
Abstract
Noise exposure is an important cause of acquired hearing loss. Studies have found that noise exposure causes dysregulated redox homeostasis in cochlear tissue, which has been recognized as a signature feature of hearing loss. Oxidative stress plays a pivotal role in many diseases via very complex and diverse mechanisms and targets. Reactive oxygen species are products of oxidative stress that exert toxic effects on a variety of physiological activities and are considered significant in noise-induced hearing loss (NIHL). Endogenous cellular antioxidants can directly or indirectly counteract oxidative stress and regulate intracellular redox homeostasis, and exogenous antioxidants can complement and enhance this effect. Therefore, antioxidant therapy is considered a promising direction for NIHL treatment. However, drug experiments have been limited to animal models of NIHL, and these experiments and related observations are difficult to translate in humans; therefore, the mechanisms and true effects of these drugs need to be further analyzed. This review outlines the effects of oxidative stress in NIHL and discusses the main mechanisms and strategies of antioxidant treatment for NIHL.
Similar content being viewed by others


Introduction
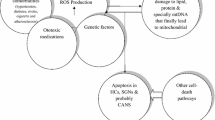
Among the numerous causes of hearing loss, noise exposure is an important contributor, particularly in noise-induced hearing loss (NIHL). Exposure to excessive noise leads to hair cell (HC) loss, auditory nerve degeneration and synaptic reduction at the microscale, and these effects manifest as loss of auditory sensitivity and a temporary or permanent hearing threshold shift [1]. Mild or moderate noise can lead to a temporary hearing threshold shift (TTS), at which time HC damage and auditory nerve fiber (ANF) degeneration are reversible [2,3,4]. However, severe or long-term noise exposure can lead to cochlear HC necrosis and apoptosis, resulting in damage that cannot be restored to the pre-trauma level, a condition called permanent threshold shift (PTS) [5, 6]. According to the World Health Organization, the prevalence of NIHL among adults worldwide is 16%, with significant regional differences [7]. In addition to hearing-related symptoms, patients with NIHL often present with headaches, hypertension, tinnitus, dizziness and other symptoms, which have a serious impact on their communication and quality of life [3, 8].
The main causes of NIHL include mechanical damage from noise itself and toxic damage from oxidative stress. At the cellular level, excessive noise extensively destroys synapses between ANFs [9], which leads to the loss of the peripheral axons of bipolar sensory neurons and eventually the degeneration of cell bodies in the spiral ganglia [9, 10]. Studies have shown that outer hair cells (OHCs) are the main targets of noise damage, and OHC loss in the basal (high frequency) cochlea is exacerbated [11]. In addition to mechanical damage, noise exerts a damaging effect on the stria vascularis, which leads to a decrease in blood flow and, together with the former, leads to a threshold elevation [10, 12]. Moreover, several pathways are activated in the pathogenesis of NIHL. Autophagy is a recently discovered highly conserved eukaryotic cell cycle process that helps maintain internal homeostasis by degrading organelles, proteins, and macromolecules and recycling materials [13,14,15]. Autophagy is involved in the development of NIHL via β-alanine metabolism and arginine and proline metabolism and exerts an inhibitory effect on NIHL [16,17,18]. As the main markers of autophagy, the mRNA expression levels of lc3b and Beclin1 increased significantly after noise exposure and were localized to cochlear spiral ganglion neurons (SGNs), as determined by immunofluorescence assay [16]. For verification, researchers used the direct autophagy inhibitor 3-MA and the mTOR signaling agonist RAP [19]. The results showed that the inhibition of autophagy significantly aggravated the noise-induced oxidative imbalance, which ultimately manifested as NIHL [20]. Some inflammatory mediators are involved in mediating NIHL. The cochlea is considered an immune-exempt organ because of its hemolymph barrier [21], but it has been proven that noise exposure can induce HC ischemia [22,23,24]. In addition, as a key nuclear factor, NF-κB is activated and translocated under the action of a variety of damage-associated molecular patterns (DAMPs), toll-like receptors (TLRs) and receptors for advanced glycation end products (RAGEs) [1], and noise-induced damage activates the NF-κB signaling cascade [25,26,27]. As an example, researchers have found that NF-κB is activated 2–6 h after PTS-inducing noise [28] and thus causes HC injury and NIHL [29]. This evidence shows that oxidative stress plays an extremely important role in NIHL.
Oxidative stress is usually defined as an imbalance between oxidant and antioxidant levels, and this definition has been refined to include the abrogation of redox signaling and regulatory controls [30, 31]. Currently, oxidative stress is the focus of research in cancer, inflammation and other diseases [32,33,34], and these studies have promoted the progressive understanding of neurodegenerative diseases to a large extent [35,36,37]. For NIHL, research on oxidative stress is not abundant, but oxidative stress has been shown to induce and aggravate the progression of NIHL [38, 39]. For example, researchers have used superoxide and lipid peroxidation markers to demonstrate the level of oxidative damage in HCs and SGNs. The results showed that HCs and SGNs in the most severely damaged medial-basal area of the noise-exposed rat cochlea emitted strong fluorescence signals [38]. Correspondingly, after the noise-exposed rat cochlea was treated with the antioxidant Q10 analog Qter, the effect of noise exposure was offset, and the immune reaction was significantly reduced at the end of the treatment period [38]. Currently, the role of oxidative stress in NIHL and targeted therapy has not been systematically described. This article reviews the mechanism of oxidative stress in NIHL and the current therapeutic antioxidant drugs, aiming to clarify the prospects of targeting oxidative stress for NIHL treatment.
Overview of the dysregulation of redox homeostasis: oxidative stress and reactive oxygen species (ROS)
Oxidative metabolism: the basis of redox homeostasis
Reactive oxygen species (ROS) are composed of free radical oxygen and nonradical oxygen formed by the partial reduction of oxygen. ROS refer to various oxygen-derived compounds, free radicals and nonradicals, including superoxide anion O2−, hydrogen peroxide H2O2 and hydroxyl radical ·HO [40]. Intracellular ROS are produced endogenously during oxidative phosphorylation in mitochondria and can be produced via interactions involving exogenous substances [41]. Electron detachment during electron transport and binding to oxygen is the main source of ROS. Modifications of biomolecules mediated through low-level ROS are reversible switches of protein function and play an important role in cell proliferation, migration and differentiation [42]. However, excessive accumulation of ROS can be detrimental to the body's function.
ROS are produced in many ways in cells, and the major pathways of cellular ROS generation and detoxification are shown in Fig. 1. Since selenocysteine(s) shows greater potential for oxidation than cysteine, the oxidative environment in the endoplasmic reticulum is supported by the expression of selenoproteins [40]. Ero1 proteins (flavoproteins) oxidize FADH2 to generate FAD and produce hydrogen peroxide with electrons and O2 [43]. Meanwhile, the protein-folding process in the endoplasmic reticulum produces immature oxidized glycoproteins, which subsequently enter the calnexin cycle [40].

Major pathways of cellular ROS generation and detoxification
The main function of NADP oxidase (NOX) is ROS production, which can affect many cellular components. The “ROS-dependent ROS generation” pathway is formed by the NOX-induced proliferation of ROS to generate other ROS-producing enzymatic systems [40]. NOXs are membrane proteins with multiple transmembrane regions, including binding sites for NADPH and FAD, and four histidine residues, which produce superoxide when generating molecular oxygen via heme [40].
In addition, the mitochondrial electron transport chain (ETC) generates highly active ·HO through a series of complex reactions, which indiscriminately oxidize proteins, lipids and DNA, resulting in DNA damage and genomic instability [44]. This critical process is described in detail in the next section.
