flow cytometry.
P12 M-Smo/EedcKO tumors showed smaller fractions of pRB + cells compared to P12 M-Smo control tumors (Fig. 5F), indicating that fewer tumor cells were proliferative. Within the pRB + fractions of P12 M-Smo/EedcKO tumors, however, more cells were in S-phase compared to the pRB + fractions of P12 M-Smo tumors (Fig. 5G), indicating more rapid progression from G1. P12 M-Smo/EedcKO tumors thus contained smaller proliferative populations, consistent with smaller tumor size. However, the cells that were proliferating in P12 M-Smo/EedcKO medulloblastomas were cycling more rapidly, suggesting a transition to faster tumor growth.
Consistent with more rapid tumor growth after P12, by P18 M-Smo/EedcKO tumors no longer showed fewer pRB + cells (Fig. 5H). Moreover, pRB + cells in M-Smo/EedcKO tumors continued to show more rapid cycling, demonstrated by greater S-phase fractions, compared to pRB + cells from P18 M-Smo tumors (Fig. 5I). Supporting the more rapid S-phase progression in M-Smo/EedcKO tumors at both P12 and P18, pRB + cells from M-Smo/EedcKO tumors showed higher EdU median fluorescence intensity (MFI), indicating increased EdU uptake within the period of EdU exposure (Fig. 5J). The smaller pRB + population in M-Smo/EedcKO tumors at P12, the increased rate of proliferation within the pRB + population, and the similar pRB + population at P18 are all consistent with a biphasic effect of Eed deletion on tumor growth, in which tumors initially grew more slowly and then accelerated, producing shorter survival times.
Up-regulation of PRC2 target genes without growth suppression in PRC2-mutant medulloblastomas
We investigated whether medulloblastomas with deletion of Eed or Ezh2 up-regulated the same PRC2 targets that were up-regulated in EedcKO CGNPs. Both M-Smo/EedcKO and M-Smo/Ezh2cKO medulloblastomas showed frequent CDKN2A+ cells which were not observed in M-Smo control tumors (Fig. 6A). CDKN2A suppressed RB phosphorylation less effectively in tumor cells than in CGNPs, as the pRB+ fractions of CDKN2A+ cells were significantly higher in M-Smo/EedcKO and M-Smo/Ezh2cKO medulloblastomas compared to P7 EedcKO cerebella (Fig. 6B). Moreover, the pRB+ fraction of CDKN2A+ cells correlated with tumor growth; M-Smo/Ezh2cKO tumors, which progressed faster than M-Smo/EedcKO tumors, showed higher pRB+ fractions of CDKN2A+ cells at P12. By P18, when proliferation accelerated in M-Smo/EedcKO tumors, the pRB+ fractions of CDKN2A+ cells was also increased, to become similar to the P12 M-Smo/Ezh2cKO tumors. CDKN2A was thus up-regulated in both Eed-deleted and Ezh2-deleted medulloblastomas but did not restrict RB phosphorylation or tumor progression.

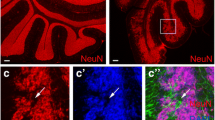
Up-regulation of PRC2 targets and myoid differentiation in both Eed-deleted and Ezh2-deleted SHH medulloblastomas. A Representative CDKN2A IHC in medulloblastomas of indicated genotypes, with quantification in replicate mice of each genotype. B Representative images of CDKN2A/pRB dual staining and quantification of pRB + fractions of CDKN2A-expressing cells in the indicated ages and genotypes. C Representative MYOG and SMYD1 IHC in medulloblastomas of indicated genotypes with quantification of MYOG + cells in replicate mice of each genotype. D Representative H&E-stained section from a medulloblastoma in a M-Smo/Ezh2cKO mouse, with myoid cells (arrows), and quantification in replicate mice of the indicated genotypes. *, **, and *** denote p < 0.05, p < 0.01 and p < 0.001 respectively, relative to controls
Analysis of muscle markers MYOG and SMYD1 showed inappropriate myoid differentiation in M-Smo/EedcKO and M-Smo/Ezh2cKO tumors (Fig. 6C), as seen in EedcKO cerebella. The expression of muscle genes in M-Smo/EedcKO and M-Smo/Ezh2cKO medulloblastomas suggested similarity to the clinically observed medulloblastoma variant, medullomyoblastoma. To determine if the M-Smo/EedcKO and M-Smo/Ezh2cKO tumors recapitulated medullomyoblastoma histopathology, we submitted H&E sections from replicate M-Smo/EedcKO, M-Smo/Ezh2cKO, and M-Smo tumors to a blinded analysis. Two experienced pediatric neuropathologists counted myoid cells, defined by key morphologic changes (Fig. 6D), in replicate sections, while blinded to the genotype. The resulting quantifications of myoid cells correctly distinguished control tumors from tumors with either Eed or Ezh2 deletion, which were not significantly different from each other (Fig. 6E). Deletion of either Eed or Ezh2 was therefore sufficient to allow myoid differentiation, reproducing the molecular and histologic features of medullomyoblastoma.
