
Abstract
Previous studies show that 3β-hydroxysterol-Δ24 reductase (DHCR24) has a remarked decline in the brain of AD patients. In brain cholesterol synthetic metabolism, DHCR24 is known as the heavily key synthetase in cholesterol synthesis. Moreover, mutations of DHCR24 gene result in inhibition of the enzymatic activity of DHCR24, causing brain cholesterol deficiency and desmosterol accumulation. Furthermore, in vitro studies also demonstrated that DHCR24 knockdown lead to the inhibition of cholesterol synthesis, and the decrease of plasma membrane cholesterol and intracellular cholesterol level. Obviously, DHCR24 could play a crucial role in maintaining cholesterol homeostasis via the control of cholesterol synthesis. Over the past two decades, accumulating data suggests that DHCR24 activity is downregulated by major risk factors for AD, suggesting a potential link between DHCR24 downregulation and AD pathogenesis. Thus, the brain cholesterol loss seems to be induced by the major risk factors for AD, suggesting a possible causative link between brain cholesterol loss and AD. According to previous data and our study, we further found that the reduced cholesterol level in plasma membrane and intracellular compartments by the deficiency of DHCR24 activity obviously was involved in β-amyloid generation, tau hyperphosphorylation, apoptosis. Importantly, increasing evidences reveal that the brain cholesterol loss and lipid raft disorganization are obviously linked to neuropathological impairments which are associated with AD pathogenesis. Therefore, based on previous data and research on DHCR24, we suppose that the brain cholesterol deficiency/loss might be involved in the pathogenesis of AD.
Similar content being viewed by others

Introduction
In 2000, Greeve et al. first found that there is a significant reduction in the expression of new gene in vulnerable brain regions in Alzheimer’s disease (AD) patients, which was named selective Alzheimer’s disease indicator 1 (Seladin-1), namely 24-dehydrocholesterol reductase (DHCR24) [45, 57]. In the post-lanosterol pathway of cholesterol synthesis, the final step in the Bloch pathway or the first step in the Kandutsch–Russell (K–R) pathway is catalyzed by the enzyme DHCR24 [132, 133]. Besides, as a link bridge between two pathways, DHCR24 can theoretically act on any intermediate from lanosterol through to desmosterol to transfer intermediates from the Bloch to the K–R pathway [30, 132]. Importantly, DHCR24 can also synergistically control the activity of 7-dehydrocholesterol reductase (DHCR7), a final key enzyme in the K–R pathway, which would ensure concerted control of cholesterol synthesis [85, 132]. From a physiological role, DHCR24 is universally regulated by sterols, dexamethasone, sex steroids, adrenocorticotropic hormone, thyroid hormone, Neurotrophins, and xenobiotics [22, 87, 112]. Furthermore, DHCR24 activity is also regulated by ubiquitination, phosphorylation, and epigenetic factors such as methylation and acetylation [64, 87]. Collectively, accumulating evidences suggest that the modulation of DHCR24 activity could be a key node in the control of cholesterol synthesis. To sum up, above findings reveal that DHCR24 could play a crucial role in maintaining the cholesterol homeostasis via the control of cholesterol synthesis (Fig. 1).

The critical role of DHCR24 in cholesterol synthesis and homeostasis. In the post-lanosterol pathway of cholesterol synthesis, the final step in the Bloch pathway or the first step in the Kandutsch–Russell pathway is catalyzed by the enzyme DHCR24. Besides, as a link bridge between two pathways, DHCR24 can theoretically act on any intermediate from lanosterol through to desmosterol to transfer intermediates from the Bloch to the K–R pathway. Thus, DHCR24 play the critical role in maintaining cholesterol homeostasis via the control of cholesterol synthesis
Nevertheless, DHCR24 activity is also obviously downregulated by major risk factors from AD, such as aging, diabetes-related factors, amyloid-β (Aβ), oxidative stress, chronic inflammation, and genetic factors [11, 20, 42, 55, 61, 63, 70, 107, 125, 135]. Thus, the above data suggest that downregulation of DHCR24 is obviously linked to the major risk factors from AD, suggesting a potential causative link between DHCR24 downregulation and major risk factors from AD. Moreover, a growing body of research has shown that deficiency of DHCR24 activity could induce lowering cholesterol level of neuronal cells and disruption of membrane lipid-raft structure and function, leading to the disregulation of cellular cholesterol homeostasis, and abnormality of cell signaling [3, 9, 24, 71, 122]. In addition, increasing evidences support that downregulation of DHCR24 could lead to Aβ production, apoptosis of neuronal or glial cells, hyperphosphorylation of microtubule-associated protein tau (tau), inhibition of autopagy, and inflammation, which are tightly associated with AD and other degenerative diseases [9, 24, 45, 71, 83, 95, 127, 155]. Therefore, previous studies strongly support that that the reduced cholesterol level in plasma membrane and/or intracellular compartments by the deficiency of DHCR24 obviously contributes to neurodegeneration such as AD (Fig. 2).

The contribution of DHCR24 to Alzheimer’s disease. The downregulation of DHCR24 could be induced by risk factors from FAD and SAD, including Aβ, aging, diabetes-related risk factors, chronic hypoxia, oxidative stress, chronic inflammation, insufficiency of brain neurotrophic substances, and metabolic syndrome, suggesting a causative link between DHCR24 downregulation and major risk factors from AD. Furthermore, DHCR24 downregulation lead to the inhibition of cholesterol synthesis and decrease of cholesterol level in the plasma membrane and intracellular organelles, resulting in cholesterol deficiency-induced pathological impairments, such as Aβ overproduction, tau hyperphosphorylation, apoptosis, synaptic impairment, and other pathological impairments, which are associated with neurodegenerative diseases such as AD. Thus, the downregulation of DHCR24 could contribute to Alzheimer’s disease and other neurodegenerative diseases
In addition, a growing body of evidence reveals that there are abnormal alterations in brain cholesterol metabolism, including the decrease of de novo cholesterol synthesis, and/or cholesterol trafficking (transportation, uptake, and intracellular transportation), and/or cholesterol catabolism, in aging human and mice, senescent-accelerated mice strain 8 (SAMP8) mice, diabetic mice, familial Alzheimer's disease (FAD) mice, genetic forms of AD animals and patients, and AD patients, suggesting the brain cholesterol loss [15, 18, 54, 66, 72, 89, 92, 107, 110, 111, 123, 129, 142, 146, 152]. The brain cholesterol loss appears to be a pervasive, prominent and common feature in these different kinds of AD models and patients. To some extent, we found that these different kinds of AD models and patients include major risk factors for AD, such as Aβ, genetic factors, aging, diabetes-related factors, chronic hypoxia, oxidative stress, chronic inflammation, and metabolic syndrome, etc. The brain cholesterol loss seems to be tightly associated with major risk factors from AD. Thus, we suppose that the brain cholesterol loss is likely to be induced by the major risk factors for AD, suggesting a possible causative link between brain cholesterol loss and AD (Fig. 3). Surprisingly, accumulating data also reveal that the brain cholesterol loss is very likely to occur in the initiation stage of AD pathology, suggesting a key role of brain cholesterol loss in initial changes of AD pathogenesis [15, 18, 28, 34, 35, 80, 92, 110, 111, 124, 141, 150]. Furthermore, the brain cholesterol deficiency seems to be an early and common driving factor in the onset and development of AD. And the brain cholesterol deficiency could be intimately linked with the generation of β-amyloid, tauopathy, synaptic loss, neuronal apoptosis and death, which are associated with the pathogenesis of AD [5, 9, 21, 24, 45, 73,74,75, 93, 95, 115]. Based on previous data and research on DHCR24, we suppose that the brain cholesterol deficiency/loss could trigger the onset and progression of AD.

A Revised Cholesterol Hypothesis of AD. There are abnormal alterations in brain cholesterol metabolism, including the decrease of de novo cholesterol synthesis, and/or cholesterol trafficking (transportation, uptake, and intracellular transportation), and/or cholesterol catabolism in aging humans and animals, SAMP8 mice, diabetic mice, FAD (5xFAD and APP/PS-1) animals, AD patients, genetic forms of AD animals and patients (ApoE4 allele, mutation of NPC1 or NPC2, polymorphism of ABC and LDL receptor family), suggesting the brain cholesterol insufficiency/loss. To some extent, we found that these different kinds of AD models and patients include major risk factors for AD, such as Aβ, genetic factors, aging, diabetes-related risk factors, chronic hypoxia, oxidative stress, chronic inflammation, and metabolic syndrome, etc. Thus, the brain cholesterol loss seems to be induced by the major risk factors for AD in these different kinds of AD models and patients, suggesting a possible causative link between brain cholesterol loss and AD. Importantly, the brain cholesterol loss might lead to the membrane lipid raft disorganization and decrease of intracellular compartments, resulting in the pathological impairments which are associated with AD pathogenesis. Therefore, based on previous data and research on DHCR24, we suppose that the brain cholesterol deficiency/loss might be involved in the onset and progression of AD
The critical role of DHCR24 in cholesterol synthesis and homeostasis
The cholesterol synthesis pathway encompasses more than 20 enzymes and can be divided into the early sterol synthesis pathway and the post-lanosterol pathway [132, 133]. In the post-lanosterol pathway, the pathway can take one of two intertwined routes, the Bloch and K–R pathway [132, 133], creating a long and complex road to cholesterol through various branch points. Theoretically speaking, DHCR24 might be a key synthetase heavily involved in cholesterol synthesis (Fig. 1).
Firstly, in the post-lanosterol pathway, lanosterol can be acted upon by Lanosterol 14-a-demethylase (LDM or CYP51A1) to enter the Bloch pathway [132, 133]. In the Bloch pathway, by reducing the double bond at carbon 24 of the last cholesterol precursor, desmosterol, the final step in the Bloch pathway is catalyzed by the enzyme DHCR24 [30, 132, 157]. And lanosterol can be also acted upon by DHCR24 to enter the K–R pathway, so DHCR24 can also control the gate of entry in the K–R pathway [30, 132, 157]. Besides, DHCR24 can theoretically act on any intermediate from lanosterol through to desmosterol to transfer intermediates from the Bloch to the K–R pathway, so it is also a link bridge between two pathways [30, 132]. Furthermore, in post-squalene pathways, DHCR7 is another important cholesterol synthetase, which controls the final step of the K–R pathway [30, 85, 132]. A previous study reveals that when the DHCR24 gene is knocked down, DHCR7 activity is also ablated. Conversely, overexpression of DHCR24 enhances DHCR7 activity [85]. So, DHCR7 activity obviously is controlled by DHCR24, which would ensure concerted control of cholesterol synthesis [85, 132]. Thus, DHCR24 obviously control the cholesterol synthesis in the post-lanosterol pathway.
In addition, mutations in DHCR24 enzyme, which converts desmosterol into cholesterol, leads to desmosterolosis, an autosomal recessive developmental disorder [3, 122, 128]. Defect in the enzyme DHCR24 causes significant elevation of the cholesterol precursor desmosterol and cholesterol deficiency [3, 6, 122]. Moreover, in DHCR24 knockout (KO) mice the brain cholesterol lack with age, and brain cholesterol deficiency in 3-week-old was associated with altered membrane composition including disrupted detergent-resistant membrane domain (DRM) structure [3, 71]. Similarly, in silencing DHCR24 cell model, it was found that cell desmosterol and 7-dehydrocholesterol (7-DHC) are significantly elevated, and cell cholesterol is greatly decreased [9, 24, 127]. So, this genetic defect manifests that the defect of DHCR24 enzyme activity leads to cholesterol deficiency, suggesting the critical role of DHCR24 in maintaining cholesterol synthesis and homeostasis.