Correspondingly, to maintain redox homeostasis, antioxidant systems in cells regulate ROS levels both temporally and spatially. Several antioxidants with enzymatic activity convert hydrogen peroxide into water, including peroxiredoxins (PRXs), catalase and glutathione peroxide (GPX) [4, 45]. Within mitochondria, NAD(P)H is the source of reducing equivalents for antioxidant GPX or PRX, and its oxidation is mediated via glutathione (GR) and thioredoxin (TR) reductase [46].
Mitochondria: the main source of ROS
As the powerhouses of eukaryotic cells, mitochondria supply most of the energy in a cell by reducing oxygen in the ETC to produce ATP. However, the process of energy production via mitochondria is accompanied by ROS generation, which poses potential threats to mitochondria and physiological processes [47]. In the ETC, oxygen is reduced to water through the addition of two electrons. Although the efficiency of this process is very high, 1–4% of oxygen is not completely reduced through one-electron transfer. In fact, superoxide dismutase 2 (SOD2) in the mitochondrial matrix and SOD1 in the mitochondrial membrane gap convert O2− to H2O2, which is converted to superoxide, the most common free radical. Then, superoxide diffuses into the cytoplasm through the mitochondrial membrane [40, 48]. Mitochondrial ROS (mtROS) are not only byproducts of the ETC; monoamine oxidase in the outer membrane and the α-ketoglutarate dehydrogenase complex are contributors to ROS production [49]. Low levels of mtROS enable metabolic adaptation and signal transduction functions. However, high levels of mtROS trigger inflammatory reactions via danger signaling and then activate autophagy and apoptosis [40]. When ROS are generated, antioxidants in the mitochondria maintain redox homeostasis under normal conditions. However, the oxidative stress generated by excessive ROS may lead to destructive consequences for mitochondria, other organelles, and cellular pathways, eventually leading to cell death [49].
Oxidative stress-related mechanisms in NIHL
Sources of ROS: destructive noise
The main sources of ROS in cells are mitochondria, and they have been thought to play a role in noise damage. On the one hand, extreme noise exposure directly causes intracellular rearrangements and mechanical damage to the cochlear structure, causing NIHL [50]. On the other hand, NIHL can be caused by more subtle forms of noise exposure [49]. Notably, ATP levels were depleted rapidly after noise exposure, indicating that mitochondria underwent metabolic changes after noise exposure [51]. Some researchers believe that the increase in the cochlear metabolic rate after noise exposure leads to excessive mitochondrial energy metabolism, which increases the rate of ROS leakage [52]. However, excessive ROS produced under stress situations show selective inhibition of the ETC, not its stimulation [53].
The effect of calcium homeostasis on ROS levels is widely recognized. ROS must pass through the mitochondrial membrane to enter the cytoplasm, and the destruction of mitochondrial membrane integrity and loss of membrane potential lead to the release of ROS into the cytoplasm [54]. Studies have shown that free Ca2+ in the cochlea increases immediately after excessive noise exposure [55], which may be a result of intracellular store release or entry via ion channels, including L-type or T-type voltage-gated calcium channels. Some experiments have proven that blocking these two types of voltage-gated calcium channels can attenuate the hearing loss caused by noise [56, 57], and extracellular Ca2+ can enhance the release of intracellular Ca2+, further increasing the level of free Ca2+ [58]. In addition, aminoglycoside antibiotics have been shown to alter the calcium homeostasis between the endoplasmic reticulum and mitochondria and thus promote the flow of Ca2+ from the endoplasmic reticulum to mitochondria [59, 60].
As another potential source of ROS, α-ketoglutarate dehydrogenase is a calcium-regulated Krebs cycle enzyme that can induce the generation of a large amount of superoxide and H2O2 [61]. In addition to increasing oxidative stress, dysfunctional α-ketoglutarate dehydrogenase increases mitochondrial free radical-induced damage and ETC inhibition, further increasing ROS production and initiating apoptotic pathways [61].
Mechanisms of ROS damage to cochlear tissue
In the cochlea, different types of cells show different sensitivities to ROS. Because of different protein expression patterns, HCs seem to be the cochlear cells most vulnerable to damage caused by free radicals [62]. As stated above, ROS levels are increased in cochlear HCs exposed to excessive noise, either indirectly via NADPH oxidase and MAPK signaling cascades or directly via changes to cell morphology and the cell cycle, programmed death, and other physiological processes.
Peroxides, such as H2O2, directly damage HCs
H2O2, a typical ROS and a key signaling molecule, is suitable for studying the effects of oxidative stress on various physiological processes [63]. Through in vitro experiments based on mouse cochlear HCs, researchers have shown that a high H2O2 level increases the level of p53 protein, leading to apoptosis and necrosis. Moreover, the number of cells with nuclear blebbing and severely altered nuclear morphology was significantly increased, fewer of these cells were in the G1 phase of the cell cycle, and the number of these cells in the G2/M phase was increased, which may indicate delays in cell cycle progression [63]. In addition, H2O2 affects the antioxidant system, as indicated by the levels of GPX4, GSH and RSH in cells significantly increasing upon exposure to high levels of H2O2, which may lead to an imbalance in redox homeostasis and eventually trigger oxidative stress [63].
MAPK-mediated pathways: a transmitter of ROS damage
Mitogen-activated protein kinases (MAPKs) are involved in cellular signaling pathways and are important mediators of injury and survival signaling. MAPKs act downstream from plasma membrane receptors, intracellular receptors, and ROS [64, 65]. They activate gene expression via intermediate signaling proteins such as Src, Ras, Rac/cdc42, and mixed lineage kinase protein families. MAPKs are categorized into several different groups, among which c-Jun N-terminal kinases (JNKs) are clearly affected by ROS [66]. The MAPK family is comprised of the following three layers: MAPK, MAPK kinase (MAPKK) and MAPKK kinase (MAPKKK) [67]. MAPKKK phosphorylates and activates MAPKK; then, MAPKK phosphorylates and activates MAPK, which phosphorylates the target substrate [68]. Intense noise has been proven to affect the phosphorylation of MAPK in the cochlea and lead to HC death [69,70,71]. In addition, enrichment analysis of the KEGG pathway confirmed that the MAPK signaling pathway was significantly enriched pathway of differentially expressed proteins in the inner ear of NIHL rats [72].
JNK is expressed as three isomers, JNK1, JNK2 and JNK3, among which JNK1 and JNK2 are extensively expressed, and JNK3 is expressed mainly in heart and nerve tissues. When exposed to extracellular stimulation, JNK signaling can reach the nucleus and mitochondria, promoting apoptosis via either pathway [66]. Specifically, a JNK signal that is transported to the nucleus phosphorylates c-Jun and other transcription factors, including p53, and then promotes the expression of proapoptotic genes (FasL, TNF α, Bim, Bak, etc.), which blocks the transcription of antiapoptotic genes (such as Bcl-2) [73,74,75,76,77,78,79,80]. In the other pathway, activated JNK can phosphorylate a variety of proteins related to mitochondrial cell death. Phosphorylated JNK indirectly triggers proapoptotic Bax-mediated mitochondrial death signaling through the cleavage of Bid. In this regard, studies have confirmed that Bax expression is upregulated in noise-exposed rats [81]. Additionally, after phosphorylation by activated JNK, the death-promoting proteins Bim and Bad stop the inhibition of the Bcl-2 and Bcl-xL proteins and promote Bax activation of the intrinsic apoptosis pathway [82,83,84,85,86,87]. The increase in ROS levels promotes the prolongation of JNK activation, which in turn leads to an increase in mitochondrial ROS production, forming a positive feedback loop [66, 88]. Correspondingly, JNK inhibitors have been proven to exert a protective effect on HC damage caused by noise exposure [89,90,91], which verifies the accuracy of the description of the aforementioned mechanisms.