Discussion
Our data show that the PRC2 maintains neuronal fate commitment in cerebellar progenitors and in SHH medulloblastoma by preventing alternative, myoid differentiation. In the postnatal cerebellum, conditional deletion of PRC2 components Ezh2 and Eed in the Atoh1 lineage disrupted PRC2-mediated H3K27 trimethylation and caused a fraction of CGNPs to differentiate along a muscle cell trajectory. Eed deletion induced myoid differentiation in more cells than Ezh2 deletion and markedly impaired cerebellar growth through a combination of decreased proliferation and increased apoptosis. In medulloblastomas, deletion of either Eed or Ezh2 resulted in myoid differentiation, but neither deletion increased apoptosis or durably prevented tumor growth. Rather, deletion of either Eed or Ezh2 accelerated tumor progression.
Single-cell transcriptomic analysis of postnatal cerebella showed that Eed-deleted CGNPs inappropriately expressed genes typically suppressed by the PRC2, including Cdkn2a, Hoxa9, and Hoxa7. Up-regulation of Cdkn2a may be sufficient to explain reduced CGNP proliferation, as seen in Eed-deleted hippocampal progenitors [11]. Up-regulation of specific BH3-only genes suggested that direct activation of BAX or BAK may mediate increased CGNP apoptosis. Co-deletion studies of Eed plus either Trp53 or Bax AND Bak confirmed that increased apoptosis occurred by p53-independent activation of the intrinsic apoptosis pathway. The partial rescue of cerebellar growth in Eed/Bax/BaktKO mice demonstrates that inappropriate apoptosis contributed to growth failure and restricted the myoid population.
PRC2 function was not required for neural differentiation, as in Eed-deleted cerebella, most CGNPs were able to complete neural differentiation, achieving the neural fates of Clusters 4_1 and 8 in EedcKO mice. The inappropriate expression of Hox genes in these clusters did not prevent a recognizable CGN-like pattern of gene expression. In contrast, the myoid differentiation of Cluster 15 demonstrates that PRC2 disruption permitted new fate possibilities.
The specific diversion of CGNPs and medulloblastoma cells into myoid fates may be related to the normal expression of the MYOD1 transcription factor during postnatal development. MYOD1 is an early myogenic transcription factor that activates MYOG in developing muscle progenitors. Prior studies show that WT P7 cerebella and SHH medulloblastomas contain populations of cells that express MYOD1 without inducing MYOG or activating a myogenic program [3, 54]. In the Eed-deleted and Ezh2-deleted cerebella and medulloblastomas, however, Myod1+ cells also expressed Myog and adopted a myogenic trajectory. These data suggest that one function of the PRC2 in CGNPs is to suppress Myog and other myoid genes, allowing CGNPs to use the MYOD1 transcription factor to regulate neural development, without risk of inappropriate differentiation. We propose more generally that by suppressing inappropriate differentiation pathways, the PRC2 allows specifically neural functions of transcriptional regulators such as MYOD1, that have non-neural functions in other types of cells.
Eed deletion resulted in cerebellar hypoplasia that was not seen in Ezh2-deleted mice. The difference in phenotype may result from a more severe PRC2 disruption caused by loss of EED. In other cell types, EED protein is required for the stability of the other components of the PRC2 [30] and for PRC2 methyltransferase activity [55]. Consistent with these prior reports, we found that EedcKO CGNPs lacked both EED and EZH2 proteins, indicating destabilization of the entire PRC2. In contrast, EZH2 is not required for PRC2 stability and can be partially compensated by the homolog EZH1 in multiple cellular contexts [56, 57]. The PRC2 components that persist in Ezh2-deleted CGNPs, possibly with compensation from EZH1, may retain sufficient function to sustain cerebellar growth, and to suppress PRC2 target gene expression in most but not all CGNPs, resulting in fewer myoid cells in Ezh2cKO cerebella compared to EedcKO cerebella. Different phenotypes were similarly noted when either Eed or Ezh2 were deleted in intestinal epithelia [44]. Conditional deletion of Eed in the intestinal crypts decreased proliferation and caused hypoplasia, while Ezh2 deletion did not cause an overt phenotype. The continued proliferation in Ezh2-deleted intestinal crypts suggests that EZH1 may compensate for EZH2 loss in these cells [44], and a similar mechanism may explain sustained proliferation of Ezh2-deleted CGNPs.
Alternatively, as a shared component of the PRC1 and PRC2 complexes, EED loss may more broadly affect chromatin repression [58]. Our finding that Eed deletion did not affect levels of H2AK119 monoubiquitylation suggests that PRC1 activity was not altered in EedcKO CGNPs or medulloblastomas. The overlapping patterns of differential gene expression in EedcKO and Ezh2cKO cerebella show that suppression of myoid differentiation in CGNPs depends on PRC2 function. We cannot, however, exclude the possibility that the growth-suppressive effects of Eed deletion are mediated by functions of EED protein outside of the PRC2.
In SHH medulloblastomas, disrupting PRC2 activity through deletion of either Eed or Ezh2 was sufficient to allow widespread expression of genes typically suppressed by the PRC2, including the CDKN2A tumor suppressor. Neither PRC2 disruption nor CDKN2A expression, however, was sufficient for sustained suppression of tumor growth. Eed deletion reduced the overall proliferation rate in each P12 M-Smo/EedcKO tumor, producing transient growth suppression. Over time, however, a fraction of Eed-deleted tumor cells that were rapidly proliferative increased, driving ultimately faster progression. The initial reduction in tumor growth in M-Smo/EedcKO tumors was consistent with previous studies that showed anti-tumor effects of PRC2 disruption using EZH2 inhibitor treatment in models SHH medulloblastoma [22, 59, 60]. However, the shorter survival times in M-Smo/EedcKO and M-Smo/Ezh2cKO mice raise concern that PRC2 disruption may not produce durable anti-tumor effects.