Furthermore, the posttranslational phosphorylation modification of DHCR24 has been identified by cell kinase signals, which is a major mode of regulating cholesterol homeostasis [86, 87]. Moreover, data have identified particular putative phosphorylated sites on DHCR24, such as T110, Y299, Y321, and Y507 [87]. In addition, protein kinase C (PKC) ablated DHCR24 activity by inhibiting a major serine/threonine kinase [87]. Thus, modulating DHCR24 activity by phosphorylation would allow for a rapid means of regulating cholesterol synthesis. In addition to phosphorylation modification, a lot of evidences supported that DHCR24 could be ubiquitinated, and 11 ubiquitination sites are identified, suggesting that DHCR24 activity may be regulated by ubiquitin-proteasomal degradation [64, 87]. To sum up, these studies indicate two important regulatory mechanisms for DHCR24 activity by cell kinase signals-mediated phosphorylation and by ubiquitin-proteasomal degradation.
As mentioned above, accumulating evidence indicates that DHCR24 play the critical role in maintaining cholesterol homeostasis via the control of cholesterol synthesis (Fig. 1). According to the above data, we conclude that the modulation of DHCR24 activity could be a key node in the control of cholesterol synthesis and homeostasis.
A causative link between DHCR24 downregulation and risk factors from AD
Amyloid-β proteins
In the amyloid cascade theory, amyloid-β protein (Aβ) is regarded as a key risk substance, which is tightly related to FAD and partly to sporadic AD (SAD) [8, 29]. In Neuro-2A cells, to be combined exposure of amyloid-β peptide 1–40 (Aβ40) or amyloid-β fragment 25–35 (Aβ25–35), the expressions of seladin-1 genes were significantly down-regulated [135]. Moreover, in vitro C6 astrocytic cell lines, we also confirmed that Aβ40 or Aβ25–35 could markedly induce the downregulation expressions of seladin-1 [14]. Additionally, Najem et al. also found amyloid-β peptide 1–42 (Aβ42) could induce the downregulation of DHCR24 and inhibited cholesterol synthesis pathway in SH-SY5Y cells [102]. Thus, above findings suggest that the downregulation of DHCR24 expression could be induced by β-amyloid proteins.
In AD patients, it has been reported that DHCR24 transcription and protein expression were selectively down-regulated in the brain areas affected in Alzheimer's disease, but the reasons for this decrease are not known [45, 57]. In APPswe/PS1deltaE9 (APP/PS1) AD mice, Vanmierlo et al. found that reduced expression of DHCR24 gene in both cortex and cerebellum as aging [57, 151].
Besides, in APP/PS1 mice, the decreased cholesterol level and increased phospholipids/cholesterol ratio might lead to the disruption of lipid raft homeostasis, which has been considered to contribute to cellular deregulation, resulting in neuronal loss in AD [35]. Further analysis found that the change of lipid raft alteration occurred in the early stage of AD pathology in APP/PS1 mice [35]. Noticeably, in 5xFAD and APP/PS1 mice brain, why is there the inhibition of cholesterol biosynthesis at the very early stage of AD? Park et al. found that Aβ production might directly correlate with cholesterol biosynthesis inhibition [110]. Furthermore, some early studies show that Aβ40/42 inhibits cholesterol synthesis and reduced cellular cholesterol levels in neuronal or glial cells by inhibiting the main cholesterol biosynthesis enzymes [14, 43, 46, 102, 135]. Intriguingly, in FAD mice, the initial increase in the production of Aβ is mutations-based and occurs relatively early, Aβ overload might induce the downregulation of cholesterol synthetic genes, resulting in the brain cholesterol loss, which could also occur in the initial stage of FAD. Does overproduction of beta-amyloid also trigger a cholesterol loss cascade leading to neurodegeneration in the early stage of FAD? In fact, APP transgenic mice exhibited lower levels of cellular cholesterol in their brains, and conversely, APP knockout mice exhibited higher levels of cellular cholesterol in their brains, suggesting that Aβ mediated regulation of cellular cholesterol synthesis [148]. Collectively, above data suggest that Aβ overproduction is likely to be a risk factor for cholesterol biosynthesis. Taken together, accumulating data support that β-amyloid proteins could lead to the downregulation of cholesterol synthetic genes, including DHCR24.
Diabetes and diabetes-related risk factors
Studies demonstrate that Diabetes mellitus (DM) enhances the risk for Alzheimer's disease [8, 13]. Moreover, diabetes-related risk factors, such as hyperglycemia, insulin insufficiency, and insulin resistance have been proposed to contribute to AD pathogenesis [13, 50, 145]. Kazkayasi et al. confirmed that constant lack of insulin for 5 days decreased DHCR24 levels in rat primary cultured neurons [61]. In addition, the intermittent high glucose concentrations also reduced the expression of DHCR24 in the human fetal neuroepithelial cells [42]. Besides, a decrease in DHCR24 was also found in the brains of rodents with streptozotocine (STZ)-induced diabetes [55, 61, 142]. Furthermore, in diabetic mice model, insulin as a regulator of DHCR24, the lack of insulin can downregulate the expression of all cholesterol synthetase, including sterol regulatory element-binding protein 2 (SREBP2), DHCR24 [61, 102, 142]. To sum up, all the above evidence indicates that the DM-related risk factors can induce the downregulation of DHCR24.
Insufficiency of neurosteroid and other neurotrophic factors
Basic and clinical evidence suggests that neurosteroid such as estrogens and androgen, and neurotrophic factors such as insulin-like growth factors (IGFs) and neurotrophins, have protective effects in the brain [17, 112]. Nevertheless, their potential role against neurodegenerative diseases, in particular Alzheimer's disease, is still a matter of debate. Accumulating data demonstrated that neurosteroid could induce the expression of DHCR24, as well as the synthesis of cell cholesterol, in neurons and astrocytes [14, 17, 112, 160]. Similarity, thyroid hormones (TH) play an important role in the development of human brain, by upregulating the expression of specific DHCR24 genes in neuronal precursors [10]. Moreover, it has been found that IGF1 and nerve growth factor (NGF) induced upregulation of DHCR24 expression, and conversely, the inhibition of IGF signaling downregulated the expression of DHCR24 [22, 42]. On the contrary, dexamethasone could obviously decrease the expression of genes involved in cholesterol synthesis genes, such as squalene epoxidase (SQLE) and DHCR24 [58]. Besides, with aging, there is a progressive, age-dependent decline in the level of many important neurotrophic factors, such as estrogen, androgen, insulin, NGF and IGFs, in the brain of aged rodents and AD patients [19, 22, 112]. Thus, evidences suggest that the downregulation of DHCR24 induced by the depletion of neurosteroids and neurotrophic factors in the brain might play a pivotal pathological role in neurodegenerative diseases. In brief, these studies suggest that insufficiency of brain neurotrophic substances might lead to the decrease of DHCR24 expression in the brain, which is involved in maintaining cholesterol synthesis and homeostasis.
Chronic hypoxia, oxidative stress and inflammation
Increasing data underscore the importance of chronic oxidative stress and inflammation in the pathogenesis of neurodegenerative diseases, including AD [23, 97, 136]. Kuehnle et al. show that DHCR24 expression is up-regulated in an acute response; conversely, upon chronic exposure to oxidative stress, the level of DHCR24 expression was lowered in SH-SY5Y cells [70, 125]. Moreover, in the hypoxia rat model, chronic hypoxia significantly induced the decrease of DHCR24 expression in the hippocampus [88]. In addition, Khuda et al. found that LPS-Induced inflammation obviously reduced the DHCR24 expression [63]. Collectively, under the pathological condition, chronic hypoxia, oxidative stress and inflammatory response could negatively modulate DHCR24 expression.
Aging and metabolic syndrome
Aging, obesity and metabolic syndrome is a cluster of risk factors that participate in the development of neurodegenerative diseases such as AD [12, 76, 139]. Interestingly, a study on bariatric surgery was performed in order to investigate whole blood gene expression profiles in obese subjects that have obvious overweight, BMI abnormality, and insulin resistance problems, including some metabolic syndrome risk factors, the study showed that expression of DHCR24 was significantly decreased [11]. Additionally, all enzyme genes in the cholesterol synthesis pathway are significantly downregulated in the aging mice brain, such as hydroxy-3-methylglutaryl-CoA reductase (HMGCR), SQLE, 7-dehydrocholesterol reductase (DHCR7), and DHCR24, compared to the adult mice [11, 106, 107]. Thus, the above dada support that aging and metabolic syndrome-related risk factors could obviously downregulate the expression of DHCR24.
Epigenetic factors
Cumulating Evidences suggest that epigenetic factors that are involved in Late-Onset Alzheimer's Disease (LOAD), such as methylation and acetylation, also obviously regulate DHCR24 expression and activity [26, 88]. Regarding epigenetic modifications, the up-to-date epigenomic findings include reported modifications in the LOAD core pathology loci DHCR24 [88]. Another epigenome-wide association study on obesity-related traits found that the novel DNA methylations were located on the DHCR24 [26, 31]. Thus, epigenetic modification could regulate DHCR24 gene expression which contributes to Late-Onset Alzheimer's Disease.
As stated above, increasing evidence reveals that the downregulation of DHCR24 could be induced by a lot of risk factors from AD, including Aβ, aging, diabetes-related factors, hypoxia, oxidative stress, chronic inflammation, insufficiency of brain neurotrophic substances, and metabolic syndrome, etc. Intriguingly, above data obviously suggests a causative link between DHCR24 downregulation and major risk factors from AD (Fig. 2). Therefore, we propose that the downregulation of DHCR24 might be an early and common regulatory pathway in the pathogenesis of FAD and SAD, which may be tightly associated with dysregulation of cholesterol homeostasis.
DHCR24 downregulation and pathological impairments related to AD
DHCR24 downregulation and Aβ metabolism
In recent years, in silencing DHCR24 SH-SY5Y cells, Sarajärvi et al. confirm that the reduced DHCR24 expression results in enhanced Golgi-localized gamma-ear-containing ARF binding protein 3 (GGA3) depletion, to further lead to augmented post-translational stabilization of beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) and increased beta-amyloidogenic processing of amyloid precursor protein (APP) and Aβ production [127]. In addition, DHCR24-deficient mice brains had reduced levels of cholesterol and disorganized cholesterol-rich membrane lipid raft, leading to membrane-dependent plasmin inactivation and the displacement of β-secretase (BACE1) from membrane lipid-raft to APP-containing membrane fractions, to increased β-cleavage of APP and high levels of Aβ production [24, 71]. Thus, the above findings suggest that DHCR24 knockdown promotes the cleavage of APP and production of Aβ through the decrease of cholesterol levels and the reorganization of lipid raft. Altogether, these data suggest that the decrease of neuronal membrane cholesterol or intracellular cholesterol might contribute to excessive Aβ production.