Antioxidant treatment of NIHL
Biological antioxidants can delay or prevent the oxidation of substrates when the concentration is lower than that of the oxidizable substrate [92], inhibiting the activity of ROS and reducing oxidative stress, DNA mutation, malignant transformation and other cell-damaging events. However, this steady state may be altered under the action of sustained free radicals, leading to disease [92, 93].
The research shows that the peroxidation reaction starts from the addition of oxygen to carbon-centered radicals, with most of the hydrogen atoms transferred to the chain carrying peroxyl radicals. Antioxidants reduce the local concentration of molecular oxygen, remove pro-oxidative metal ions, capture aggressive ROS (superoxide anion radicals and hydrogen peroxide, etc.), remove chain-starting free radicals (hydroxyl, peroxyl, alkoxyl, etc.), inhibit the sequential production of radical chains, and quench singlet oxygen [94, 95]. The redox homeostasis of cells is maintained by complex endogenous antioxidant defense systems, including endogenous antioxidant enzymes (catalase, superoxide dismutase, glutathione peroxidase, etc.) and nonenzymatic compounds (glutathione, metal-chelating proteins, etc.) [96]. In addition, exogenous antioxidants are capable of synergistically reducing oxidative stress and impacting related disease processes [96, 97]. Hence, based on these mechanisms, antioxidants and their analogs have become the main components in antioxidant treatment strategies. There have been many drug studies targeting NIHL based on these findings, and we will discuss some potential drugs.
Activation of endogenous antioxidants: the core antioxidant system
The Kelch‐like ECH‐associated protein 1 (Keap1)–nuclear factor erythroid 2‐related factor 2 (Nrf2)–antioxidant response element (ARE) pathway is the center of the biological response to oxidative stress, regulating a variety of cell-protecting proteins (also known as phase 2 enzymes) to maintain the homeostasis of ROS levels in cells [98,99,100,101,102]. Under oxidative stress conditions, the Nrf2 protein is stabilized and transferred to the nucleus, where it activates a variety of ARE-response genes, including antioxidant genes, heme oxygenase-1 (HO-1) and nicotinamide adenine dinucleotide phosphate (NAD(P)H) [101,102,103,104]. The Nrf2 pathway is also activated by the interruption of the interaction between Nrf2 and Keap1 [105]. p62 is a ubiquitin-binding protein that activates Nrf2 in autophagy-deficient cells by disrupting the Keap1-Nrf2 interaction [106]. p62 is upregulated under oxidative stress, leading to the sequestration of Keap1 and activating Nrf2 and Nrf2-dependent antioxidant defense gene expression [107]. The Nrf2-ARE pathway targets a wide range of downstream proteins, which are listed in Table 1. In this section, we describe in detail several major endogenous antioxidant enzymes that have been studied and shown to exert effects in NIHL.
Fluorescence analysis showed that Nrf2, HO-1 and SOD levels were slightly increased in cells damaged by noise, revealing an adaptive stress response. In the same HC and SGN samples used for the fluorescence assay, Nrf2 translocation paralleled the increase in expression of HO-1 [108]. This means that Nrf2-induced upregulation of HO-1 affects OHC signal amplification and transmission from primary afferent neurons to the central acoustic pathway [4]. The inducible isoform of HO-1 plays an antioxidative role by removing the oxidation-promoting molecule haem, thereby acting as a microsomal enzyme in haem catabolism [109]. The catabolic products of haem include biliverdin, which is converted into bilirubin after reduction via biliverdin reductase. Both haem and biliverdin exert significant antioxidant effects, contribute to resistance to oxidative stress, and protect cells [109,110,111]. Iron is released during haem catabolism, which leads to the upregulation of ferritin. As an iron-storing protein, ferritin participates in HO-1 activity against oxidative stress [111, 112].
In addition, an increase in GSH production and the activity of its scavenger has been shown to be induced by Nrf2 [108, 113, 114]. Glutathione is the most abundant antioxidant and plays a major role in oxidative stress resistance [115]. When Nrf2 and HO-1 levels are increased, the amount of GSH also increases. Nrf2 not only regulates GSH synthesis but also coordinates GSH reductase transcription. Nrf2 leverages the reducing equivalents of NAD(P)H to reduce oxidized GSH, thus maintaining it in the reduced state [116,117,118].
Furthermore, Nrf2 regulates the major NAD(P)H-generating enzyme, aldehyde dehydrogenase, via transcription, inducing aldehyde dehydrogenase expression in microsomes, the cytoplasm and mitochondria and generating NAD(P)H with the cofactor NAD(P) [102]. Compared with that in Keap1-wild-type cells, the total mitochondrial NADH pool in Keap1-KO cells was significantly increased, and it was significantly decreased in Nrf2-KO cells [119]. Researchers analyzed the NAD(P)H reduction capacity of OHCs and found that after noise exposure, the oxidation of NAD(P)H increased, the plasma membrane peroxidation rate increased, and plasma membrane fluidity decreased concomitantly [120,121,122]. The accumulation of free radicals leads to plasma membrane peroxidation and then triggers lipid peroxidation, indicating that effective antioxidant treatment may enhance the protection of OHCs from cell death [4].
Exogenous antioxidants: prospects for drug intervention
Endogenous antioxidants play an extremely important role in maintaining redox homeostasis. When the concentration of endogenous antioxidants is insufficient to maintain redox homeostasis, exogenous antioxidants are considered effective for inducing oxidative stress resistance. On this basis, antioxidants that regulate cellular oxidative stress to treat NIHL by targeting free radicals have been developed [123]; some of these antioxidants are listed in Table 2. In the remainder of this section, we describe several drugs that have been extensively studied.
N-Acetylcysteine (NAC)
A precursor of cysteine, NAC is a substrate antioxidant in glutathione synthesis [124,125,126] that may inhibit oxidative stress and reduce cochlear damage caused by noise exposure by providing a substrate for GSH synthesis, inhibiting the activation of the MAPK pathway, and scavenging free radicals [125, 127]. As a drug approved by FAD, oral NAC has been used clinically for more than 30 years, even in very large doses (8 g per day for 8 weeks), and it causes only mild side effects [125, 128]. However, as the synthesis of glutathione decreases with age, the effectiveness of NAC decreases with age [126]. Since ROS are considered necessary for physiological cellular activities, a certain ROS level must be maintained for drugs such as NAC to be used as an effective treatment [41].
Performing animal experiments, researchers found that NAC minimized the apoptosis rate of sensory HCs and significantly reduced low- and medium-frequency auditory threshold shifts [124, 129]. When used in humans, oral NAC is metabolized into cysteine in the intestine, and this cysteine is oxidized to cystine, which is then transported into cells to be consumed as an essential precursor for glutathione synthesis [125]. In fact, the effect of the cysteine product is more significant than that of NAC treatment. However, due to differences in intestinal and visceral metabolism, the bioavailability of cysteine after oral administration of NAC is very high, which makes NAC a better supplement than cysteine for consumption as a GSH precursor [126, 130, 131].
In addition, NAC has been shown to inhibit JNK, p38 MAP kinase and NF-κB pathway activation, and the inhibition of these signaling pathways reduces the corresponding cell damage and apoptosis rate [132, 133]. Moreover, NAC has also been found to be a mitochondrial protectant in different models of stress induction [134, 135]. Therefore, NAC is thought to attenuate NIHL via multiple antioxidant mechanisms.