Our genetic studies identify a role of PRC2 in regulating neural fate commitment of cerebellar progenitors and medulloblastoma cells, and implicate PRC2 disruption in the pathogenesis of medullomyoblastoma, a subtype of medulloblastomas characterized by myogenic differentiation of tumor cells [61, 62]. Our data also caution that pharmacologically inhibiting PRC2 function in medulloblastoma may hasten, rather than slow, tumor growth.
Methods and materials
Mice
We generated Ezh2cKO mice by breeding Math1-Cre mice (Jackson Labs Stock #011104), which express Cre recombinase in CGNPs, with Ezh2LoxP/LoxP (Jackson Labs Stock #022616). We generated EedcKO mice by breeding Math1-Cre mice with EedLoxP/LoxP mice (generously donated by Dr. Terry Magnuson). To generate Eed/Tp53dKO mice, we interbred Math1-Cre/EedLoxP/LoxP and Trp53LoxP/LoxP mice (Jackson Labs stock # 008462). To generate Eed/Bax/BaktKO mice, we interbred Math1-Cre/EedLoxP/LoxP and BaxLoxP/LoxP/Bak−/− mice (Jackson Labs stock # 006329).
To generate M-Smo mice, we crossed Math1-Cre mice with SmoM2 mice (Jackson Labs stock #005131) that harbor a Cre-conditional transgene comprising of an oncogenic allele of Smo, fused to the YFP coding sequence. We then crossed Math1-Cre/Ezh2LoxP/LoxP and Ezh2LoxP/LoxP/SmoM2LoxP/LoxP mice to produce M-Smo/Ezh2cKO mice and Math1-Cre/EedLoxP/LoxP and EedLoxP/LoxP/SmoM2LoxP/LoxP mice to produce M-Smo/EedcKO mice.
All mice were of species Mus musculus and crossed into the C57BL/6 background through at least five generations.
Histology and immunohistochemistry
Mouse brains were processed, immunostained, and quantified as previously described [67,68,69]. In brief, mice were placed under isoflurane anesthesia and decapitated. Harvested brains were fixed by immersion in 4% formaldehyde for 24 h and then transferred to a graded ethanol series and embedded in paraffin and sectioned along the sagittal midline. Samples were stained and imaged using an Aperio Scanscope and quantified via automated cell counting using Tissue Studio (Definiens).
Primary antibodies used were: H3K27me3 diluted 1:200 (Cell Signaling, #9733), pRB diluted 1:3000 (Cell Signaling, #8516), cC3 diluted 1:400 (Biocare Medical, #CP229C), NeuN diluted 1:10,000 (Millipore, MAB377), Myogenin (MYOG) diluted 1:500 (Abcam, ab124800), SMYD1 diluted 1:100 (ThermoFisher, PA5-84544), and CDKN2A diluted 1:500 (Abcam, ab241543). Stained images were counterstained with DAPI.
Western blot
Whole cerebella or tumors were harvested and homogenized in an SDS lysis buffer, which included SDS Solution (20%) (ThermoFisher 151-21-3), UltraPure™ 1 M Tris-HCI Buffer, pH 7.5 (ThermoFisher 15567027), UltraPure™ 0.5 M EDTA, pH 8.0 (ThermoFisher 15575038), Phenylmethanesulfonyl fluoride (≥ 98.5%; powder) (Millipore Sigma 329-98-6), Isopropyl alcohol (Mallinckrodt 3037), and purified water. Equal total protein concentrations were loaded from each sample and run on SDS-polyacrylamide gels (BioRad, #4561105, #4568094), transferred onto polyvinylidene difluoride membranes, and membranes blotted using a SNAP i.d. 2.0 Protein Detection System (Millipore). The following antibodies were used: H3K27me3 (Cell Signaling, #9733; 1:500 dilution), H3K4me3 (Cell Signaling, #9751; 1:500 dilution), H2AK119ub (Cell Signaling, #8240; 1:500 dilution), H3K27Ac (Cell Signaling, #8173; 1:500 dilution), NeuN (Millipore, MAB377; 1:500 dilution), β-actin (Cell Signaling, #3700; 1:5000 dilution), Anti-rabbit IgG, HRP-linked antibody (Cell Signaling, #7074; 1.5:1000 dilution), and Anti-mouse IgG, HRP-linked antibody (Cell Signaling, #7076; 1.5:1000 dilution). Membranes were imaged using a chemiluminescent SuperSignal West Femto Maximum Sensitivity Substrate (34095, Thermo Fisher Scientific) and the C-DiGit blot scanner (LI-COR Biosciences). Blots were then quantified using Image Studio Lite software (LI-COR).