DHCR24 downregulation and tauopathy
Interestingly, in APP/PS1 transgenic animals, it is found that that the downregulation of seladin-1 expression in vulnerable AD brain areas is paralleled by an increase in the amount of hyperhosphorylated microtubule-associated protein tau (tau) [57]. Thus, Iivonen et al. suppose that the downregulation of DHCR24 expression might be associated with hyperphosphorylated tau in AD. Furthermore, in our study, after silencing DHCR24 by lentivirus-mediated DHCR24 short hairpin RNA (shRNA) in SH-SY5Y cells, we found silencing DHCR24 could markedly induce hyperphosphorylation of tau at some specific sites, including Thr181, Ser199, Thr231, Ser262, Ser396, and Ser422 [9, 119]. Besides, we further found that DHCR24 knockdown lead to the decrease of plasma membrane cholesterol and disruption of lipid raft/caveolae, resulting in inhibition of lipid raft-dependent phosphoinositide 3-kinase (PI3-K)/ protein kinase B (Akt) signaling, Protein phosphatase 2A (PP2A) signaling, as well as the overactivation of glycogen synthase kinases-3beta (GSK3β) and mammalian target of rapamycin (mTOR) signaling [9, 103, 119]. Similarly, defects in the cholesterol trafficking are associated with enhanced generation of hyperphosphorylated Tau and Amyloid-β protein [131]. Moreover, previous studies confirm that these sites are tightly correlated with a possible toxic effect of phosphorylated tau, which is involved in AD and other tauopathies [2, 4, 104]. Overall, these data suggest that cholesterol loss by DHCR24 knockdown could play a crucial role in tau hyperphosphorylation.
DHCR24 downregulation and synaptopathy
Desmosterolosis is caused by mutations in DHCR24, lead to the elevated desmosterol levels and decreased level of cholesterol in the patient's brain, resulting in multiple congenital anomalies including white matter atrophy and synaptic abnormality [3, 71, 122, 159]. Furthermore, in a mouse model of desmosterolosis, DHCR24-KO mice brains showed complex changes in expression of lipid and sterol transcripts and synaptic plasticity transcripts, and the decrease of membrane cholesterol and disruption of membrane lipid raft and increased arborization synapse [3, 24, 71, 122]. On the contrary, the overexpression of DHCR24 significantly increased the total number of dendritic spines and the mushroom spines in mature mouse hippocampal neurons, facilitating synapse formation [95]. Very importantly, a body of evidences support that cholesterol reduction can trigger dysfunction of synaptic structure and function, and possible mechanisms by which cholesterol content in the plasma membrane influences synaptic processes [69, 78, 114]. Therefore, the cholesterol loss by DHCR24 downregulation may impair synapse formation, maturation, and function.
DHCR24 downregulation and apoptosis
AD is characterized by severe neuronal and/or glial cells loss; however, the mechanisms by which neurons or glial cells die remain elusive [20, 105]. A line of studies has shown that over-expression of DHCR24 protected the cells from apoptotic cell death by amyloid-β-mediated toxicity or other stresses, and low-expression of DHCR24 obviously induced an apoptosis upon exposure to different stress conditions [45, 70, 82,83,84, 95, 127, 155]. Thus, these findings support that cholesterol deficiency by DHCR24 knockdown leads to a cell apoptosis under different pathological or stress conditions.
DHCR24 downregulation and other pathological injuries
In our study, we found that DHCR24 knockdown could obviously induce the inhibition of autophagy [9]. In addition to Aβ pathology, tauopathy, synaptopathy, and apoptosis, thus, we suppose that cholesterol loss by DHCR24 knockdown also might be involved in other pathological injuries which are related to AD, such as autophagy, mitochondrial injuries, inflammation, neurosteroid synthesis, and other metabolic abnormalities. Certainly, further study is still to be performed in order to elucidate to complex relationship between DHCR24 and pathological impairments.
In conclusion, based on current knowledge about DHCR24, accumulating data support that there is an obvious link between DHCR24 downregulation and major risk factors from FAD and SAD (Fig. 1). Furthermore, compelling evidences support the deficiency of DHCR24 activity lead to the inhibition of cholesterol synthesis and decrease of cholesterol level in the plasma membrane and intracellular organelles, coupled with disruption of membrane lipid raft, resulting in cholesterol deficiency-induced pathological impairments [9, 45, 70, 82,83,84, 95, 112, 113, 127, 155]. Thus, accumulating evidences strongly reveal that cholesterol loss by DHCR24 downregulation could lead to Aβ overproduction, tau hyperphosphorylation, and other pathological impairments which are associated with neurodegenerative diseases such as AD (Fig. 2). Regretfully, because desmosterolosis by DHCR24 mutation is a lethal disorder, the mice model lacking one or both alleles of DHCR24 gene is still lack [3]. So, we still need to further investigate the role of DHCR24 in AD pathogenesis in in vitro or in vivo model systems.
Alteration of cholesterol metabolism in different kinds of AD models and patients
Alteration of cholesterol metabolism in aging humans and rodents, SAMP8 mice, and diabetic mice
Age-related brain aging is regarded as a major risk factor in the initiation and progression of Alzheimer's disease [98, 106, 130]. Compared to the adult mice, all enzyme genes in the cholesterol synthesis pathway are significantly downregulated in the aging brain, such as HMGCR, SQLE, DHCR7, and DHCR24, suggesting decreased de novo cholesterol biosynthesis in the aging brain [15, 106, 107]. On the contrary, genes involved in cholesterol-transporting proteins such as Apolipoprotein E (ApoE), are obviously upregulated, suggesting a compensatory response due to the decrease of cholesterol synthesis and decreased cholesterol level in the aging brain [15, 107]. Moreover, aging shows an age-dependent decrease of cholesterol level, and is accompanied by the decrease of synaptic cholesterol levels in the hippocampus [138]. This is consistent with the cholesterol loss observed in the cortex of aged rodents and humans [91, 92, 137, 138, 143]. However, others have found that although cholesterol synthesis is decreased in the hippocampus, the total brain cholesterol content remains stable [138, 146]. Thus, the reduction of cholesterol in the brain could present regional specificity during aging. Similarly, in SAMP8, it has been found that the hippocampus of SAMP8 mice presents reduced cholesterol levels at 6 months of age [111]. Further, although cholesterol levels did not differ in 2-month-old mice, a significant 35% decrease was observed in hippocampal extracts from 6-month-old SAMP8 mice [111]. The extent of the change was similar to that observed in the hippocampus of aged mice compared with young wild-type mice [91, 92, 138]. Pérez-Cañamás et al. confirm that cholesterol loss in the hippocampus of SAMP8 mice is an aging hallmark directly involved in cognitive decline. Overall, above data supports that these aging-related risk factors could induce the decrease of brain cholesterol synthesis and cholesterol level.
In recent years, it is confirmed that there is a significant reduction in expression of SREBP-2, and its downstream cholesterol synthesis genes in the diabetic mice brain, leading to a reduction in brain cholesterol synthesis in type 1 and type 2 diabetic mice [72, 91, 92, 124, 138]. Moreover, altered insulin signaling also modulates the expression of molecules involved in cholesterol biosynthesis, resulting in inhibition of brain cholesterol synthesis and decreased level of free cholesterol in diabetic mice brain [68, 72, 142]. And the lowering-expression of cholesterol synthesis genes is due, at least in part, to diabetes-related risk factors, such as insulin-deficiency, insulin resistance, lower or high glucose [72, 91, 92, 124, 138]. Thus, inhibition of brain cholesterol synthesis and cholesterol insufficiency could be induced by diabetes and diabetes-related risk factors.
Alteration of cholesterol metabolism in 5xFAD and APP/PS-1 mice
In a recent study, Ye and colleagues found that in vitro cultured primary astrocytes stimulated with Aβ exhibited higher expression of ABC transporters that is involved in cholesterol efflux. Unsurprisingly, detection of this sterol revealed that the intracellular cholesterol level was significantly reduced in astrocytes [156]. The same expression pattern of ABC transporters was also found in 5xFAD mice, an AD mice model with early onset of Aβ pathology [156]. Park et al. confirmed that the majority of genes involved in cholesterol biosynthesis are obviously dysregulated in 5xFAD mice, suggesting the inhibition of the cholesterol biosynthesis genes and decrease of cholesterol biosynthesis in mice brain [110, 156]. Similarly, the main cholesterol synthetic genes were markedly downregulated in AD astrocytes of APP/PS1 [106, 107]. Conversely, further analysis found that main genes which mediate cholesterol transportation were significantly upregulated, such as apoE, ATP binding cassette A1 (ABCA1), low-density lipoprotein receptor (LDLR), and sterol O-acyltransferase 1 (SOAT1) in the APP/PS1 and 5xFAD mice brain, suggesting an increasing ability of cholesterol trafficking in order to compensate for cholesterol loss by decreasing cholesterol synthesis [107, 110, 156]. What's more, Park et al. also found that cholesterol synthetic genes were downregulated in FAD mice brain as a consequence of the chronically stimuli, such as Aβ, which is consistent with the previous studies [14, 43, 46, 102, 107, 110, 135, 148, 156]. Therefore, above data suggest that Aβ could lead to the inhibition of the cholesterol synthesis and the decrease of brain cholesterol in FAD mice brain.
Alteration of cholesterol metabolism in AD patients
Intriguingly, many evidences suggest that de novo synthesis of cholesterol in the brain decline in AD patients [68, 117, 123, 129, 146]. In addition, compared with non-demented controls, the cerebrospinal fluid (CSF) levels of cholesterol and its precursors (lanosterol, lathosterol and desmosterol) are lower in the brain of AD patients, suggesting that cholesterol de novo synthesis within the brain of AD patients might be reduced [68]. And cholesterol synthesis is decreased in the hippocampus, while absolute cholesterol content remains at a stable level in the AD, suggesting a brain region-specific decrease of cholesterol synthesis [92, 123, 146]. Furthermore, in AD, the level of cholesterol is reduced in the hippocampus, lipid raft fraction in the whole brain, and white matter, coupled with membrane lipid structure perturbation; the brain cholesterol deficit/loss play a major role in the disruption of AD membrane lipid structure [1, 96, 101]. Consequently, above data suggest that the human AD brain could have decreased cholesterol levels, and a region-dependence of cholesterol synthesis is influenced [123, 129, 146]
Intriguingly, in a new study, Varma et al. found that the majority of genes (14/15) within the de novo cholesterol biosynthesis pathway, including 3 in pre-squalene and 12 in post-squalene, showed significantly lower gene expression in the entorhinal cortex and hippocampus of AD patients compared to the non AD control, and no alterations were detected in the visual cortex, suggesting a regional-specific reduction of cholesterol biosynthesis [152]. Very importantly, these alterations of differential and region-specific genes expression in the entorhinal cortex and hippocampus appears to provide insights into cholesterol homeostasis dysregulation in AD pathogenesis, which might be tightly related to the initiation and progression of AD [62, 152]. Notably, Varma et al. found that gene expression alterations identified in AD brain were not observed in PD brain, suggesting that these changes may be specific to AD. Thus, the author supposes that the decrease of brain cholesterol synthesis likely reflects fundamental features of AD pathogenesis [152].
In addition, it is striking that gene expression of cytochrome P450 46A1 (CYP46A1), is also significantly lowered in the entorhinal cortex and hippocampus in AD. Inactivation of CYP46A1 has been shown to lower cholesterol efflux from the brain, leading to a compensatory response due to the decrease in de novo cholesterol biosynthesis [152]. Furthermore, Varma et al. also show that the principal cholesterol precursor lanosterol and catabolic product 24S-hydroxycholesterol (24-OHC) is lower in AD, suggesting that both de novo cholesterol biosynthesis and catabolism are impaired by the disease [152]. Interestingly, a growing bulk of evidence reveals that CYP46A1 expression and its catabolic product 24-OHC content significantly decreased in late AD compared to control and early AD brains [41]. However, a few studies suggest that the increase of Cyp46A1 activity might be partly responsible for cholesterol loss in aged and AD brain [138]. Collectively, the decrease of CYP46A1 expression in AD brains is likely to be due to the compensatory response to brain cholesterol loss, or a selective loss of neurons expressing the enzyme CYP46A1 during AD development [41, 152]. Therefore, increasing evidence suggest that there is a decrease of the cholesterol biosynthesis and/or decrease of cholesterol catabolism, resulting in brain cholesterol loss in AD brain.