Ebselen
Ebselen is a selenium-based organic compound with antioxidant properties that can mimic and enhance the activity of GPX1 [123, 136]. Immunofluorescence assays of rat cochlea showed that the weighted intensity of the GPX1-expressing area in the rat cochlea decreased by 8.8% after noise exposure compared with a decrease in the no-noise controls, but the weighted intensity of the GPX1-expressing area in the noise-exposed rat cochlea after ebselen treatment increased by 9.9% compared with the level in the no-noise controls [136]. This study shows that ebselen plays a role in enhancing GPX1 expression.
In fact, ebselen shows multifunctional ear protection activity. In other experiments, ebselen was found to exert a significant inhibitory effect on ROS production and lipid peroxidation [137]. Additionally, ebselen enhanced the activity of the cellular antioxidant system. At a certain concentration, ebselen significantly increased the transcriptional activity of ARE and the expression level of the HO-1 protein, contributing to the fight against oxidative stress [137].
Previous studies have shown that the immediate injection of ebselen into rats before and after NIHL induction can effectively reduce auditory thresholds and HC death [136]. The safety and limited but significant efficacy of ebselen in humans have also been proven, providing a basis for the future use of ebselen in clinical treatment [138].
Vitamins
Some micronutrients, such as vitamins A, C, E and B12, exhibit antioxidant properties. They are considered to exert synergistic effects and have been used either individually or in combination for the experimental treatment of NIHL.
The main antioxidant effect of vitamin A is the removal of singlet oxygen. Since singlet oxygen and lipids are substrates of lipid peroxide, removing singlet oxygen can prevent lipid peroxidation [139,140,141,142,143]. When retinoic acid (the most active metabolite of vitamin A) was injected into noise-exposed mice, the apoptosis rate of HCs was significantly reduced, the cochlea was protected, and the auditory threshold was also restored rapidly [144,145,146].
Vitamin C is a water-soluble molecule; therefore, the elimination of oxygen radicals by vitamin C occurs in the aqueous phase and is very effective in blocking and/or reverting lipid peroxidation at the plasma membrane [147, 148]. Studies have shown that taking vitamin C before excessive noise exposure significantly reduced the TTS in rats [149]. Furthermore, vitamin C protects cell membranes by regenerating vitamin E from its oxidized form, and vitamins C and E play opposite roles, which shows that the oxidation of vitamin C and E is coupled [141, 143].
The antioxidation mechanism of vitamin E is closely related to vitamin C. Vitamin E exists in lipids in cells and is a main free radical scavenger of cell membranes. It inhibits the proliferation cycle of lipid peroxidation by reacting with peroxyl radicals and reducing their number [140,141,142,143]. Researchers gave vitamins A, C, and E and magnesium to animals exposed to noise and found that the threshold of the audit brain response (ABR) in the animals treated with combined administration was significantly reduced, and the HCs of these animals survived [141].
Vitamin B12 has been found to be associated with NIHL. Researchers found that the incidence of vitamin B12 deficiency in NIHL patients was higher than that in subjects with normal hearing. Further studies showed that the serum vitamin B12 and folic acid levels in NIHL patients were significantly lower than those in subjects without NIHL [150,151,152]. Deficiency in vitamin B12 and folic acid increases the level of homocysteine, which may lead to a decrease in the intracellular glutathione concentration, thus increasing lipid peroxidation [151, 153].
Comprehensive research has shown that the combined treatment with vitamins A, C, and E and magnesium taken one hour before noise exposure significantly reduced NIHL and cell death, and long-term high-dose intake of these micronutrients showed no obvious adverse effects [141, 153]. These studies provide a direction and basis for the use of vitamins in the treatment of NIHL.
HK-2
HK-2 is a multifunctional antioxidant with the characteristics of a free radical scavenger and metal chelator [154,155,156]. Therefore, in addition to reducing the oxidative stress generated by free radicals, HK-2 synthesizes bioactive transition metals, including Fe2+, thus reducing the availability of these ions for Fenton reactions, which produce highly toxic hydroxyl radicals [156].
In an in vitro oxidative stress experiment, HK-2 showed a more significant protective effect than Trolox in reducing hydrogen peroxide damage, hydroxyl damage and peroxynitrite damage. With respect to superoxide damage, both Trolox and HK-2 were effective in inhibiting mitochondrial superoxide production in rat cochlear cells [156]. This finding suggests that HK-2 significantly inhibits damage caused by a variety of NIHL-related peroxides [157, 158].
Taking HK-2 before noise exposure can have a significant and particularly substantial protective effect. A dose-dependent reduction in hearing threshold shifts was obtained in rats after continuous treatment with HK-2 for 5 days prior to noise exposure, and this protection lasted for 10 days postexposure [156]. In addition, preventive treatment with HK-2 significantly reduced the pathological changes in OHCs. Notably, the magnitude of the protective effect of HK-2 orally ingested 10 days after exposure was profoundly lower than that of the preventive treatment [156].
In conclusion, HK-2 has been shown to significantly reduce NIHL, attenuate HC loss in rats and significantly reduce ROS-induced cell loss and damage. Furthermore, HK-2 did not show any adverse effects in rats [154,155,156], suggesting that HK-2 is a new therapeutic option for NIHL.
Conclusions and perspectives
Oxidative stress plays a pivotal role in many diseases, and its complex and diverse mechanisms indicate a variety of targets for possible treatments. In contrast, therapeutic options for NIHL, a disease with a wide impact, are rare. Oxidative stress-related pathways have been found to be significantly variable and specific in NIHL. Therefore, in-depth study of oxidative stress and NIHL is warranted. In this paper, we provide a review of the mechanisms of NIHL and oxidative stress and describe analyses that link pathways in NIHL and oxidative stress, establishing a starting point to develop new and promising drug treatments.
Antioxidants are promising therapeutic modalities for NIHL because they can counteract oxidative stress directly or indirectly. A variety of antioxidants have been shown to protect animal HCs from damage, thereby reducing hearing threshold shifts and attenuating NIHL [159,160,161,162], and because noise exposure is often predictable, prophylactic treatment with NIHL is a highly feasible option that has been tested with varying degrees of success. Prophylactic treatment allows for the utilization of many micronutrients that are usually ingested daily and induce very few side effects [143], providing a significant advancement in NIHL treatment options.
However, because of the difficulty of performing biopsies of cochlear tissue, drug trials for NIHL have been limited to animal models, and these experiments or direct observations are difficult to translate to humans. Therefore, antioxidants that have been validated in animal studies still need to be subjected to follow-up experiments to determine their side effects in humans when administered alone or in combination. While these problems have limited the research on oxidative stress-related drugs, the progress to date portends great potential for these treatments.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and references.
Abbreviations
- ROS:
-
Reactive oxygen species
- NIHL:
-
Noise-induced hearing loss
- HC:
-
Hair cell
- TTS:
-
Temporary hearing threshold shift
- ANF:
-
Auditory nerve fiber
- PTS:
-
Permanent threshold shift
- OHC:
-
Outer hair cell
- SGN:
-
Spiral ganglion neuron
- DAMP:
-
Damage-associated molecular pattern
- TLR:
-
Toll-like receptor
- RAGE:
-
Receptor for advanced glycation end product
- NOX:
-
NADP oxidase
- ETC:
-
Electron transport chain
- PRX:
-
Peroxiredoxin
- GPX:
-
Glutathione peroxide
- SOD2:
-
Superoxide dismutase 2
- mtROS:
-
Mitochondrial ROS
- MAPK:
-
Mitogen-activated protein kinase
- JNK:
-
C-Jun N-terminal kinase
- MAPKK:
-
MAPK kinase
- MAPKKK:
-
MAPKK kinase
- Keap1:
-
Kelch‐like ECH‐associated protein 1
- Nrf2:
-
Nuclear factor erythroid 2‐related factor 2
- ARE:
-
Antioxidant response element
- HO-1:
-
Haem oxygenase-1
- NAD(P)H:
-
Nicotinamide adenine dinucleotide phosphate
- ABR:
-
Audit brain response
References
Kurabi A, Keithley EM, Housley GD, Ryan AF, Wong AC. Cellular mechanisms of noise-induced hearing loss. Hear Res. 2017;349:129–37.