Flow cytometry: cell cycle analysis
P12 and P18 M-Smo and M-Smo/EedcKO mice were injected with Edu 1 h prior to harvest. Mice were anesthetized with isoflurane and decapitated. Tumors samples were dissociated using the Cell Dissociation Kit (Worthington Biochemical Corporation, #LK003150), which included dissociation with papain at 37 °C for 15 min, and isolation using an ovomucoid inhibitor density gradient. Tumor cells were then treated with the Fixation and Permeabilization Kit (Life Technologies, #GAS004), and stained using the following antibodies: 647-conjugated pRB diluted 1:50 (Cell Signaling, #8974) and FxCycle Violet at 1:100 (Life Technologies, #F10347). Edu was detected using a Click-iT EdU Alexa Fluor 488 Imaging Kit (catalogue number C10337; Life Sciences). Samples were run on an LSRFortessa (BD Biosciences) at the UNC Flow Cytometry Core. Data was analyzed using Flow Jo v10.
Single cell sequencing (scRNA-seq): sample collection
Brains were harvested and cut along the sagittal midline. One half of the cerebellum from each mouse was dissociated and processed for scRNA-seq, and the other half of the brain was fixed, sectioned, and analyzed to confirm phenotype. Half cerebella were processed using the Cell Dissociation Kit (Worthington Biochemical Corporation, #LK003150), in which samples were treated with papain at 37 °C for 15 min and then separated by centrifugation of an ovomucoid inhibitor density gradient. Cells were then subjected to bead pairing by microfluidics, cDNA synthesis, and library construction using the Drop-seq V3 method [66] as in our prior studies [70,71,72,73].
scRNA-seq: processing data
Data analysis was performed using the Seurat R package version 3.1.1 [74]. Data were subjected to several filtering steps. Genes detected in > 30 cells were filtered out, to prevent misaligned reads appearing as rare transcripts in the data. Putative cells with fewer than 500 detected RNA molecules (nCount) or 200 different genes (nFeature) were considered to have too little information to be useful, and potentially to contain mostly ambient mRNA reads. Putative cells with greater than 4 standard deviations above the median nCount or nFeature were suspected to be doublets, improperly merged barcodes, or sequencing artifacts, and were excluded. As in our previously published work, putative cells with more than 10% mitochondrial transcripts were suspected to be dying cells and also excluded [70].
In total, 86% of putative cells from WT mice and 74% of putative cells from EedcKO mice met QC criteria and were included in the analysis. From the 5 WT mice, we included a total of 6558 cells with a range of 673–1852 cells per animal and a median of 1138 cells. From the 3 EedcKO mice, we included a total of 2576 cells, with a range of 692–1036 cells per animal and a median of 847 cells.
scRNA-seq: data normalization, clustering, differential gene expression, and cell type identification
The data was normalized using the SCTransform method as implemented in Seurat. The function then selected the top 3000 most highly variable genes. PCA was performed on the subset of highly variable genes using the RunPCA function. We used 15 PCs in downstream analysis, based on examining the elbow in the elbow plot as implemented by Seurat. We identified cell clusters using the FindNeighbors and FindClusters functions.
To identify differential genes between clusters of cells, we used the Wilcoxon rank sum test to compare gene expression of cells within the cluster of interest to all cells outside that cluster, implemented by the FindMarkers function. Uniform Manifold Approximation and Projection (UMAP) was used to reduce the PCs to two dimensions for data visualization using the RunUMAP function. For re-iterated analysis of the Clusters 4 and 6, the same procedures were used. We then determined the type of cell within each cluster by analyzing cluster-specific gene expression patterns.
Pathology scoring
Sagittal H&E sections of P12 M-Smo, M-Smo/EedcKO, and M-Smo/Ezh2cKO mouse brains were analyzed by neuropathologists (MS and JV) while blinded to the genotype, and the number of myoid cells per sample were manually counted.
Survival curves
Tumor-bearing mice were monitored daily and harvested according to a pre-determined humane endpoint, which included a decrease of weight > 10% overnight, a hunched posture, decreased mobility or inability to eat, and ataxia.
Statistical analyses
Two-tailed Student’s t-tests were used to compare IHC and western blot quantifications between Ezh2cKO and EedcKO genotypes. One-tailed Student’s t-tests were used to compare these genotypes versus controls for markers that were absent in controls. The Barnard’s exact test was used to make comparisons between categorical variables in comparisons of markers that were determined to be present or absent in individual replicates. Survival curves were compared using the Log-rank (Mantel-Cox) test. Dirichelet regression analysis was performed in R using the DirchletReg 0.7-1 package [63].
Availability of data and materials
The scRNA-seq data are publicly available in the GEO database under accession number GSE129730 for controls and GSE207451 for EedcKO cerebella.