Alteration of cholesterol trafficking in animals/patients with genetic forms of AD
The apoE4 allele is the dominant genetic risk factor for late-onset AD, and apoE4 has great influence in Aβ aggregation and clearance, tau pathogenesis, neuroinflammation, synaptic dysfunction, and neuronal loss [56, 89, 147]. However, the association between apoE4 and AD pathogenesis remains ambiguous. Though much of the research has focused on the ability of the apoE4 to increase the aggregation and decrease the clearance of Aβ, a lot of evidences show that apoE4 obviously impacts cholesterol transport and homeostasis in the brain [39, 89]. ApoE isoforms exert a central role in controlling the transport of brain lipid, including cholesterol, and maintaining cholesterol homeostasis in the brain [89]. Moreover, the accelerated degradation of apoE4 by astrocytes and neurons, resulting in decreased apoE4 levels in the brain [39, 89, 147]. In apoE4 mice, with the reduced secretion of apoE4 by astrocytes, Astrocytes secreted 34% less cholesterol than those from wild-type mice, the amounts of total cholesterol were significantly decreased compared with the wild-type littermates [48, 158]. ApoE4-expressing cultured astrocytes and neurons have reduced cholesterol and phospholipid secretion, decreased affinity for lipids, and increased intracellular degradation [56, 89]. In addition, in cultured neurons, cholesterol uptake is lower when the lipid is bound to apoE4 compared to apoE2 and apoE3 [56, 121]. ApoE4 is less efficient than other forms in promoting cholesterol efflux from both neurons and astrocytes [100]. Importantly, low membrane cholesterol was observed in hippocampal membranes of apoE4 AD cases [74]. To sum up, the structural differences among different apoE isoforms may account for the alterations in cholesterol trafficking. Therefore, above findings support that apoE4 markedly lead to the decrease of brain cholesterol levels in the apoE4 mice and patients, which may be involved in AD pathogenesis.
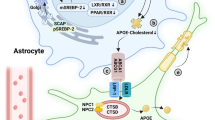
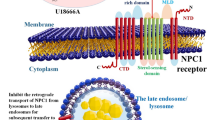
Interestingly, another direct association between cell cholesterol loss in the brain and neurodegeneration has been clearly demonstrated in Niemann–Pick C disease (NPC) [93, 150]. Niemann–Pick C disease is an autosomal recessive disorder caused by mutations in the NPC1 or NPC2 genes, which present clinical and neuropathological signs of Alzheimer's disease dementia [93, 150]. Besides, at the histological level, NPC-deficient brains present with amyloid-β deposition, neurofibrillary tangles, neuroinflammation, neuroaxonal dystrophy, and loss of neurons [93, 150]. NPC1 and NPC2 each bind to cholesterol and act in tandem in late endosomes and/or lysosomes to mediate the egress of unesterified cholesterol derived from endocytosed lipoproteins [116, 120, 150]. Thus, in NPC1- or NPC2-deficient cells, including neurons and glial cells, unesterified cholesterol and other lipids become sequestered in late endosomes and/or lysosomes [60, 93]. Accordingly, in addition to late endosomes and/or lysosomes, the amount of cholesterol in the plasma membrane, endoplasmic reticulum and axons is reduced in Npc1−/− or Npc2−/− neurons, suggesting cholesterol deficiency in the brain of Niemann–Pick C disease [60, 79, 93]. Particularly, NPC and AD share some similar molecular pathological features, including abnormal cholesterol metabolism, and involvement of amyloid-β and tau pathology [67, 93]. Obviously, further studies of similarities between AD and NPC may be useful to increase the understanding of AD pathogenesis. Taking together, the neurological deficits in NPC disease might be attributable to a deficiency, rather than an excess, of cholesterol in plasma membrane and intracellular organelles, which might be associated with AD pathogenesis [93, 150].
In addition to apoE and NPC, other genes involved in the transportation of cholesterol have been suggested as putative risk factors for AD [18, 65]. ATP-binding cassette transporters (ABC) are essential component for mediating lipid transport in brain, especially in the formation of apoE-containing lipoproteins [18, 65, 144]. Neuron and glia specific ABCA1 deficiency leads to poor lipidation of apoE, and significant decrease of cholesterol level, decrease of apoE level in brain, leading to the pathological injuries that are tightly associated with degenerative diseases neurodegenerative diseases [18, 54]. Intriguingly, many lipoprotein receptors of LDL receptor family have been identified in brain including LDLR, Low density lipoprotein receptor-related protein 1 (LRP1), very low-density lipoprotein (VLDL)-receptor, Apolipoprotein E receptor 2 (apoER2/LRP8), and the sortilin-related receptor 1 (SORL1/LP11) [18, 51]. Conditional deletion of lipoprotein receptors genes in mouse brain significantly decreases apoE and cholesterol level, resulting in related-AD neuropathological damages [51, 80, 118]. Collectively, genetic defects in the genes of ABC transporters and lipoprotein receptors of LDL receptor family are related to decrease of brain cholesterol transport and uptake, resulting in decreased brain cholesterol level. Based on the above data, we found that genetic defects in cholesterol trafficking (transport and uptake) also obviously lead to decreased brain cholesterol level which might be involved in neurodegenerative diseases, such as AD.
Summary: brain cholesterol deficiency and AD
As stated above, a growing body of research has revealed that there is abnormal brain cholesterol metabolism in the brain in aging human and animals, SAMP8 mice, diabetic mice, FAD (5xFAD and APP/PS-1) animals, AD patients, genetic forms of AD animals and patients (ApoE4 allele, mutation of NPC1 or NPC2, polymorphism or mutations of ABC transporter and LDL receptor family). Furthermore, we found that dysregulation of cholesterol metabolism may be involved in cholesterol synthesis, trafficking and catabolism, including: 1) the decrease of de novo cholesterol synthesis; 2) and/or the decrease of cholesterol trafficking (transportation, uptake, and intracellular transportation); 3) and/or the decrease of cholesterol catabolism, suggesting the brain cholesterol loss in these different kinds of AD animals and patients (Fig. 3). Interestingly, we found that the brain cholesterol deficiency appears to be a pervasive and prominent pathological feature in these different kinds of AD models and patients (Table 1). Therefore, above data strongly suggest a new idea that there may be the brain cholesterol insufficiency or loss in the brain of AD models and patients.
Widen the view on AD: brain cholesterol level and AD
Over the last decades, increasing biochemical and molecular biological evidences reveal that altered cholesterol metabolism appears to play fundamental roles in amyloid plaque formation, tau hyperphosphorylation, synaptic loss, and apoptosis, suggesting a key role of cholesterol in the initiation and progression of AD [1, 5, 21, 74, 75, 91, 93, 115]. However, the role of cholesterol for neurodegeneration such as AD, remains still controversial [1, 5, 81, 93, 115, 154].
High plasma cholesterol level and AD
Early epidemiological studies suggest that increased level of plasma cholesterol is a risk factor for the development of AD [77, 140, 149]. Several studies show that lipophilic statins (brain-permeant statin) which can cross the blood–brain barrier, present a reduced incidence of AD [7, 37, 38, 59]. However, randomized double-blind placebo-controlled studies have shown no beneficial effect of statins on the progression of symptoms in subjects with AD [126, 134]. On the contrary, the lipophilic statins induce high amyloid production and senile plaque deposition in mice brain [74, 109]. And recent studies show that lipophilic statins could increase the risk of developing dementia, coupled with the decrease of brain cholesterol level, suggesting brain cholesterol loss have the increasing risk of dementia [47, 108]. Because the blood–brain barrier prevents entry of cholesterol-rich lipoproteins, all cholesterol in the brain is made locally. Thus, causal correlations between high blood cholesterol and AD are controversial.
High brain cholesterol level and AD
Early-study shows that brain cholesterol is high in the brains of patients with AD [25]. However, many studies suggest that cholesterol levels don’t differ in hippocampal region and the cerebral cortex tissue of AD patients compared with control subjects [32, 52]. Further, there are inconsistent outcomes in brain cholesterol levels of AD patients, but the variability of cholesterol level amongst the studies might obviously pertain to brain tissue sample selection, tissue preparation and assay methods [153]. Consequently, there is no enough evidence to prove that high brain cholesterol level contributes to AD.
Low brain cholesterol level and AD
Based on the data in the part 5, increasing evidences support that there may be obvious cholesterol loss in the brain of different kinds of AD animals and patients. Intriguingly, we found that the different kinds of AD models and patients included the major risk factors for AD, such as Aβ, aging, diabetes and diabetes-related factors, oxidative stress, chronic inflammation, and genetic risk factors, metabolic syndrome, etc. (Fig. 3). Furthermore, the brain cholesterol loss seems to be tightly associated with major risk factors from the different kinds of AD model animals and patients. Thus, we suppose that the brain cholesterol loss seems to be induced by the major risk factors for AD in the different kinds of AD models and patients. Thus, accumulating evidences suggest that there may be a direct link between brain cholesterol loss and major risk factors for AD.
Although there are conflicting reports on the role of cholesterol in AD, it is not difficult to envision how reduced neuronal cholesterol levels can lead to the neuropathological impairments which are associated with AD, resulting in the brain dysfunction [16, 27, 33, 40, 49, 53]. Very importantly, increasing evidences reveal that the neuronal cholesterol deficit/loss could induce the disruption of membrane lipid rafts and/or intracellular organelles, and eventually leads to the formation of pathological impairments, which are obviously linked to the pathological changes which are associated with the pathogenesis of AD and other neurodegenerative diseases [1, 5, 9, 16, 21, 24, 27, 33, 36, 40, 44, 74, 75, 91, 93, 99, 115, 119]. Taking together, compelling evidences suggest that the brain cholesterol deficiency could contribute to AD pathogenesis.
Conclusion and future perspective
In this paper, we try to provide a current state of research on the role of DHCR24 in the pathogenesis of Alzheimer's disease. Importantly, based on previous studies and our research on DHCR24, the decreased cholesterol level by DHCR24 knockdown could contribute to neurodegenerative diseases such as AD, thus, these findings suggest that augmentation of DHCR24 in the affected brain areas might provide a potential therapeutic approach to intervene in AD and other neurodegenerative diseases. As a key node in the control of cholesterol synthesis and homeostasis, targeting DHCR24, careful modulation of brain cholesterol metabolism may provide an alternative or complementary interventional approach in order to test whether modulating brain neuronal cholesterol metabolism is a viable strategy for preventing AD.
With the continuously growing body of knowledge in this field, a body of studies has pinpointed that brain cholesterol deficiency is very likely to be an early and common driving factor in the onset and development of AD, and seems to be intimately linked with the generation of amyloid plaques, tauopathy, synaptic injuries, neuronal loss, which are central to the pathogenesis of AD. To sum up, based on previous data and research on DHCR24, we suppose that the brain cholesterol deficiency/loss could trigger the onset and progression of AD (Fig. 3). In addition, although there are many acceptable hypotheses, such as amyloid-β, tau, and inflammatory hypotheses, the pathogenetic mechanism of Alzheimer's disease is still elusive. Furthermore, uncovering the key causative alterations of AD can be valuable in developing models for AD treatment. In order to gain a better understanding of cholesterol’s role in AD pathogenesis, we hope that this new proposal will stimulate further experimental research in this direction that allows the testing of our hypothesis. In the review, we only chose some topics for in depth discussions. Unfortunately, a lot of important research topics were left with little or with no discussion. Certainly, in order to gain a more comprehensive recognition of cholesterol’s role in AD pathogenesis, we still need to investigate more the role of cholesterol metabolism in AD patient and animal brains, including the brain cholesterol amount, specific regional changes, as well as the distribution of cholesterol within neurons such as lipid rafts or intracellular organelles.