Lynch ED, Kil J. Compounds for the prevention and treatment of noise-induced hearing loss. Drug Discov Today. 2005;10(19):1291–8.
Verbeek JH, Kateman E, Morata TC, Dreschler W, Sorgdrager B. Interventions to prevent occupational noise induced hearing loss. Cochrane Database Syst Rev. 2009;7(3):Cd006396.
Fetoni AR, Paciello F, Rolesi R, Paludetti G, Troiani D. Targeting dysregulation of redox homeostasis in noise-induced hearing loss: oxidative stress and ROS signaling. Free Radic Biol Med. 2019;135:46–59.
Liberman MC. Noise-induced hearing loss: permanent versus temporary threshold shifts and the effects of hair cell versus neuronal degeneration. Adv Exp Med Biol. 2016;875:1–7.
Tikka C, Verbeek JH, Kateman E, Morata TC, Dreschler WA, Ferrite S. Interventions to prevent occupational noise-induced hearing loss. Cochrane Database Syst Rev. 2017;7(7):Cd006396.
Nelson DI, Nelson RY, Concha-Barrientos M, Fingerhut M. The global burden of occupational noise-induced hearing loss. Am J Ind Med. 2005;48(6):446–58.
Clark WW, Bohne BA. Effects of noise on hearing. JAMA. 1999;281(17):1658–9.
Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci: Off J Soc Neurosci. 2009;29(45):14077–85.
Yan W, Liu W, Qi J, Fang Q, Fan Z, Sun G, Han Y, Zhang D, Xu L, Wang M, Li J, Chen F, Liu D, Chai R, Wang H. A Three-dimensional culture system with matrigel promotes purified spiral ganglion neuron survival and function in vitro. Mol Neurobiol. 2018;55(3):2070–84.
Wu PZ, O’Malley JT, de Gruttola V, Liberman MC. Primary neural degeneration in noise-exposed human cochleas: correlations with outer hair cell loss and word-discrimination scores. J Neurosci: Off J Soc Neurosci. 2021;41(20):4439–47.
Wang Y, Hirose K, Liberman MC. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J Assoc Res Otolaryngol: JARO. 2002;3(3):248–68.
Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20(3):460–73.
Li H, Song Y, He Z, Chen X, Wu X, Li X, Bai X, Liu W, Li B, Wang S, Han Y, Xu L, Zhang D, Li J, Chai R, Wang H, Fan Z. Meclofenamic acid reduces reactive oxygen species accumulation and apoptosis, inhibits excessive autophagy, and protects hair cell-like HEI-OC1 cells from cisplatin-induced damage. Front Cell Neurosci. 2018;12:139.
Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383(16):1564–76.
Miao L, Zhang J, Yin L, Pu Y. Metabolomics analysis reveals alterations in cochlear metabolic profiling in mice with noise-induced hearing loss. Biomed Res Int. 2022;2022:9548316.
Schwarz C, Stekovic S, Wirth M, Benson G, Royer P, Sigrist SJ, Pieber T, Dammbrueck C, Magnes C, Eisenberg T, Pendl T, Bohlken J, Köbe T, Madeo F, Flöel A. Safety and tolerability of spermidine supplementation in mice and older adults with subjective cognitive decline. Aging. 2018;10(1):19–33.
Pekar T, Bruckner K, Pauschenwein-Frantsich S, Gschaider A, Oppliger M, Willesberger J, Ungersbäck P, Wendzel A, Kremer A, Flak W, Wantke F, Jarisch R. The positive effect of spermidine in older adults suffering from dementia : first results of a 3-month trial. Wien Klin Wochenschr. 2021;133(9–10):484–91.
Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 2008;90(2):313–23.
Yuan H, Wang X, Hill K, Chen J, Lemasters J, Yang SM, Sha SH. Autophagy attenuates noise-induced hearing loss by reducing oxidative stress. Antioxid Redox Signal. 2015;22(15):1308–24.
Fujioka M, Okano H, Ogawa K. Inflammatory and immune responses in the cochlea: potential therapeutic targets for sensorineural hearing loss. Front Pharmacol. 2014;5:287.
Fujioka M, Kanzaki S, Okano HJ, Masuda M, Ogawa K, Okano H. Proinflammatory cytokines expression in noise-induced damaged cochlea. J Neurosci Res. 2006;83(4):575–83.
Tornabene SV, Sato K, Pham L, Billings P, Keithley EM. Immune cell recruitment following acoustic trauma. Hear Res. 2006;222(1–2):115–24.
Wakabayashi K, Fujioka M, Kanzaki S, Okano HJ, Shibata S, Yamashita D, Masuda M, Mihara M, Ohsugi Y, Ogawa K, Okano H. Blockade of interleukin-6 signaling suppressed cochlear inflammatory response and improved hearing impairment in noise-damaged mice cochlea. Neurosci Res. 2010;66(4):345–52.
Zhang G, Zheng H, Pyykko I, Zou J. The TLR-4/NF-κB signaling pathway activation in cochlear inflammation of rats with noise-induced hearing loss. Hear Res. 2019;379:59–68.
Sai N, Shi X, Zhang Y, Jiang QQ, Ji F, Yuan SL, Sun W, Guo WW, Yang SM, Han WJ. Involvement of cholesterol metabolic pathways in recovery from noise-induced hearing loss. Neural Plast. 2020;2020:6235948.
Paciello F, Di Pino A, Rolesi R, Troiani D, Paludetti G, Grassi C, Fetoni AR. Anti-oxidant and anti-inflammatory effects of caffeic acid: in vivo evidences in a model of noise-induced hearing loss. Food Chem Toxicol: Int J Publ Br Ind Biol Res Assoc. 2020;143:111555.
Masuda M, Nagashima R, Kanzaki S, Fujioka M, Ogita K, Ogawa K. Nuclear factor-kappa B nuclear translocation in the cochlea of mice following acoustic overstimulation. Brain Res. 2006;1068(1):237–47.
Vethanayagam RR, Yang W, Dong Y, Hu BH. Toll-like receptor 4 modulates the cochlear immune response to acoustic injury. Cell Death Dis. 2016;7(6):e2245.
Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8(9–10):1865–79.
Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–3.
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radical Biol Med. 2010;49(11):1603–16.
Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. 2017;19(11):42.
Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–88.
Giordano S, Darley-Usmar V, Zhang J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014;2:82–90.
Pal R, Bajaj L, Sharma J, Palmieri M, Di Ronza A, Lotfi P, Chaudhury A, Neilson J, Sardiello M, Rodney GG. NADPH oxidase promotes Parkinsonian phenotypes by impairing autophagic flux in an mTORC1-independent fashion in a cellular model of Parkinson’s disease. Sci Rep. 2016;6:22866.
Zhang C, Li Q, Chen M, Lu T, Min S, Li S. The role of oxidative stress in the susceptibility of noise-impaired cochleae to synaptic loss induced by intracochlear electrical stimulation. Neuropharmacology. 2021;196:108707.