References
Roussel MF, Hatten ME (2011) Cerebellum development and medulloblastoma. Curr Top Dev Biol 94:235–282. https://doi.org/10.1016/B978-0-12-380916-2.00008-5
Englund C, Kowalczyk T, Daza RA, Dagan A, Lau C, Rose MF, Hevner RF (2006) Unipolar brush cells of the cerebellum are produced in the rhombic lip and migrate through developing white matter. J Neurosci 26(36):9184–9195. https://doi.org/10.1523/JNEUROSCI.1610-06.2006
Ocasio J, Babcock B, Malawsky D, Weir SJ, Loo L, Simon JM, Zylka MJ, Hwang D, Dismuke T, Sokolsky M, Rosen EP, Vibhakar R, Zhang J, Saulnier O, Vladoiu M, El-Hamamy I, Stein LD, Taylor MD, Smith KS, Northcott PA, Colaneri A, Wilhelmsen K, Gershon TR (2019) scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat Commun 10(1):5829. https://doi.org/10.1038/s41467-019-13657-6
Yang Z-J, Ellis T, Markant SL, Read T-A, Kessler JD, Bourboulas M, Schüller U, Machold R, Fishell G, Rowitch DH, Wainwright BJ, Wechsler-Reya RJ (2008) Medulloblastoma can be initiated by deletion of patched in lineage-restricted progenitors or stem cells. Cancer Cell 14(2):135–145. https://doi.org/10.1016/j.ccr.2008.07.003
Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TTT, Lin SM, Wechsler-Reya RJ (2005) Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development 132(10):2425–2439
Schüller U, Heine VM, Mao J, Kho AT, Dillon AK, Han Y-G, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL (2008) Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14(2):123–134. https://doi.org/10.1016/j.ccr.2008.07.005
Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, Olson JM (2004) The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Can Res 64(21):7794–7800. https://doi.org/10.1158/0008-5472.CAN-04-1813
Hatten ME, Roussel MF (2011) Development and cancer of the cerebellum. Trends Neurosci 34(3):134–142. https://doi.org/10.1016/j.tins.2011.01.002
Bailey P, Cushing H (1925) Medulloblastoma cerebelli: a common type of mid-cerebellar glioma of childhood. Arch Neurol Psychiatry 14:192–224
Yao M, Ventura PB, Jiang Y, Rodriguez FJ, Wang L, Perry JSA, Yang Y, Wahl K, Crittenden RB, Bennett ML, Qi L, Gong CC, Li XN, Barres BA, Bender TP, Ravichandran KS, Janes KA, Eberhart CG, Zong H (2020) Astrocytic trans-differentiation completes a multicellular paracrine feedback loop required for medulloblastoma tumor growth. Cell. 180(3):502–20. https://doi.org/10.1016/j.cell.2019.12.024
Liu PP, Xu YJ, Dai SK, Du HZ, Wang YY, Li XG, Teng ZQ, Liu CM (2019) Polycomb protein EED regulates neuronal differentiation through targeting SOX11 in hippocampal dentate gyrus. Stem Cell Rep 13(1):115–131. https://doi.org/10.1016/j.stemcr.2019.05.010
Shi X, Zhang Z, Zhan X, Cao M, Satoh T, Akira S, Shpargel K, Magnuson T, Li Q, Wang R, Wang C, Ge K, Wu J (2014) An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nat Commun 5:5425. https://doi.org/10.1038/ncomms6425
Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron 22(1):103–114. https://doi.org/10.1016/s0896-6273(00)80682-0
Kenney AM, Rowitch DH (2000) Sonic hedgehog promotes G1 cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol 20(23):9055–9067. https://doi.org/10.1128/mcb.20.23.9055-9067.2000
Goodrich LV, Milenkovic L, Higgins KM, Scott MP (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277(5329):1109–1113
Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, Squire JA, Rutka JT, Hogg D (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31(3):306–310. https://doi.org/10.1038/ng916
Vorechovsky I, Tingby O, Hartman M, Stromberg B, Nister M, Collins VP, Toftgard R (1997) Somatic mutations in the human homologue of Drosophila patched in primitive neuroectodermal tumours. Oncogene 15(3):361–366. https://doi.org/10.1038/sj.onc.1201340
Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G (1997) Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res 57(13):2581–2585
Pirrotta V, Gross DS (2005) Epigenetic silencing mechanisms in budding yeast and fruit fly: different paths, same destinations. Mol Cell 18(4):395–398. https://doi.org/10.1016/j.molcel.2005.04.013
Obier N, Lin Q, Cauchy P, Hornich V, Zenke M, Becker M, Muller AM (2015) Polycomb protein EED is required for silencing of pluripotency genes upon ESC differentiation. Stem Cell Rev Rep 11(1):50–61. https://doi.org/10.1007/s12015-014-9550-z
Yeo GW, Coufal N, Aigner S, Winner B, Scolnick JA, Marchetto MC, Muotri AR, Carson C, Gage FH (2008) Multiple layers of molecular controls modulate self-renewal and neuronal lineage specification of embryonic stem cells. Hum Mol Genet 17(R1):R67-75. https://doi.org/10.1093/hmg/ddn065
Zhang H, Zhu D, Zhang Z, Kaluz S, Yu B, Devi NS, Olson JJ, Van Meir EG (2020) EZH2 targeting reduces medulloblastoma growth through epigenetic reactivation of the BAI1/p53 tumor suppressor pathway. Oncogene 39(5):1041–1048. https://doi.org/10.1038/s41388-019-1036-7