Availability of data and materials
Not applicable.
Abbreviations
- 5xFAD:
-
Five familial AD
- 7-DHC:
-
7-Dehydrocholesterol
- AD:
-
Alzheimer’s disease
- Akt:
-
Protein kinase
- APP/PS1:
-
APPswe/PS1deltaE9
- APP:
-
Aamyloid-β precursor protein
- Aβ:
-
Amyloid-β protein
- BACE1:
-
Beta-site amyloid precursor protein cleaving enzyme 1 or β-secretase
- DHCR24:
-
3β-Hydroxysterol-Δ24 reductase or 24-dehydrocholesterol reductase
- DHCR7:
-
7-Dehydrocholesterol reductase
- DM:
-
Diabetes mellitus
- DRM:
-
Detergent-resistant membrane domain
- FAD:
-
Familial Alzheimer's disease
- GGA3:
-
Golgi-localized gamma-ear-containing ARF binding protein 3
- GSK3β:
-
Glycogen synthase kinases-3beta
- HMGCR:
-
Hydroxy-3-methylglutaryl-CoA reductase
- IGFs:
-
Insulin-like growth factors
- KO:
-
Knockout
- K–R pathway:
-
Kandutsch–Russell pathway
- LDM:
-
CYP51A1 or Lanosterol 14-a-demethylase
- LOAD:
-
Late-Onset Alzheimer's Disease
- mTOR:
-
Mammalian target of rapamycin
- NGF:
-
Nerve growth factor
- PI3-K:
-
Phosphoinositide 3-kinase
- PP2A:
-
Protein phosphatase 2A
- SAD:
-
Sporadic AD
- Seladin-1:
-
Selective Alzheimer’s disease indicator 1
- shRNA:
-
Short hairpin RNA
- SQLE:
-
Squalene epoxidase
- STZ:
-
Streptozotocine
- Tau:
-
Microtubule-associated protein tau
- TH:
-
Thyroid hormones
References
Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, Dingwall C, De Strooper B, Dotti CG (2004) Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol 167(5):953–960. https://doi.org/10.1083/jcb.200404149
Alejandra DA, John DC, Bin L, Christopher PC, Alaniz ME, Grundke-Iqbal I (2010) Phosphorylation of tau at Thr212 Thr231 and Ser262 combined causes neurodegeneration. J Biol Chem 285(40):30851–30860. https://doi.org/10.1074/jbc.M110.110957
Allen LB, Genaro-Mattos TC, Porter NA, Mirnics K, Korade Z (2019) Desmosterolosis and desmosterol homeostasis in the developing mouse brain. J Inherit Metab Dis 42(5):934–943. https://doi.org/10.1002/jimd.12088
Alonso AD, Cohen LS, Corbo C, Morozova V, ElIdrissi A, Phillips G, Kleiman FE (2018) Hyperphosphorylation of tau associates with changes in its function beyond microtubule stability. Front Cell Neurosci 12:338. https://doi.org/10.3389/fncel.2018.00338
Anchisi L, Dessì S, Pani A, Mandas A (2013) Cholesterol homeostasis: a key to prevent or slow down neurodegeneration. Front Physiol 3:486. https://doi.org/10.3389/fphys.2012.00486
Andersson HC, Kratz L, Kelley R (2002) Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay. Am J Med Genet 113(4):315–319. https://doi.org/10.1002/ajmg.b.10873
Austen B, Christodoulou G, Terry JE (2002) Relation between cholesterol levels, statins and Alzheimer’s disease in the human population. J Nutr Health Aging 6:377–382
Baglietto-Vargas D, Shi J, Yaeger DM, Ager R, LaFerla FM (2016) Diabetes and Alzheimer’s disease crosstalk. Neurosci Biobehav Rev 64:272–287. https://doi.org/10.1016/j.neubiorev.2016.03.005
Bai X, Wu J, Zhang M, Xu Y, Duan L, Yao K, Zhang J, Bo J, Zhao Y, Xu G, Zu H (2021) DHCR24 knock-down induced Tau hyperphosphorylation at Thr181, Ser199, Thr231, Ser262, Ser396 epitopes and inhibition of autophagy by overactivation of GSK3β/mTOR signaling. Front Aging Neurosci 13:513605. https://doi.org/10.3389/fnagi.2021.513605
Benvenuti S, Luciani P, Cellai I, Deledda C, Baglioni S, Saccardi R, Urbani S, Francini F, Squecco R, Giuliani C, Vannelli GB, Serio M, Pinchera A, Peri A (2008) Thyroid hormones promote cell differentiation and up-regulate the expression of the seladin-1 gene in in vitro models of human neuronal precursors. J Endocrinol 197(2):437–446. https://doi.org/10.1677/JOE-07-0324
Berisha SZ, Serre D, Schauer P, Kashyap SR, Smith JD (2011) Changes in whole blood gene expression in obese subjects with type 2 diabetes following bariatric surgery: a pilot study. PLoS ONE 6(3):e16729. https://doi.org/10.1371/journal.pone.0016729
Bhatti JS, Bhatti GK (1863) Reddy PH (2017) Mitochondrial dysfunction and oxidative stress in metabolic disorders—a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis 5:1066–1077. https://doi.org/10.1016/j.bbadis.2016.11.010
Biessels GJ, Despa F (2018) Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol 14(10):591–604. https://doi.org/10.1038/s41574-018-0048-7
Bing L, Wu J, Zhang J, Chen Y, Hong Z, Zu H (2015) DHT inhibits the Aβ25-35-induced apoptosis by regulation of seladin-1, survivin, XIAP, bax, and bcl-xl expression through a rapid PI3-K/Akt signaling in C6 glial cell lines. Neurochem Res 40(1):41–48. https://doi.org/10.1007/s11064-014-1463-3
Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ (2018) The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep 22(1):269–285. https://doi.org/10.1016/j.celrep.2017.12.039
Bok E, Leem E, Lee BR, Lee JM, Yoo CJ, Lee EM, Kim J (2021) Role of the lipid membrane and membrane proteins in Tau pathology. Front Cell Dev Biol 9:653815. https://doi.org/10.3389/fcell.2021.653815
Bonaccorsi L, Luciani P, Nesi G, Mannucci E, Deledda C, Dichiara F, Paglierani M, Rosati F, Masieri L, Serni S, Carini M, Proietti-Pannunzi L, Monti S, Forti G, Danza G, Serio M, Peri A (2008) Androgen receptor regulation of the seladin-1/DHCR24 gene: altered expression in prostate cancer. Lab Invest 88(10):1049–1056. https://doi.org/10.1038/labinvest.2008.80
Bu G (2009) Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10(5):333–344. https://doi.org/10.1038/nrn2620
Budni J, Bellettini-Santos T, Mina F, Garcez ML, Zugno AI (2015) The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis 6(5):331–41. https://doi.org/10.14336/AD.2015.0825
Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, Belfiore R, Winslow W, Oddo S (2017) Necroptosis activation in Alzheimer’s disease. Nat Neurosci 20(9):1236–1246. https://doi.org/10.1038/nn.4608
Calleros L, Lasa M, Toro MJ, Chiloeches A (2006) Low cell cholesterol levels increase NFkappaB activity through a p38 MAPK-dependent mechanism. Cell Signal 18:2292–2301. https://doi.org/10.1016/j.cellsig.2006.05.012
Colardo M, Martella N, Pensabene D, Siteni S, Bartolomeo SD, Pallottini V, Segatto M (2021) Neurotrophins as key regulators of cell metabolism: implications for cholesterol homeostasis. Int J Mol Sci 22(11):5692. https://doi.org/10.3390/ijms22115692
Collin F (2019) Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int J Mol Sci 20(10):2407. https://doi.org/10.3390/ijms20102407
Crameri A, Biondi E, Kuehnle K, Lütjohann D, Thelen KM, Perga S, Dotti CG, Nitsch RM, Maria Ledesma MD, Mohajeri MH (2006) The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J 25(2):432–443. https://doi.org/10.1038/sj.emboj.7600938
Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA 101:2070–2075. https://doi.org/10.1073/pnas.0305799101
Dhana K, Braun KVE, Nano J, Voortman T, Demerath EW, Guan W, Fornage M, van Meurs JBJ, Uitterlinden AG, Hofman A, Franco OH, Dehghan A (2018) An epigenome-wide association study of obesity-related traits. Am J Epidemiol 187(8):1662–1669. https://doi.org/10.1093/aje/kwy025
Díaz M, Fabelo N, Ferrer I, Marín R (2018) “Lipid raft aging” in the human frontal cortex during nonpathological aging: gender influences and potential implications in Alzheimer’s disease. Neurobiol Aging 67:42–52. https://doi.org/10.1016/j.neurobiolaging.2018.02.022
Díaz M, Fabelo N, MartínV FI, GómezT MR (2015) Biophysical alterations in lipid rafts from human cerebral cortex associate with increased BACE1/AβPP interaction in early stages of Alzheimer’s disease. J Alzheimers Dis 43(4):1185–1198. https://doi.org/10.3233/JAD-141146
Dorszewska J, Prendecki M, Oczkowska A, Dezor M, Kozubski W (2016) Molecular basis of familial and sporadic Alzheimer’s disease. Curr Alzheimer Res 13(9):952–963. https://doi.org/10.2174/1567205013666160314150501
Drzewińska J, Pułaski L, Soszyński M, Bartosz G (2009) Seladin-1/DHCR24: a key protein of cell homeostasis and cholesterol biosynthesis. Postepy Hig Med Dosw 63:318–330 (Polish)
Drzewinska J, Walczak-Drzewiecka A, Ratajewski M (2011) Identification and analysis of the promoter region of the human DHCR24 gene: involvement of DNA methylation and histone acetylation. Mol Biol Rep 38:1091–1101. https://doi.org/10.1007/s11033-010-0206-z
Eckert GP, Cairns NJ, Maras A, Gattaz WF, Müller WE (2000) Cholesterol modulates the membrane disordering effects of β-amyloid peptides in the hippocampus: specific changes in Alzheimer’s disease. Dement Geriatr Cogn Disord 11:181–186. https://doi.org/10.1159/000017234
Egawa J, Pearn ML, Lemkuil BP, Patel PM, Head BP (2016) Membrane lipid rafts and neurobiology: age-related changes in membrane lipids and loss of neuronal function. J Physiol 594(16):4565–4579. https://doi.org/10.1113/JP270590
Fabelo N, Martín V, Marín R, Moreno D, Ferrer I, Díaz M (2014) Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol Aging 35(8):1801–1812. https://doi.org/10.1016/j.neurobiolaging.2014.02.005
Fabelo N, Martín V, Marín R, Santpere G, Aso E, Ferrer I, Díaz M (2012) Evidence for premature lipid raft aging in APP/PS1 double-transgenic mice, a model of familial Alzheimer disease. J Neuropathol Exp Neurol 71(10):868–881. https://doi.org/10.1097/NEN.0b013e31826be03c
Fantini J, Barrantes FJ (2013) How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front Physiol 4:31. https://doi.org/10.3389/fphys.2013.00031
Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller DP, Run H, Kuhl S, Bertsch T, Von Bergmann K, Hennerici M, Beyreuther K, Hartmann T (2001) Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA 98:5856–5861. https://doi.org/10.1073/pnas.081620098
Fassbender K, Stroick M, Bertsch T, Ragoschke A, Kuehl S, Walter S, Walter J, Brechtel K, Muehlhauser F, Von Bergmann K, Lütjohann D (2002) Effects of statins on human cerebral cholesterol metabolism and secretion of Alzheimer amyloid peptide. Neurology 59(8):1257–1258. https://doi.org/10.1212/wnl.59.8.1257