Fetoni AR, De Bartolo P, Eramo SL, Rolesi R, Paciello F, Bergamini C, Fato R, Paludetti G, Petrosini L, Troiani D. Noise-induced hearing loss (NIHL) as a target of oxidative stress-mediated damage: cochlear and cortical responses after an increase in antioxidant defense. J Neurosci: Off J Soc Neurosci. 2013;33(9):4011–23.
Kopke RD, Coleman JK, Liu J, Campbell KC, Riffenburgh RH. Candidate’s thesis: enhancing intrinsic cochlear stress defenses to reduce noise-induced hearing loss. Laryngoscope. 2002;112(9):1515–32.
Tretter V, Hochreiter B, Zach ML, Krenn K, Klein KU. Understanding cellular redox homeostasis: a challenge for precision medicine. Int J Mol Sci. 2021;23(1):106.
Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–90.
Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363–83.
Benham AM. Endoplasmic reticulum redox pathways: in sickness and in health. FEBS J. 2019;286(2):311–21.
Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol: CB. 2014;24(10):R453–62.
Hamanaka RB, Weinberg SE, Reczek CR, Chandel NS. The mitochondrial respiratory chain is required for organismal adaptation to hypoxia. Cell Rep. 2016;15(3):451–9.
Blacker TS, Duchen MR. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic Biol Med. 2016;100:53–65.
Lee HC, Wei YH. Mitochondria and aging. Adv Exp Med Biol. 2012;942:311–27.
Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal. 2005;7(9–10):1140–9.
Böttger EC, Schacht J. The mitochondrion: a perpetrator of acquired hearing loss. Hear Res. 2013;303:12–9.
Spoendlin H. Primary structural changes in the organ of Corti after acoustic overstimulation. Acta Otolaryngol. 1971;71(2):166–76.
Chen FQ, Zheng HW, Hill K, Sha SH. Traumatic noise activates Rho-family GTPases through transient cellular energy depletion. J Neurosci: Off J Soc Neurosci. 2012;32(36):12421–30.
Henderson D, Bielefeld EC, Harris KC, Hu BH. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 2006;27(1):1–19.
Sipos I, Tretter L, Adam-Vizi V. Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem. 2003;84(1):112–8.
Batandier C, Leverve X, Fontaine E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J Biol Chem. 2004;279(17):17197–204.
Fridberger A, Flock A, Ulfendahl M, Flock B. Acoustic overstimulation increases outer hair cell Ca2+ concentrations and causes dynamic contractions of the hearing organ. Proc Natl Acad Sci USA. 1998;95(12):7127–32.
Heinrich UR, Maurer J, Mann W. Ultrastructural evidence for protection of the outer hair cells of the inner ear during intense noise exposure by application of the organic calcium channel blocker diltiazem. ORL; J Oto-rhino-laryngol Relat Spec. 1999;61(6):321–7.
Shen H, Zhang B, Shin JH, Lei D, Du Y, Gao X, Wang Q, Ohlemiller KK, Piccirillo J, Bao J. Prophylactic and therapeutic functions of T-type calcium blockers against noise-induced hearing loss. Hear Res. 2007;226(1–2):52–60.
Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4(7):552–65.
Esterberg R, Hailey DW, Coffin AB, Raible DW, Rubel EW. Disruption of intracellular calcium regulation is integral to aminoglycoside-induced hair cell death. J Neurosci: Off J Soc Neurosci. 2013;33(17):7513–25.
Esterberg R, Hailey DW, Rubel EW, Raible DW. ER-mitochondrial calcium flow underlies vulnerability of mechanosensory hair cells to damage. J Neurosci: Off J Soc Neurosci. 2014;34(29):9703–19.
Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci: Off J Soc Neurosci. 2004;24(36):7771–8.
Sha SH, Taylor R, Forge A, Schacht J. Differential vulnerability of basal and apical hair cells is based on intrinsic susceptibility to free radicals. Hear Res. 2001;155(1–2):1–8.
Gentilin E, Cani A, Simoni E, Chicca M, Di Paolo ML, Martini A, Astolfi L. Hydrogen peroxide toxicity on auditory cells: an in vitro study. Chem Biol Interact. 2021;345:109575.
Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003;8:d369–91.
Wortzel I, Seger R. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer. 2011;2(3):195–209.
Dinh CT, Goncalves S, Bas E, Van De Water TR, Zine A. Molecular regulation of auditory hair cell death and approaches to protect sensory receptor cells and/or stimulate repair following acoustic trauma. Front Cell Neurosci. 2015;9:96.
Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68(2):320–44.
Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol (Clifton, NJ). 2010;661:3–38.
Murai N, Kirkegaard M, Järlebark L, Risling M, Suneson A, Ulfendahl M. Activation of JNK in the inner ear following impulse noise exposure. J Neurotrauma. 2008;25(1):72–7.
Maeda Y, Fukushima K, Omichi R, Kariya S, Nishizaki K. Time courses of changes in phospho- and total- MAP kinases in the cochlea after intense noise exposure. PLoS ONE. 2013;8(3):e58775.
Anttonen T, Herranen A, Virkkala J, Kirjavainen A, Elomaa P, Laos M, Liang X, Ylikoski J, Behrens A, Pirvola U. c-Jun N-terminal phosphorylation: biomarker for cellular stress rather than cell death in the injured cochlea. Eneuro. 2016;3(2).
Zhang AR, Ma KF, She XJ, Liu HT, Cui B, Wang R. Preliminary observation on the differential expression of metformin in preventing noise-induced hearing loss in inner ear protein group of rats. Zhonghua lao dong wei sheng zhi ye bing za zhi = Zhonghua laodong weisheng zhiyebing zazhi = Chin J Ind Hyg Occup Dis. 2022;40(4):248–54.
Faris M, Kokot N, Latinis K, Kasibhatla S, Green DR, Koretzky GA, Nel A. The c-Jun N-terminal kinase cascade plays a role in stress-induced apoptosis in Jurkat cells by up-regulating Fas ligand expression. J Immunol (Baltimore, Md: 1950). 1998;160(1):134–44.
Budhram-Mahadeo V, Morris PJ, Smith MD, Midgley CA, Boxer LM, Latchman DS. p53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J Biol Chem. 1999;274(21):15237–44.
Eichhorst ST, Müller M, Li-Weber M, Schulze-Bergkamen H, Angel P, Krammer PH. A novel AP-1 element in the CD95 ligand promoter is required for induction of apoptosis in hepatocellular carcinoma cells upon treatment with anticancer drugs. Mol Cell Biol. 2000;20(20):7826–37.
Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29(3):629–43.
Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15(1):36–42.
Oleinik NV, Krupenko NI, Krupenko SA. Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene. 2007;26(51):7222–30.
Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245–51.
Amaral JD, Xavier JM, Steer CJ, Rodrigues CM. The role of p53 in apoptosis. Discov Med. 2010;9(45):145–52.
Jia H, Yu Z, Ge X, Chen Z, Huang X, Wei Y. Glucocorticoid-induced leucine zipper protects noise-induced apoptosis in cochlear cells by inhibiting endoplasmic reticulum stress in rats. Med Mol Morphol. 2020;53(2):73–81.
Ottilie S, Diaz JL, Horne W, Chang J, Wang Y, Wilson G, Chang S, Weeks S, Fritz LC, Oltersdorf T. Dimerization properties of human BAD. Identification of a BH-3 domain and analysis of its binding to mutant BCL-2 and BCL-XL proteins. J Biol Chem. 1997;272(49):30866–72.
Puthalakath H, Huang DC, O’Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3(3):287–96.
Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277(43):40944–9.
Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol. 2002;22(13):4929–42.
Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA. 2003;100(5):2432–7.