Yi J, Kim B, Shi X, Zhan X, Lu QR, Xuan Z, Wu J (2022) PRC2 heterogeneity drives tumor growth in medulloblastoma. Cancer Res 82(16):2874–2886. https://doi.org/10.1158/0008-5472.CAN-21-4313
Huang J, Gou H, Yao J, Yi K, Jin Z, Matsuoka M, Zhao T (2021) The noncanonical role of EZH2 in cancer. Cancer Sci 112(4):1376–1382. https://doi.org/10.1111/cas.14840
Vo BT, Li C, Morgan MA, Theurillat I, Finkelstein D, Wright S, Hyle J, Smith SMC, Fan Y, Wang YD, Wu G, Orr BA, Northcott PA, Shilatifard A, Sherr CJ, Roussel MF (2017) Inactivation of Ezh2 upregulates Gfi1 and drives aggressive Myc-driven group 3 medulloblastoma. Cell Rep 18(12):2907–2917. https://doi.org/10.1016/j.celrep.2017.02.073
Scelfo A, Piunti A, Pasini D (2015) The controversial role of the Polycomb group proteins in transcription and cancer: how much do we not understand Polycomb proteins? FEBS J 282(9):1703–1722. https://doi.org/10.1111/febs.13112
Feng X, Juan AH, Wang HA, Ko KD, Zare H, Sartorelli V (2016) Polycomb Ezh2 controls the fate of GABAergic neurons in the embryonic cerebellum. Development 143(11):1971–1980. https://doi.org/10.1242/dev.132902
Wallace VA (1999) Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 9(8):445–448. https://doi.org/10.1016/s0960-9822(99)80195-x
Machold R, Fishell G (2005) Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron 48(1):17–24. https://doi.org/10.1016/j.neuron.2005.08.028
Montgomery ND, Yee D, Chen A, Kalantry S, Chamberlain SJ, Otte AP, Magnuson T (2005) The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr Biol 15(10):942–947. https://doi.org/10.1016/j.cub.2005.04.051
Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L, Skotte J, Wutz A, Porse B, Jensen ON, Helin K (2010) Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res 38(15):4958–4969. https://doi.org/10.1093/nar/gkq244
Lavarone E, Barbieri CM, Pasini D (2019) Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat Commun 10(1):1679. https://doi.org/10.1038/s41467-019-09624-w
Dahmane N, Ruiz-i-Altaba A (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126(14):3089–3100
Garcia I, Crowther AJ, Gama V, Miller CR, Deshmukh M, Gershon TR (2013) Bax deficiency prolongs cerebellar neurogenesis, accelerates medulloblastoma formation and paradoxically increases both malignancy and differentiation. Oncogene 32(18):2304–2314. https://doi.org/10.1038/onc.2012.248
Veleta KA, Cleveland AH, Babcock BR, He YW, Hwang D, Sokolsky-Papkov M, Gershon TR (2021) Antiapoptotic Bcl-2 family proteins BCL-xL and MCL-1 integrate neural progenitor survival and proliferation during postnatal cerebellar neurogenesis. Cell Death Differ 28(5):1579–1592. https://doi.org/10.1038/s41418-020-00687-7
Luecken MD, Theis FJ (2019) Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol 15(6):e8746. https://doi.org/10.15252/msb.20188746
Lim C, Dismuke T, Malawsky D, Ramsey JD, Hwang D, Godfrey VL, Kabanov AV, Gershon TR, Sokolsky-Papkov M (2022) Enhancing CDK4/6 inhibitor therapy for medulloblastoma using nanoparticle delivery and scRNA-seq-guided combination with sapanisertib. Sci Adv. 8(4):eabl5838. https://doi.org/10.1126/sciadv.abl5838
Malawsky DS, Weir SJ, Ocasio JK, Babcock B, Dismuke T, Cleveland AH, Donson AM, Vibhakar R, Wilhelmsen K, Gershon TR (2021) Cryptic developmental events determine medulloblastoma radiosensitivity and cellular heterogeneity without altering transcriptomic profile. Commun Biol 4(1):616. https://doi.org/10.1038/s42003-021-02099-w
Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W, Pun SH, Sellers DL, Tasic B, Seelig G (2018) Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360(6385):176–182. https://doi.org/10.1126/science.aam8999
Malawsky D, Gershon TR (2023) scRNA-seq for microcephaly research [IV]: dirichlet regression for single-cell population differences. Methods Mol Biol 2583:123–125. https://doi.org/10.1007/978-1-0716-2752-5_11
Stojic L, Jasencakova Z, Prezioso C, Stutzer A, Bodega B, Pasini D, Klingberg R, Mozzetta C, Margueron R, Puri PL, Schwarzer D, Helin K, Fischle W, Orlando V (2011) Chromatin regulated interchange between polycomb repressive complex 2 (PRC2)-Ezh2 and PRC2-Ezh1 complexes controls myogenin activation in skeletal muscle cells. Epigenetics Chromatin 4:16. https://doi.org/10.1186/1756-8935-4-16
Guo J, Cai J, Yu L, Tang H, Chen C, Wang Z (2011) EZH2 regulates expression of p57 and contributes to progression of ovarian cancer in vitro and in vivo. Cancer Sci 102(3):530–539. https://doi.org/10.1111/j.1349-7006.2010.01836.x
Yang X, Karuturi RK, Sun F, Aau M, Yu K, Shao R, Miller LD, Tan PB, Yu Q (2009) CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLOS ONE 4(4):e5011. https://doi.org/10.1371/journal.pone.0005011
Koppens MA, Bounova G, Gargiulo G, Tanger E, Janssen H, Cornelissen-Steijger P, Blom M, Song JY, Wessels LF, van Lohuizen M (2016) Deletion of polycomb repressive complex 2 from mouse intestine causes loss of stem cells. Gastroenterology. 151(4):684–97. https://doi.org/10.1053/j.gastro.2016.06.020
Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M (1999) The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397(6715):164–168. https://doi.org/10.1038/16476
Jürgens G (1985) A group of genes controlling the spatial expression of the bithorax complex in Drosophila. Nature 316(6024):153–155. https://doi.org/10.1038/316153a0
Nagel S, Venturini L, Marquez VE, Meyer C, Kaufmann M, Scherr M, MacLeod RA, Drexler HG (2010) Polycomb repressor complex 2 regulates HOXA9 and HOXA10, activating ID2 in NK/T-cell lines. Mol Cancer 9:151. https://doi.org/10.1186/1476-4598-9-151
Lee Y, McKinnon PJ (2000) ATM dependent apoptosis in the nervous system. Apoptosis 5(6):523–529
Crowther AJ, Ocasio JK, Fang F, Meidinger J, Wu J, Deal AM, Chang SX, Yuan H, Schmid R, Davis I, Gershon TR (2016) Radiation sensitivity in a preclinical mouse model of medulloblastoma relies on the function of the intrinsic apoptotic pathway. Cancer Res 76(11):3211–3223. https://doi.org/10.1158/0008-5472.CAN-15-0025
Crowther AJ, Gama V, Bevilacqua A, Chang SX, Yuan H, Deshmukh M, Gershon TR (2013) Tonic activation of Bax primes neural progenitors for rapid apoptosis through a mechanism preserved in medulloblastoma. J Neurosci 33(46):18098–18108. https://doi.org/10.1523/JNEUROSCI.2602-13.2013
Lang PY, Gershon TR (2018) A new way to treat brain tumors: targeting proteins coded by microcephaly genes? Brain tumors and microcephaly arise from opposing derangements regulating progenitor growth. Drivers of microcephaly could be attractive brain tumor targets. Bioessays. 40(5):e1700243. https://doi.org/10.1002/bies.201700243
Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, McMahon AP (2006) A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res 66(20):10171–10178. https://doi.org/10.1158/0008-5472.CAN-06-0657
Riemondy KA, Venkataraman S, Willard N, Nellan A, Sanford B, Griesinger AM, Amani V, Mitra S, Hankinson TC, Handler MH, Sill M, Ocasio J, Weir SJ, Malawsky DS, Gershon TR, Garancher A, Wechsler-Reya RJ, Hesselberth JR, Foreman NK, Donson AM, Vibhakar R (2021) Neoplastic and Immune single cell transcriptomics define subgroup-specific intra-tumoral heterogeneity of childhood medulloblastoma. Neuro Oncol. 24:273–286. https://doi.org/10.1093/neuonc/noab135
Dey J, Dubuc AM, Pedro KD, Thirstrup D, Mecham B, Northcott PA, Wu X, Shih D, Tapscott SJ, LeBlanc M, Taylor MD, Olson JM (2013) MyoD is a tumor suppressor gene in medulloblastoma. Cancer Res 73(22):6828–6837. https://doi.org/10.1158/0008-5472.CAN-13-0730-T
Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ (2009) Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461(7265):762–767. https://doi.org/10.1038/nature08398
Yoo KH, Oh S, Kang K, Hensel T, Robinson GW, Hennighausen L (2015) Loss of EZH2 results in precocious mammary gland development and activation of STAT5-dependent genes. Nucleic Acids Res 43(18):8774–8789. https://doi.org/10.1093/nar/gkv776
Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH (2008) EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 32(4):491–502. https://doi.org/10.1016/j.molcel.2008.10.016
Cao Q, Wang X, Zhao M, Yang R, Malik R, Qiao Y, Poliakov A, Yocum AK, Li Y, Chen W, Cao X, Jiang X, Dahiya A, Harris C, Feng FY, Kalantry S, Qin ZS, Dhanasekaran SM, Chinnaiyan AM (2014) The central role of EED in the orchestration of polycomb group complexes. Nat Commun 5:3127. https://doi.org/10.1038/ncomms4127
Cheng Y, Liao S, Xu G, Hu J, Guo D, Du F, Contreras A, Cai KQ, Peri S, Wang Y, Corney DC, Noronha AM, Chau LQ, Zhou G, Wiest DL, Bellacosa A, Wechsler-Reya RJ, Zhao Y, Yang ZJ (2020) NeuroD1 dictates tumor cell differentiation in medulloblastoma. Cell Rep 31(12):107782. https://doi.org/10.1016/j.celrep.2020.107782
Miele E, Valente S, Alfano V, Silvano M, Mellini P, Borovika D, Marrocco B, Po A, Besharat ZM, Catanzaro G, Battaglia G, Abballe L, Zwergel C, Stazi G, Milite C, Castellano S, Tafani M, Trapencieris P, Mai A, Ferretti E (2017) The histone methyltransferase EZH2 as a druggable target in SHH medulloblastoma cancer stem cells. Oncotarget 8(40):68557–68570. https://doi.org/10.18632/oncotarget.19782
Benson R, Mallick S, Bhanu Prasad V, Haresh KP, Gupta S, Julka PK, Rath GK (2015) Medullomyoblastoma treated with craniospinal radiation and adjuvant chemotherapy: report of 4 cases and review of the literature. J Egypt Natl Canc Inst 27(2):109–111. https://doi.org/10.1016/j.jnci.2015.03.001
Sachdeva MU, Vankalakunti M, Rangan A, Radotra BD, Chhabra R, Vasishta RK (2008) The role of immunohistochemistry in medullomyoblastoma–a case series highlighting divergent differentiation. Diagn Pathol 3:18. https://doi.org/10.1186/1746-1596-3-18