Fernandez CG, Hamby ME, McReynolds ML, Ray WJ (2019) The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front Aging Neurosci 11:14. https://doi.org/10.3389/fnagi.2019.00014
Ferris HA, Perry RJ, Moreira GV, Shulman GI, Horton JD, Kahn CR (2017) Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc Natl Acad Sci USA 114(5):1189–1194. https://doi.org/10.1073/pnas.1620506114
Gamba P, Giannelli S, Staurenghi E, Testa G, Sottero B, Biasi F, Poli G, Leonarduzzi G (2021) The controversial role of 24-S-hydroxycholesterol in Alzheimer’s disease. Antioxidants 10(5):740. https://doi.org/10.3390/antiox10050740
Giannini S, Benvenuti S, Luciani P, Manuelli C, Cellai I, Deledda C, Pezzatini A, Vannelli GB, Maneschi E, Rotella CM, Serio M, Peri A (2008) Intermittent high glucose concentrations reduce neuronal precursor survival by altering the IGF system: the involvement of the neuroprotective factor DHCR24 (Seladin-1). J Endocrinol 198(3):523–532. https://doi.org/10.1677/JOE-07-0613
Gong JS, Sawamura N, Zou K, Sakai J, Yanagisawa K, Michikawa M (2002) Amyloid beta-protein affects cholesterol metabolism in cultured neurons: implications for pivotal role of cholesterol in the amyloid cascade. J Neurosci Res 70(3):438–446
Grassi S, Giussani P, Mauri L, Prioni S, Sonnino S, Prinetti A (2020) Lipid rafts and neurodegeneration: structural and functional roles in physiologic aging and neurodegenerative diseases. J Lipid Res 61(5):636–654. https://doi.org/10.1194/jlr.TR119000427
Greeve I, Hermans-Borgmeyer I, Brellinger C, Kasper D, Gomez-Isla T, Behl C, Levkau B, Nitsch RM (2000) The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress. J Neurosci 20(19):7345–7352. https://doi.org/10.1523/JNEUROSCI.20-19-07345.2000
Grimm MO, Grimm HS, Pätzold AJ, Zinser EG, Halonen R, Duering M, Tschäpe JA, De Strooper B, Müller U, Shen J, Hartmann T (2005) Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol 7(11):1118–1123
Guo Y, Zou G, Qi K, Jin J, Yao L, Pan Y, Xiong W (2021) Simvastatin impairs hippocampal synaptic plasticity and cognitive function in mice. Mol Brain 14(1):41. https://doi.org/10.1186/s13041-021-00758-x
Hamanaka H, Katoh-Fukui Y, Suzuki K, Kobayashi M, Suzuki R, Motegi Y, Nakahara Y, Takeshita A, Kawai M, Ishiguro K, Yokoyama M, Fujita SC (2000) Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum Mol Genet 9(3):353–361. https://doi.org/10.1093/hmg/9.3.353
Head BP, Patel HH (1838) Insel PA (2014) Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta 2:532–545. https://doi.org/10.1016/j.bbamem.2013.07.018
Hegde V, Dhurandhar NV, Reddy PH (2019) Hyperinsulinemia or insulin resistance: What impacts the progression of Alzheimer’s disease? J Alzheimers Dis 72(s1):S71–S79. https://doi.org/10.3233/JAD-190808
Herz J (2009) Apolipoprotein E receptors in the nervous system. Curr Opin Lipidol 20:190–196. https://doi.org/10.1097/MOL.0b013e32832d3a10
Heverin M, Bogdanovic N, Lutjohann D, Bayer TA, Pikuleva I, Bretillon L, Diczfalusy U, Winblad B, Björkhem I (2004) Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J Lipid Res 45:186–193. https://doi.org/10.1194/jlr.M300320-JLR200
Hicks DA, Nalivaeva NN, Turner AJ (2012) Lipid rafts and Alzheimer’s disease: protein-lipid interactions and perturbation of signaling. Front Physiol 3:189. https://doi.org/10.3389/fphys.2012.00189
Hirsch-Reinshagen V, Zhou S, Burgess BL, Bernier L, McIsaac SA, Chan JY, Tansley GH, Cohn JS, Hayden MR, Wellington CL (2004) Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem 279:41197–41207. https://doi.org/10.1074/jbc.M407962200
Hosseinzadeh S, Zahmatkesh M, Heidari M, Hassanzadeh GR, Karimian M, Sarrafnejad A, Zarrindast MR (2015) Hippocampal DHCR24 down regulation in a rat model of streptozotocin-induced cognitive decline. Neurosci Lett 587:107–112. https://doi.org/10.1016/j.neulet.2014.12.039
Husain MA, Laurent B, Plourde M (2021) APOE and Alzheimer’s disease: from lipid transport to physiopathology and therapeutics. Front Neurosci 15:630502. https://doi.org/10.3389/fnins.2021.630502
Iivonen S, Hiltunen M, Alafuzoff I, Mannermaa A, Kerokoski P, Puoliväli J, Salminen A, Helisalmi S, Soininen H (2002) Seladin-1 transcription is linked to neuronal degeneration in Alzheimer’s disease. Neuroscience 113(2):301–310. https://doi.org/10.1016/s0306-4522(02)00180-x
Ing NH, Forrest DW, Riggs PK, Loux S, Love CC, Brinsko SP, Varner DD, Welsh TH Jr (2014) Dexamethasone acutely down-regulates genes involved in steroidogenesis in stallion testes. J Steroid Biochem Mol Biol 143:451–459. https://doi.org/10.1016/j.jsbmb.2014.07.003
Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA (2000) Statins and the risk of dementia. Lancet 356(9242):1627–1631. https://doi.org/10.1016/s0140-6736(00)03155-x
Karten B, Vance DE, Campenot RB, Vance JE (2002) Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann–Pick C1-deficient neurons. J Neurochem 83:1154–1163. https://doi.org/10.1046/j.1471-4159.2002.01220.x
Kazkayasi I, Ismail MA, Parrado-Fernandez C, Björkhem I, Pekiner C, Uma S, Cedazo-Minguez A, Burul-Bozkurt N (2016) Lack of insulin results in reduced seladin-1 expression in primary cultured neurons and in cerebral cortex of STZ-induced diabetic rats. Neurosci Lett 633:174–181. https://doi.org/10.1016/j.neulet.2016.09.018
Khan UA, Liu L, Provenzano FA, Berman DE, Profaci P, CP, Sloan R, Mayeux R, Duff KE, Small SA, (2014) Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat Neurosci 17(2):304–311. https://doi.org/10.1038/nn.3606
Khuda II, Koide N, Noman AS, Dagvadorj J, Tumurkhuu G, Naiki Y, Komatsu T, Yoshida T, Yokochi T (2010) Seladin-1 is a novel lipopolysaccharide (LPS)-responsive gene and inhibits the tumour necrosis factor-alpha production and osteoclast formation in response to LPS. Immunology 131(1):59–66. https://doi.org/10.1111/j.1365-2567.2010.03274.x
Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 44:325–340. https://doi.org/10.1016/j.molcel.2011.08.025
Kim WS, Weickert CS, Garner B (2008) Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J Neurochem 104(5):1145–1166. https://doi.org/10.1111/j.1471-4159.2007.05099.x
Kleinridders A, Ferris HA, Cai W, Kahn CR (2014) Insulin action in brain regulates systemic metabolism and brain function. Diabetes 63(7):2232–2243. https://doi.org/10.2337/db14-0568
Kodam A, Maulik M, Peake K, Amritraj A, Vetrivel KS, Thinakaran G, Vance JE, Kar S (2010) Altered levels and distribution of amyloid precursor protein and its processing enzymes in Niemann–Pick type C1-deficient mouse brains. Glia 58(11):1267–1281. https://doi.org/10.1002/glia.21001
Kölsch H, Heun R, Jessen F, Popp J, Hentschel F, Maier W (1801) Lütjohann D (2010) Alterations of cholesterol precursor levels in Alzheimer’s disease. Biochim Biophys Acta 8:945–950. https://doi.org/10.1016/j.bbalip.2010.03.001
Koudinov AR, Koudinova NV (2005) Cholesterol homeostasis failure as a unifying cause of synaptic degeneration. J Neurol Sci 229–230:233–240. https://doi.org/10.1016/j.jns.2004.11.036
Kuehnle K, Crameri A, Kälin RE, Luciani P, Benvenuti S, Peri A, Ratti F, Rodolfo M, Kulic L, Heppner FL, Nitsch RM, Mohajeri MH (2008) Prosurvival effect of DHCR24/Seladin-1 in acute and chronic responses to oxidative stress. Mol Cell Biol 28(2):539–550. https://doi.org/10.1128/MCB.00584-07
Kuehnle K, Ledesma MD, Kalvodova L, Smith AE, Crameri A, Skaanes-Brunner F, Thelen KM, Kulic L, Lütjohann D, Heppner FL, Nitsch RM, Mohajeri MH (2009) Age-dependent increase in desmosterol restores DRM formation and membrane-related functions in cholesterol-free DHCR24−/− mice. Neurochem Res 34(6):1167–1182. https://doi.org/10.1007/s11064-009-9994-8
Kulas JA, Weigel TK, Ferris HA (2020) Insulin resistance and impaired lipid metabolism as a potential link between diabetes and Alzheimer’s disease. Drug Dev Res 81(2):194–205. https://doi.org/10.1002/ddr.21643
Ledesma MD, Abad-Rodriguez J, Galvan C, Biondi E, Navarro P, Delacourte A, Dingwall C, Dotti CG (2003) Raft disorganization leads to reduced plasmin activity in Alzheimer’s disease brains. EMBO Rep 4:1190–1196. https://doi.org/10.1038/sj.embor.7400021
Ledesma MD, Dotti CG (2005) The conflicting role of brain cholesterol in Alzheimer’s disease: lessons from the brain plasminogen system. Biochem Soc Symp 72:129–138. https://doi.org/10.1042/bss0720129
Ledesma MD, Martin MG, Dotti CG (2012) Lipid changes in the aged brain: effect on synaptic function and neuronal survival. Prog Lipid Res 51(1):23–35. https://doi.org/10.1016/j.plipres.2011.11.004
Lemche E (2018) Early life stress and epigenetics in late-onset Alzheimer’s dementia: a systematic review. Curr Genomics 19(7):522–602. https://doi.org/10.2174/1389202919666171229145156
Leoni V, Solomon A, Kivipelto M (2010) Links between ApoE, brain cholesterol metabolism, tau and amyloid beta-peptide in patients with cognitive impairment. Biochem Soc Trans 1021–1025. https://doi.org/10.1042/BST0381021
Linetti A, Fratangeli A, Taverna E, Valnegri P, Francolini M, Cappello V, Matteoli M, Passafaro M, Rosa P (2010) Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci 123(Pt 4):595–605. https://doi.org/10.1242/jcs.060681
Liscum L, Ruggiero RM, Faust JR (1989) The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann–Pick type C fibroblasts. J Cell Biol 108:1625–1636. https://doi.org/10.1083/jcb.108.5.1625
Liu Q, Trotter J, Zhang J, Peters MM, Cheng H, Bao J, Han X, Weeber EJ, Bu G (2010) Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci 30:17068–17078. https://doi.org/10.1523/JNEUROSCI.4067-10.2010