Wang XT, Pei DS, Xu J, Guan QH, Sun YF, Liu XM, Zhang GY. Opposing effects of Bad phosphorylation at two distinct sites by Akt1 and JNK1/2 on ischemic brain injury. Cell Signal. 2007;19(9):1844–56.
Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22(15):3898–909.
Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, Walton KM, Ylikoski J. Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci: Off J Soc Neurosci. 2000;20(1):43–50.
Wang X, Truong T, Billings PB, Harris JP, Keithley EM. Blockage of immune-mediated inner ear damage by etanercept. Otol Neurotol: Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2003;24(1):52–7.
Wang J, Ruel J, Ladrech S, Bonny C, van de Water TR, Puel JL. Inhibition of the c-Jun N-terminal kinase-mediated mitochondrial cell death pathway restores auditory function in sound-exposed animals. Mol Pharmacol. 2007;71(3):654–66.
Godic A, Poljšak B, Adamic M, Dahmane R. The role of antioxidants in skin cancer prevention and treatment. Oxid Med Cell Longev. 2014;2014:860479.
Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: a review. Eur J Med Chem. 2015;97:55–74.
Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111(10):5944–72.
Martysiak-Żurowska D, Wenta W. A comparison of ABTS and DPPH methods for assessing the total antioxidant capacity of human milk. Acta Sci Pol Technol Aliment. 2012;11(1):83–9.
Poljsak B, Šuput D, Milisav I. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxid Med Cell Longev. 2013;2013:956792.
Willett WC. The Mediterranean diet: science and practice. Public Health Nutr. 2006;9(1a):105–10.
Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99(18):11908–13.
Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, Williams C, Risingsong R, Honda T, Gribble GW, Sporn MB, Talalay P. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci USA. 2005;102(12):4584–9.
Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–40.
Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39(4):199–218.
Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88(Pt B):179–88.
Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic Biol Med. 2004;37(4):433–41.
Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010;13(11):1665–78.
Tu W, Wang H, Li S, Liu Q, Sha H. The anti-inflammatory and anti-oxidant mechanisms of the keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 2019;10(3):637–51.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–23.
Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress, Biochimica et biophysica acta. Mol Cell Res. 2018;1865(5):721–33.
Fetoni AR, Paciello F, Rolesi R, Eramo SL, Mancuso C, Troiani D, Paludetti G. Rosmarinic acid up-regulates the noise-activated Nrf2/HO-1 pathway and protects against noise-induced injury in rat cochlea. Free Radic Biol Med. 2015;85:269–81.
Maines MD. The heme oxygenase system: past, present, and future. Antioxid Redox Signal. 2004;6(5):797–801.
Clark JE, Foresti R, Green CJ, Motterlini R. Dynamics of haem oxygenase-1 expression and bilirubin production in cellular protection against oxidative stress. Biochem J. 2000;348(Pt 3):615–9.
Motterlini R, Foresti R. Heme oxygenase-1 as a target for drug discovery. Antioxid Redox Signal. 2014;20(11):1810–26.
Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Jacob HS, Eaton JW, Balla G. Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid Redox Signal. 2007;9(12):2119–37.
Stefanson AL, Bakovic M. Dietary regulation of Keap1/Nrf2/ARE pathway: focus on plant-derived compounds and trace minerals. Nutrients. 2014;6(9):3777–801.
Lu SC. Regulation of glutathione synthesis. Mol Asp Med. 2009;30(1–2):42–59.
Calabrese G, Morgan B, Riemer J. Mitochondrial glutathione: regulation and functions. Antioxid Redox Signal. 2017;27(15):1162–77.
Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011;30(3):546–55.
Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194(1):7–15.
Lenaz G, Genova ML. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv Exp Med Biol. 2012;748:107–44.
Holmström KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, Stanyer L, Yamamoto M, Dinkova-Kostova AT, Abramov AY. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open. 2013;2(8):761–70.
Chen GD, Zhao HB. Effects of intense noise exposure on the outer hair cell plasma membrane fluidity. Hear Res. 2007;226(1–2):14–21.
Tiede L, Steyger PS, Nichols MG, Hallworth R. Metabolic imaging of the organ of corti—a window on cochlea bioenergetics. Brain Res. 2009;1277:37–41.
Tekpli X, Landvik NE, Anmarkud KH, Skaug V, Haugen A, Zienolddiny S. DNA methylation at promoter regions of interleukin 1B, interleukin 6, and interleukin 8 in non-small cell lung cancer. Cancer Immunol Immunother: CII. 2013;62(2):337–45.
Alvarado JC, Fuentes-Santamaría V, Juiz JM. Antioxidants and vasodilators for the treatment of noise-induced hearing loss: are they really effective? Front Cell Neurosci. 2020;14:226.
Kopke RD, Weisskopf PA, Boone JL, Jackson RL, Wester DC, Hoffer ME, Lambert DC, Charon CC, Ding DL, McBride D. Reduction of noise-induced hearing loss using L-NAC and salicylate in the chinchilla. Hear Res. 2000;149(1–2):138–46.
Kopke RD, Jackson RL, Coleman JK, Liu J, Bielefeld EC, Balough BJ. NAC for noise: from the bench top to the clinic. Hear Res. 2007;226(1–2):114–25.
Schmitt B, Vicenzi M, Garrel C, Denis FM. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: a comparative crossover study. Redox Biol. 2015;6:198–205.
Kopke R, Slade MD, Jackson R, Hammill T, Fausti S, Lonsbury-Martin B, Sanderson A, Dreisbach L, Rabinowitz P, Torre P 3rd, Balough B. Efficacy and safety of N-acetylcysteine in prevention of noise induced hearing loss: a randomized clinical trial. Hear Res. 2015;323:40–50.
De Rosa SC, Zaretsky MD, Dubs JG, Roederer M, Anderson M, Green A, Mitra D, Watanabe N, Nakamura H, Tjioe I, Deresinski SC, Moore WA, Ela SW, Parks D, Herzenberg LA, Herzenberg LA. N-acetylcysteine replenishes glutathione in HIV infection. Eur J Clin Invest. 2000;30(10):915–29.
Lu J, Li W, Du X, Ewert DL, West MB, Stewart C, Floyd RA, Kopke RD. Antioxidants reduce cellular and functional changes induced by intense noise in the inner ear and cochlear nucleus. J Assoc Res Otolaryngol: JARO. 2014;15(3):353–72.
Hong SY, Gil HW, Yang JO, Lee EY, Kim HK, Kim SH, Chung YH, Lee EM, Hwang SK. Effect of high-dose intravenous N-acetylcysteine on the concentration of plasma sulfur-containing amino acids. Korean J Intern Med. 2005;20(3):217–23.
Baker DH. Comparative species utilization and toxicity of sulfur amino acids. J Nutr. 2006;136(6 Suppl):1670s–5s.
De Flora S, Izzotti A, D’Agostini F, Balansky RM. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis. 2001;22(7):999–1013.
Hashimoto S, Gon Y, Matsumoto K, Takeshita I, Horie T. N-acetylcysteine attenuates TNF-alpha-induced p38 MAP kinase activation and p38 MAP kinase-mediated IL-8 production by human pulmonary vascular endothelial cells. Br J Pharmacol. 2001;132(1):270–6.
Cocco T, Sgobbo P, Clemente M, Lopriore B, Grattagliano I, Di Paola M, Villani G. Tissue-specific changes of mitochondrial functions in aged rats: effect of a long-term dietary treatment with N-acetylcysteine. Free Radic Biol Med. 2005;38(6):796–805.
Grattagliano I, Portincasa P, Cocco T, Moschetta A, Di Paola M, Palmieri VO, Palasciano G. Effect of dietary restriction and N-acetylcysteine supplementation on intestinal mucosa and liver mitochondrial redox status and function in aged rats. Exp Gerontol. 2004;39(9):1323–32.