Maier M (2014) DirichletReg: Dirichlet regression for compositional data in R. Research Report Series/Department of Statistics and Mathematics, WU Vienna University of Economics and Business, Vienna, p 125
Scelfo A, Piunti A, Pasini D (2015) The controversial role of the Polycomb group proteins in transcription and cancer: how much do we not understand Polycomb proteins? FEBS J 282(9):1703–1722. https://doi.org/10.1111/febs.13112
Deb G, Singh AK, Gupta S (2014) EZH2: not EZHY (easy) to deal. Mol Cancer Res 12(5):639–653. https://doi.org/10.1158/1541-7786.MCR-13-0546
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA (2015) Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161(5):1202–1214. https://doi.org/10.1016/j.cell.2015.05.002
Malawsky DS, Weir SJ, Ocasio JK, Babcock B, Dismuke T, Cleveland AH, Donson AM, Vibhakar R, Wilhelmsen K, Gershon TR (2021) Cryptic developmental events determine medulloblastoma radiosensitivity and cellular heterogeneity without altering transcriptomic profile. Commun Biol 4(1):616. https://doi.org/10.1038/s42003-021-02099-w
Lim C, Dismuke T, Malawsky D, Ramsey JD, Hwang D, Godfrey VL, Kabanov AV, Gershon TR, Sokolsky-Papkov M (2022) Enhancing CDK4/6 inhibitor therapy for medulloblastoma using nanoparticle delivery and scRNA-seq-guided combination with sapanisertib. Sci Adv. 8(4):eabl5838. https://doi.org/10.1126/sciadv.abl5838
Lang PY, Nanjangud GJ, Sokolsky-Papkov M, Shaw C, Hwang D, Parker JS, Kabanov AV, Gershon TR (2016) ATR maintains chromosomal integrity during postnatal cerebellar neurogenesis and is required for medulloblastoma formation. Development 143(21):4038–4052. https://doi.org/10.1242/dev.139022
Ocasio J, Babcock B, Malawsky D, Weir SJ, Loo L, Simon JM, Zylka MJ, Hwang D, Dismuke T, Sokolsky M, Rosen EP, Vibhakar R, Zhang J, Saulnier O, Vladoiu M, El-Hamamy I, Stein LD, Taylor MD, Smith KS, Northcott PA, Colaneri A, Wilhelmsen K, Gershon TR (2019) scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat Commun 10(1):5829. https://doi.org/10.1038/s41467-019-13657-6
Babcock B, Weir S (2023) scRNA-seq for microcephaly research [I]: single-cell droplet encapsulation, mRNA capture, and cDNA synthesis. Methods Mol Biol 2583:83–97. https://doi.org/10.1007/978-1-0716-2752-5_8
Babcock B, Weir S (2023) scRNA-seq for microcephaly research [II]: preparation of single-cell cDNA libraries. Methods Mol Biol 2583:99–104. https://doi.org/10.1007/978-1-0716-2752-5_9
Babcock B, Malawsky D (2023) scRNA-seq for microcephaly research [III]: computational analysis of scRNA-seq data. Methods Mol Biol 2583:105–121. https://doi.org/10.1007/978-1-0716-2752-5_10
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36(5):411–420. https://doi.org/10.1038/nbt.4096
Acknowledgements
We thank the UNC CGBID Histology Core supported by P30 DK 034987, and the UNC Tissue Pathology Laboratory Core supported by NCI CA016086 and UNC UCRF. TRG is an Editor for Acta Neuropathologica Communications. And has been excluded from the editorial process.
Funding
This work was supported the NINDS (R01NS088219, R01NS102627, R01NS106227).
Author information
Authors and Affiliations
Contributions
Conceptualization: AHC, ID, TRG; Methodology: AHK, DM, MS, JEVV, TRG; Investigation: AHK, DM, MC, CR, FR, MS, JEVV; Visualization: AHC, DM, TRG; Funding acquisition: TRG; Supervision: TRG; Writing—original draft: AHK, DM, TRG; Writing—review and editing: AHK, DM, ID, TRG. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal studies were reviewed by the University of North Carolina Institutional Animal Care and Use Committee and conducted under IACUC protocols 19-098 and 21-011.
Consent for publication
All co-authors consent to the publication of this work.
Competing interests
The authors report no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
. Initial uniform manifold approximation and projection (UMAP) qualitative map of cells dissociated from harvested cerebella from 5 WT and 3 EedcKO mice. Cells were subdivided into 20 color-coded clusters. Figure S2 Disabling apoptosis increased the myoid population. Representative images of MYOG IHC in cerebella of indicated genotypes show that a population of myoid cells persisted in EedcKO cerebella at P21, and that the myoid population increased when apoptosis was blocked by deletion of both Bax AND Bak. The increased myoid cells in Eed/Bax/BaktKO cerebella indicates that apoptosis decreases the myoid population in EedcKO cerebella
Additional file 2
. Cluster-specific sets of differentially expressed genes, with statistical analyses.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cleveland, A.H., Malawsky, D., Churiwal, M. et al. PRC2 disruption in cerebellar progenitors produces cerebellar hypoplasia and aberrant myoid differentiation without blocking medulloblastoma growth. acta neuropathol commun 11, 8 (2023). https://doi.org/10.1186/s40478-023-01508-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-023-01508-x