Loera-Valencia R, Goikolea J, Parrado-Fernandez C, Merino-Serrais P, Maioli S (2019) Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: potential novel targets for treatment. J Steroid Biochem Mol Biol 190:104–114. https://doi.org/10.1016/j.jsbmb.2019.03.003
Lu X, Kambe F, Cao X, Kozaki Y, Kaji T, Ishii T, Seo H (2008) 3beta-Hydroxysteroid-delta24 reductase is a hydrogen peroxide scavenger, protecting cells from oxidative stress-induced apoptosis. Endocrinology 149(7):3267–3273. https://doi.org/10.1210/en.2008-0024
Lu X, Kambe F, Cao X, Yoshida T, Ohmori S, Murakami K, Kaji T, Ishii T, Zadworny D, Seo H (2006) DHCR24-knockout embryonic fibroblasts are susceptible to serum withdrawal-induced apoptosis because of dysfunction of caveolae and insulin-Akt-Bad signaling. Endocrinology 147(6):3123–3132. https://doi.org/10.1210/en.2005-1426
Lu X, Li Y, Wang W, Chen S, Liu T, Jia D, Quan X, Sun D, Chang AK, Gao B (2014) 3 β-hydroxysteroid-Δ 24 reductase (DHCR24) protects neuronal cells from apoptotic cell death induced by endoplasmic reticulum (ER) stress. PLoS ONE 9(1):e86753. https://doi.org/10.1371/journal
Luu W, Hart-Smith G, Sharpe L, Brown A (2015) The terminal enzymes of cholesterol synthesis, DHCR24 and DHCR7, interact physically and functionally. J Lipid Res 56(4):888–897. https://doi.org/10.1194/jlr.M056986
Luu W, Sharpe LJ, Gelissen IC, Brown AJ (2013) The role of signalling in cellular cholesterol homeostasis. IUBMB Life 65:675–684. https://doi.org/10.1002/iub.1182
Luu W, Zerenturk EJ, Kristiana I, Bucknall MP, Sharpe LJ, Brown AJ (2014) Signaling regulates activity of DHCR24, the final enzyme in cholesterol synthesis. J Lipid Res 55(3):410–420. https://doi.org/10.1194/jlr.M043257
Mahakizadeh S, Mokhtari T, Navaee F, Poorhassan M, Armin Tajik A, Hassanzadeh G (2020) Effects of chronic hypoxia on the expression of seladin-1/Tuj1 and the number of dark neurons of hippocampus. J Chem Neuroanat 104:101744. https://doi.org/10.1016/j.jchemneu.2020.101744
Mahley RW (2016) Central nervous system lipoproteins ApoE and regulation of cholesterol metabolism. Arterioscler Thromb Vasc Biol 36(7):1305–1315. https://doi.org/10.1161/ATVBAHA.116.307023
Malnar M, Hecimovic S, Mattsson N, Zetterberg H (2014) Bidirectional links between Alzheimer’s disease and Niemann–Pick type C disease. Neurobiol Dis 72(Pt A):37–47. https://doi.org/10.1016/j.nbd.2014.05.033
Martin M, Dotti CG, Ledesma MD (2010) Brain cholesterol in normal and pathological aging. Biochim Biophys Acta 1801:934–944. https://doi.org/10.1016/j.bbalip.2010.03.011
Martin MG, Ahmed T, Korovaichuk A, Venero C, Menchón SA, Salas I, Munck S, Herreras O, Balschun D, Dotti CG (2014) Constitutive hippocampal cholesterol loss underlies poor cognition in old rodents. EMBO Mol Med 6:902–917. https://doi.org/10.15252/emmm.201303711
Martín MG, FrankPfrieger F, Dotti CG (2014) Cholesterol in brain disease: sometimes determinant and frequently implicated. EMBO Rep 15(10):1036–1052. https://doi.org/10.15252/embr.201439225
Martin MG, Perga S, Trovo L, Rasola A, Holm P, Rantamaki T, Dotti CG (2008) Cholesterol loss enhances TrkB signaling in hippocampal neurons aging in vitro. Mol Biol Cell 19:2101–2112. https://doi.org/10.1091/mbc.e07-09-0897
Martiskainen H, Paldanius KMA, Natunen T, Takalo M, Marttinen M, Leskelä S, Huber N, Mäkinen P, Bertling E, Dhungana H, Huuskonen M, Honkakoski P, Hotulainen P, Rilla K, Koistinaho J, Soininen H, Malm T, Haapasalo A, Hiltunen M (2017) DHCR24 exerts neuroprotection upon inflammation-induced neuronal death. J Neuroinflammation 14(1):215. https://doi.org/10.1186/s12974-017-0991-6
Mason RP, Shoemaker WJ, Shajenko L, Chambers TE, Herbette LG (1992) Evidence for changes in the Alzheimer’s disease brain cortical membrane structure mediated by cholesterol. Neurobiol Aging 13:413–419. https://doi.org/10.1016/0197-4580(92)90116-f
McGeer PL, Rogers J, McGeer EG (2016) Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. J Alzheimers Dis 54(3):853–857. https://doi.org/10.3233/JAD-160488
Mecocci P, Boccardi V, Cecchetti R, Bastiani P, Scamosci M, Ruggiero C, Baroni M (2018) A long journey into aging, brain aging, and Alzheimer’s Disease Following The Oxidative Stress Tracks. J Alzheimers Dis 62(3):1319–1335. https://doi.org/10.3233/JAD-170732
Mesa-Herrera F, Taoro-González L, Valdés-Baizabal C, Diaz M, Marín R (2019) Lipid and lipid raft alteration in aging and neurodegenerative diseases: a window for the development of new biomarkers. Int J Mol Sci 20(15):3810. https://doi.org/10.3390/ijms20153810
Michikawa M, Fan QW, Isobe I, Yanagisawa K (2000) Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem 74:1008–16. https://doi.org/10.1046/j.1471-4159.2000.0741008.x
Molander-Melin M, Blennow K, Bogdanovic N, Dellheden B, Månsson J-E, Fredman P (2005) Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J Neurochem 92:171–182. https://doi.org/10.1111/j.1471-4159.2004.02849.x
Najem D, Bamji-Mirza M, Yang Z, Zhang W (2016) Aβ-induced insulin resistance and the effects of insulin on the cholesterol synthesis pathway and Aβ secretion in neural cells. Neurosci Bull 32(3):227–238. https://doi.org/10.1007/s12264-016-0034-9
Nanjundaiah S, Chidambaram H, Chandrashekar M, Chinnathambi S (2021) Role of microglia in regulating cholesterol and Tau pathology in Alzheimer’s disease. Cell Mol Neurobiol 41(4):651–668. https://doi.org/10.1007/s10571-020-00883-6
Neddens J, Temmel M, Flunkert S, Kerschbaumer B, Hoeller C, Loeffler T, Niederkofler V, Daum G, Attems J, Hutter-Paier B (2018) Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol Commun 6(1):52. https://doi.org/10.1186/s40478-018-0557-6
Obulesu M, Lakshmi MJ (2014) Apoptosis in Alzheimer’s disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem Res 39(12):2301–2312. https://doi.org/10.1007/s11064-014-1454-4
Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, Hol EM (2014) Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging 35(12):2746–2760. https://doi.org/10.1016/j.neurobiolaging.2014.06.004
Orre M, Kamphuis W, Osborn LM, Melief J, Kooijman L, Huitinga I, Klooster J, Bossers K, Hol EM (2014) Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging 35:1–14. https://doi.org/10.1016/j.neurobiolaging.2013.07.008
Padmanabham S, Liu S, Silverman D (2021) Lipophilic statins in subjects with early mild cognitive impairement: associations with conversion to dementia and decline in posterior cingulate brain metabolism in a long-term prospective longitudinal multi-center study. J Nucl Med 62(supplement 1):102
Park IH, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J, Jung MW, Bang OY, Kim SU, Mook-Jung I (2003) Lovastatin enhances Abeta production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging 24(5):637–643. https://doi.org/10.1016/s0197-4580(02)00155-0
Park J, Kim H, Kim J, Cheon M (2020) A practical application of generative adversarial networks for RNA-seq analysis to predict the molecular progress of Alzheimer’s disease. PLoS Comput Biol 16(7):e1008099. https://doi.org/10.1371/journal.pcbi.1008099
Pérez-Cañamás A, Sarroca S, Melero-Jerez C, Porquet D, Sansa J, Knafo S, Esteban JA, Sanfeliu C, Ledesma MD (2016) A diet enriched with plant sterols prevents the memory impairment induced by cholesterol loss in senescence-accelerated mice. Neurobiol Aging 48:1–12. https://doi.org/10.1016/j.neurobiolaging.2016.08.009
Peri A, Danza G, Benvenuti S, Luciani P, Deledda C, Rosati F, Cellai I, Serio M (2009) New insights on the neuroprotective role of sterols and sex steroids: the seladin-1/DHCR24paradigm. Front Neuroendocrinol 30(2):119–129. https://doi.org/10.1016/j.yfrne.2009.03.006
Peri A (2016) Neuroprotective effects of estrogens: the role of cholesterol. J Endocrinol Invest 39(1):11–18. https://doi.org/10.1007/s40618-015-0332-5
Petrov AM, Kasimov MR, Zefirov AL (2016) Brain cholesterol metabolism and its defects: linkage to neurodegenerative diseases and synaptic dysfunction. Acta Naturae 8(1):58–73
Petrov AM, Kasimov MR, Zefirov AL (2017) Cholesterol in the pathogenesis of Alzheimer’s, Parkinson’s diseases and autism: link to synaptic dysfunction. Acta Naturae 9(1):26–37
Pfeffer SR (2019) NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J Biol Chem 294(5):1706–1709. https://doi.org/10.1074/jbc.TM118.004165
Popp J, Meichsner S, Kölsch H, Lewczuk P, Maier W, Kornhuber J, Jessen F, Lütjohann D (2013) Cerebral and extracerebral cholesterol metabolism and CSF markers of Alzheimer’s disease. Biochem Pharmacol 86(1):37–42. https://doi.org/10.1016/j.bcp.2012.12.007
Pottier C, Hannequin D, Coutant S, Rovelet-Lecrux A, Wallon D, Rousseau S, Legallic S, Paquet C, Bombois S, Pariente J, Thomas-Anterion C, Michon A, Croisile B, Etcharry-Bouyx F, Berr C, Dartigues JF, Amouyel P, Dauchel H, Boutoleau-Bretonnière C, Thauvin C, Frebourg T, Lambert JC, Campion D, Collaborators PHRCGMAJ (2012) High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiarty 17:875–879. https://doi.org/10.1038/mp.2012.15
Qi Z, Zhang Y, Yao K, Zhang M, Xu Y, Zhang J, Bai X, Zu H (2021) DHCR24 knockdown lead to hyperphosphorylation of Tau at Thr181, Thr231, Ser262, Ser396, and Ser422 Sites by membrane lipid-raft dependent PP2A signaling in SH-SY5Y cells. Neurochem Res 46(7):1627–1640. https://doi.org/10.1007/s11064-021-03273-6
Qian H, Wu X, Du X, Yao X, Zhao X, Lee J, Yang H, Yan N (2020) Structural basis of low-pH-dependent lysosomal cholesterol egress by NPC1 and NPC2. Cell 182(1):98–111. https://doi.org/10.1016/j.cell.2020.05.020
Rapp A, Gmeiner B, Hüttinger M (2006) Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie 88(5):473–483. https://doi.org/10.1016/j.biochi.2005.10.007
Rohanizadegan M, Sacharow S (2018) Desmosterolosis presenting with multiple congenital anomalies. Eur J Med Genet 61(3):152–156. https://doi.org/10.1016/j.ejmg.2017.11.009