Kil J, Pierce C, Tran H, Gu R, Lynch ED. Ebselen treatment reduces noise induced hearing loss via the mimicry and induction of glutathione peroxidase. Hear Res. 2007;226(1–2):44–51.
Kim SJ, Park C, Han AL, Youn MJ, Lee JH, Kim Y, Kim ES, Kim HJ, Kim JK, Lee HK, Chung SY, So H, Park R. Ebselen attenuates cisplatin-induced ROS generation through Nrf2 activation in auditory cells. Hear Res. 2009;251(1–2):70–82.
Kil J, Lobarinas E, Spankovich C, Griffiths SK, Antonelli PJ, Lynch ED, Le Prell CG. Safety and efficacy of ebselen for the prevention of noise-induced hearing loss: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet (London, England). 2017;390(10098):969–79.
Sies H, Stahl W. Vitamins E and C, beta-carotene, and other carotenoids as antioxidants. Am J Clin Nutr. 1995;62(6 Suppl):1315s–21s.
Schafer FQ, Wang HP, Kelley EE, Cueno KL, Martin SM, Buettner GR. Comparing beta-carotene, vitamin E and nitric oxide as membrane antioxidants. Biol Chem. 2002;383(3–4):671–81.
Le Prell CG, Hughes LF, Miller JM. Free radical scavengers vitamins A C, and E plus magnesium reduce noise trauma. Free Radic Biol Med. 2007;42(9):1454–63.
Alvarado JC, Fuentes-Santamaría V, Melgar-Rojas P, Valero ML, Gabaldón-Ull MC, Miller JM, Juiz JM. Synergistic effects of free radical scavengers and cochlear vasodilators: a new otoprotective strategy for age-related hearing loss. Front Aging Neurosci. 2015;7:86.
Alvarado JC, Fuentes-Santamaría V, Gabaldón-Ull MC, Juiz JM. An oral combination of vitamins A, C, E, and Mg(++) improves auditory thresholds in age-related hearing loss. Front Neurosci. 2018;12:527.
Ahn JH, Kang HH, Kim YJ, Chung JW. Anti-apoptotic role of retinoic acid in the inner ear of noise-exposed mice. Biochem Biophys Res Commun. 2005;335(2):485–90.
Ahn JH, Shin JE, Chung BY, Lee HM, Kang HH, Chung JW, Pak JH. Involvement of retinoic acid-induced peroxiredoxin 6 expression in recovery of noise-induced temporary hearing threshold shifts. Environ Toxicol Pharmacol. 2013;36(2):463–71.
Prasad KN, Bondy SC. Increased oxidative stress, inflammation, and glutamate: potential preventive and therapeutic targets for hearing disorders. Mech Ageing Dev. 2020;185:111191.
Evans P, Halliwell B. Free radicals and hearing. Cause, consequence, and criteria. Ann N Y Acad Sci. 1999;884:19–40.
McFadden SL, Woo JM, Michalak N, Ding D. Dietary vitamin C supplementation reduces noise-induced hearing loss in guinea pigs. Hear Res. 2005;202(1–2):200–8.
Loukzadeh Z, Hakimi A, Esmailidehaj M, Mehrparvar AH. Effect of ascorbic acid on noise induced hearing loss in rats. Iran J Otorhinolaryngol. 2015;27(81):267–72.
Shemesh Z, Attias J, Ornan M, Shapira N, Shahar A. Vitamin B12 deficiency in patients with chronic-tinnitus and noise-induced hearing loss. Am J Otolaryngol. 1993;14(2):94–9.
Gok U, Halifeoglu I, Canatan H, Yildiz M, Gursu MF, Gur B. Comparative analysis of serum homocysteine, folic acid and Vitamin B12 levels in patients with noise-induced hearing loss. Auris Nasus Larynx. 2004;31(1):19–22.
Martínez-Vega R, Garrido F, Partearroyo T, Cediel R, Zeisel SH, Martínez-Álvarez C, Varela-Moreiras G, Varela-Nieto I, Pajares MA. Folic acid deficiency induces premature hearing loss through mechanisms involving cochlear oxidative stress and impairment of homocysteine metabolism. FASEB J: Off Publ Fed Am Soc Exp Biol. 2015;29(2):418–32.
Abbasi M, Pourrajab B, Tokhi MO. Protective effects of vitamins/antioxidants on occupational noise-induced hearing loss: a systematic review. J Occup Health. 2021;63(1):e12217.
Kawada H, Blessing K, Kiyota T, Woolman T, Winchester L, Kador PF. Effects of multifunctional antioxidants on mitochondrial dysfunction and amyloid-β metal dyshomeostasis. J Alzheimer’s Dis: JAD. 2015;44(1):297–307.
Kawada H, Kador PF. Orally bioavailable metal chelators and radical scavengers: multifunctional antioxidants for the coadjutant treatment of neurodegenerative diseases. J Med Chem. 2015;58(22):8796–805.
Chen GD, Daszynski DM, Ding D, Jiang H, Woolman T, Blessing K, Kador PF, Salvi R. Novel oral multifunctional antioxidant prevents noise-induced hearing loss and hair cell loss. Hear Res. 2020;388:107880.
Bielefeld EC, Hu BH, Harris KC, Henderson D. Damage and threshold shift resulting from cochlear exposure to paraquat-generated superoxide. Hear Res. 2005;207(1–2):35–42.
Möhrle D, Reimann K, Wolter S, Wolters M, Varakina K, Mergia E, Eichert N, Geisler HS, Sandner P, Ruth P, Friebe A, Feil R, Zimmermann U, Koesling D, Knipper M, Rüttiger L. NO-sensitive guanylate cyclase isoforms NO-GC1 and NO-GC2 contribute to noise-induced inner hair cell synaptopathy. Mol Pharmacol. 2017;92(4):375–88.
Bielefeld EC, Kopke RD, Jackson RL, Coleman JK, Liu J, Henderson D. Noise protection with N-acetyl-l-cysteine (NAC) using a variety of noise exposures, NAC doses, and routes of administration. Acta oto-laryngolog. 2007;127(9):914–9.
Fetoni AR, Mancuso C, Eramo SL, Ralli M, Piacentini R, Barone E, Paludetti G, Troiani D. In vivo protective effect of ferulic acid against noise-induced hearing loss in the guinea-pig. Neuroscience. 2010;169(4):1575–88.
Fetoni AR, Eramo S, Troiani D, Paludetti G. Therapeutic window for ferulic acid protection against noise-induced hearing loss in the guinea pig. Acta Otolaryngol. 2011;131(4):419–27.
Le Prell CG, Gagnon PM, Bennett DC, Ohlemiller KK. Nutrient-enhanced diet reduces noise-induced damage to the inner ear and hearing loss. Transl Res: J Lab Clin Med. 2011;158(1):38–53.
Acknowledgements
Not applicable.
Funding
This work was supported by Grants from the Project Supported by Zhejiang Provincial Natural Science Foundation of China (LY22H090020).
Author information
Authors and Affiliations
Contributions
YZ, CF and LY designed the review and wrote the manuscript. MG, XX, and AS conceived the artwork and performed the bibliographical research. AZ and DZ supervised the writing. All the authors revised and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, Y., Fang, C., Yuan, L. et al. Redox homeostasis dysregulation in noise-induced hearing loss: oxidative stress and antioxidant treatment. J of Otolaryngol - Head & Neck Surg 52, 78 (2023). https://doi.org/10.1186/s40463-023-00686-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40463-023-00686-x