Roher AE, Weiss N, Kokjohn TA, Kuo Y-M, Kalback W, Anthony J, Watson D, Luehrs DC, Sue L, Walker D, Emmerling M, Goux W, Beach T (2002) Increased a beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry 41:11080–11090. https://doi.org/10.1021/bi026173d
Romano S, Mitro N, Giatti S, Diviccaro S, Pesaresi M, Spezzano R, Audano M, Garcia-Segura LM, Caruso D, Melcangi RC (2018) Diabetes induces mitochondrial dysfunction and alters cholesterol homeostasis and neurosteroidogenesis in the rat cerebral cortex. J Steroid Biochem Mol Biol 178:108–116. https://doi.org/10.1016/j.jsbmb.2017.11.009
Samara A, Galbiati M, Luciani P, Deledda C, Messi E, Peri A, Maggi R (2014) Altered expression of 3-betahydroxysterol delta-24-reductase/selective Alzheimer’s disease indicator-1 gene in Huntington’s disease models. J Endocrinol Invest 37(8):729–737. https://doi.org/10.1007/s40618-014-0098-1
Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH, Aisen PS (2011) A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 77(6):556–563. https://doi.org/10.1212/WNL.0b013e318228bf11
Sarajärvi T, Haapasalo A, Viswanathan J, Mäkinen P, Laitinen M, Soininen H, Hiltunen M (2009) Down-regulation of seladin-1 increases BACE1 levels and activity through enhanced GGA3 depletion during apoptosis. J Biol Chem 284(49):34433–34443. https://doi.org/10.1074/jbc.M109.036202
Schaaf CP, Koster J, Katsonis P, Kratz L, Shchelochkov OA, Scaglia F, Kelley RI, Lichtarge O, Waterham HR, Shinawi M (2011) Desmosterolosis-phenotypic and molecular characterization of a third case and review of the literature. Am J Med Genet A 155A(7):1597–1604. https://doi.org/10.1002/ajmg.a.34040
Segatto M, Leboffe L, Trapani L, Pallottini V (2014) Cholesterol homeostasis failure in the brain: implications for synaptic dysfunction and cognitive decline. Curr Med Chem 21(24):2788–2802. https://doi.org/10.2174/0929867321666140303142902
Sengoku R (2020) Aging and Alzheimer’s disease pathology. Neuropathology 40(1):22–29. https://doi.org/10.1111/neup.12626
Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K (1998) Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys 357(2):299–309. https://doi.org/10.1006/abbi.1998.0813
Sharpe LJ, Coates HW, Brown AJ (2020) Post-translational control of the long and winding road to cholesterol. J Biol Chem 295(51):17549–17559. https://doi.org/10.1074/jbc.REV120.010723
Sharpe LJ, Brown AJ (2013) Controlling cholesterol synthesis beyond 3-Hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem 288(26):18707–18715. https://doi.org/10.1074/jbc.R113.479808
Shepardson NE, Shankar GM, Selkoe DJ (2011) Cholesterol level and statin use in Alzheimer disease: II. Review of human trials and recommendations. Arch Neurol 68(11):1385–92. https://doi.org/10.1001/archneurol.2011.242
Shih PH, Wu CH, Yeh CT, Yen GC (2011) Protective effects of anthocyanins against amyloid β-peptide-induced damage in neuro-2A cells. J Agric Food Chem 59(5):1683–1689. https://doi.org/10.1021/jf103822h
Singh A, Kukreti R, Saso L, Kukreti S (2019) Oxidative stress: a key modulator in neurodegenerative diseases. Molecules 24(8):1583. https://doi.org/10.3390/molecules24081583
Soderberg M, Edlund C, Kristensson K, Dallner G (1990) Lipid compositions of different regions of the human brain during aging. J Neurochem 54:415–423. https://doi.org/10.1111/j.1471-4159.1990.tb01889.x
Sodero AO, Trovò L, Iannilli F, Veldhoven PV, Dotti CG, Martin MG (2011) Regulation of tyrosine kinase B activity by the Cyp46/cholesterol loss pathway in mature hippocampal neurons: relevance for neuronal survival under stress and in aging. J Neurochem 116(5):747–755. https://doi.org/10.1111/j.1471-4159.2010.07079.x
Solfrizzi V, Scafato E, Capurso C, D’Introno A, Colacicco AM, Frisardi V, Vendemiale G, Baldereschi M, Crepaldi G, Di Carlo A, Galluzzo L, Gandin C, Inzitari D, Maggi S, Capurso A, Panza F (2011) Metabolic syndrome, mild cognitive impairment, and progression to dementia: the Italian Longitudinal Study on Aging. Neurobiol Aging 32(11):1932–1941. https://doi.org/10.1016/j.neurobiolaging.2009.12.012
Sparks DL, Scheff SW, Hunsaker JC, Liu H, Landers T, Gross DR (1994) Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol 126:88–94. https://doi.org/10.1006/exnr.1994.1044
Stefani M, Liguri G (2009) Cholesterol in Alzheimer’s disease: unresolved questions. Curr Alzheimer Res 6(1):15–29. https://doi.org/10.2174/156720509787313899
Suzuki R, Lee K, Jing E, Biddinger SB, McDonald JG, Montine TJ, Craft S, Kahn CR (2010) Diabetes and insulin in regulation of brain cholesterol metabolism. Cell Metab 12(6):567–579. https://doi.org/10.1016/j.cmet.2010.11.006
Svennerholm L, Boström K, Jungbjer B, Olsson L (1994) Membrane lipids of adult human brain: lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J Neurochem 63:1802–1811. https://doi.org/10.1046/j.1471-4159.1994.63051802.x
Tachikawa M, Watanabe M, Hori S, Fukaya M, Ohtsuki S, Asashima T, Terasaki T (2005) Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J Neurochem 95:294–304. https://doi.org/10.1111/j.1471-4159.2005.03369.x
Tamarai K, Bhatti JS, Reddy PH (2019) Molecular and cellular bases of diabetes: focus on type 2 diabetes mouse model-TallyHo. Biochim Biophys Acta Mol Basis Dis 1865(9):2276–2284. https://doi.org/10.1016/j.bbadis.2019.05.004
Thelen KM, Falkai P, Bayer TA, Lutjohann D (2006) Cholesterol synthesis rate in human hippocampus declines with aging. Neurosci Lett 403:15–19. https://doi.org/10.1016/j.neulet.2006.04.034
Uddin MS, Kabir MT, Mamun AA, Abdel-Daim MM, Barreto GE, Ashraf GM (2019) APOE and Alzheimer’s disease: evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Mol Neurobiol 56(4):2450–2465. https://doi.org/10.1007/s12035-018-1237-z
Umeda T, Mori H, Zheng H, Tomiyama T (2010) Regulation of cholesterol efflux by amyloid beta secretion. J Neurosci Res 88(9):1985–1994
Umeda T, Tomiyama T, Kitajima E, Idomoto T, Nomura S, Lambert MP, Klein WL, Mori H (2012) Hypercholesterolemia accelerates intraneuronal accumulation of Aβ oligomers resulting in memory impairment in Alzheimer’s disease model mice. Life Sci 91(23–24):1169–1176. https://doi.org/10.1016/j.lfs.2011.12.022
Vance JE (2012) Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis Model Mech 5(6):746–755. https://doi.org/10.1242/dmm.010124
Vanmierlo T, Bloks VW, van Vark-van der Zee LC, Rutten K, Kerksiek A, Friedrichs S, Sijbrands E, Steinbusch HW, Kuipers F, Lütjohann D, Mulder M (2010) Alterations in brain cholesterol metabolism in the APPSLxPS1mut mouse, a model for Alzheimer's disease. J Alzheimers Dis 19(1):117–127. https://doi.org/10.3233/JAD-2010-1209
Varma VR, Lüleci HB, Oommen AM, Varma S, Blackshear CT, Griswold ME, An Y, Roberts JA, O’Brien R, Pletnikova O, Troncoso JC, Bennett DA, Çakır T, Legido-Quigley C, Thambisetty M (2021) Abnormal brain cholesterol homeostasis in Alzheimer’s disease—a targeted metabolomic and transcriptomic study. NPJ Aging Mech Dis 7:11. https://doi.org/10.1038/s41514-021-00064-9
Wood WG, Igbavboa U, Eckert GP, Johnson-Anuna LN, Müller WE (2005) Is hypercholesterolemia a risk factor for Alzheimer’s disease? Mol Neurobiol 31:185–192. https://doi.org/10.1385/MN:31:1-3:185
Wood WG, Li L, Müller WE, Eckert GP (2014) Cholesterol as a causative factor in Alzheimer’s disease: a debatable hypothesis. J Neurochem 129(4):559–572. https://doi.org/10.1111/jnc.12637
Wu C, Miloslavskaya I, Demontis S, Maestro R, Galaktionov K (2004) Regulation of cellular response to oncogenic and oxidative stress by Seladin-1. Nature 432(7017):640–645. https://doi.org/10.1038/nature03173
Ye B, Shen H, Zhang J, Zhu YG, Ransom BR, Chen XC, Ye ZC (2015) Dual pathways mediate β-amyloid stimulated glutathione release from astrocytes. Glia 63(12):2208–2219. https://doi.org/10.1002/glia.22886
Zerenturk E, Sharpe L, Ikonen E, Brown A (2013) Desmosterol and DHCR24: unexpected new directions for a terminal step in cholesterol synthesis. Prog Lipid Res 52(4):666–680. https://doi.org/10.1016/j.plipres.2013.09.002
Zhong N, Scearce-Levie K, Ramaswamy G, Weisgraber KH (2008) Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimers Dement 4(3):179–192. https://doi.org/10.1016/j.jalz.2008.01.006
Zolotushko J, Flusser H, Markus B, Shelef I, Langer Y, Heverin M, Björkhem I, Sivan S, Birk OS (2011) The desmosterolosis phenotype: spasticity, microcephaly and micrognathia with agenesis of corpus callosum and loss of white matter. Eur J Hum Genet 19(9):942–946. https://doi.org/10.1038/ejhg.2011.74
Zu H, Wu J, Zhang J, Yu M, Hong Z (2012) Testosterone up-regulates seladin-1 expression by iAR and PI3-K/Akt signaling pathway in C6 cells. Neurosci Lett 514(1):122–126. https://doi.org/10.1016/j.neulet.2012.02.072
Funding
This review was supported by the Shanghai Health and Medical Development Foundation [201740209]; and the Shanghai Jinshan District Key Medical Foundation [JSZK2015A05 and JSZK2019A06].
Author information
Authors and Affiliations
Contributions
BX, MM, and YK make the same contribution to the article. BX, MM, and YK partly write the article. LD, JB, and JZ draw the model diagram. MZ, YH, WZ, XG, YX, YZ and AQ participate in the study of DHCR24; JW, YZ and XY provide critical feedback and revise the manuscript. ZH designs this review (conceptualization of the idea), gives an interpretation of data, and writes the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xiaojing Bai, Meiting Mai and Kai Yao are the co-first authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bai, X., Mai, M., Yao, K. et al. The role of DHCR24 in the pathogenesis of AD: re-cognition of the relationship between cholesterol and AD pathogenesis. acta neuropathol commun 10, 35 (2022). https://doi.org/10.1186/s40478-022-01338-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-022-01338-3