
Abstract
Adoptive cell therapies (ACTs) have revolutionized cancer immunotherapy, prompting exploration into their application against oncoviruses. Oncoviruses such as human papillomavirus (HPV), hepatitis B virus (HBV), hepatitis C virus (HCV), and Epstein-Barr virus (EBV) contribute significantly (12-25%) to human malignancies through direct or indirect oncogenic mechanisms. These viruses persistently or latently infect cells, disrupt cellular homeostasis and pathways, challenging current antiviral treatment paradigms. Moreover, viral infections pose additional risks in the setting of long-term cancer therapy and lead to morbidity and mortality. Virally encoded oncoproteins, which are tumor-restricted, immunologically foreign, and even uniformly expressed, represent promising targets for patient-tailored ACTs. This review elucidates the rationale for leveraging viral antigen-specific ACTs in combating viral-associated malignancies. On this basis, ongoing preclinical studies consolidate our understanding of harnessing ACTs against viral malignancies, underscoring their potential to eradicate viruses implicated in cancer progression. Furthermore, we scrutinize the current landscape of clinical trials focusing on virus-specific ACTs and discuss their implications for therapeutic advancement.
Similar content being viewed by others

Introduction
The International Agency for Research on Cancer (IARC) recognizes 7 major human viruses as direct oncogenic agents, including human papilloma virus (HPV), hepatitis B virus (HBV), hepatitis C virus (HCV), Epstein-Barr virus (EBV), Kaposi’s sarcoma-associated herpesvirus (KSHV), Merkel cell polyomavirus (MCPV), and human immunodeficiency virus type 1 (HIV-1) [1]. Additionally, cytomegalovirus (CMV) reactivation and infection are frequently observed in immunocompromised individuals such as transplant recipients or HIV-1 carriers [2], with mounting evidence suggesting CMV’s potential as an oncogenic virus [3,4,5,6].
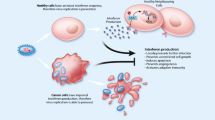
Despite the typically robust immune response to viral antigens in most infected individuals, persistent or latent infection of oncoviruses enables to evade the immune system and induce immune tolerance through mechanisms such as downregulating major histocompatibility complex (MHC) molecules, producing immunosuppressive proteins, and directly infecting immune cells [7,8,9], which increases the risk of virus-driven or associated cancers (Fig. 1a). For instance, HPV, EBV and CMV encode viral oncoproteins that mimic or interfere with host regulatory mechanisms, disrupt cellular homeostasis, and impact cellular proteins, such as the tumor suppressor proteins p53 and pRb (Table 1) [2, 10]. In some other cases involving either DNA or RNA viruses (HPV, HBV, EBV, CMV and HIV-1), the viral genome can integrate into the host genome, remaining dormant until conditions favor reactivation, thereby contributing to viral persistence and disease manifestation [11]. Importantly, the “hit-and-run’’ theory also posits that viruses induce a series of cellular changes, promoting normal cells to become cancer cells, after which the virus leaves while the cancer cells develop [12]. However, HBV and HCV can create a microenvironment conducive to tumorigenesis through chronic inflammation. Persistent inflammation leads to the production of cytokines and growth factors, that promote cell proliferation, angiogenesis, and genomic instability [13].

a Viral persistent or latent infection and host cellular immunity against oncogenic infection (By Figdraw). A variety of virally oncogenic mechanisms determine the transformation and maintenance of the malignancy. Virus-infected cells can be recognized and eliminated by host cellular immunity. b Schematic diagram of ACTs on viral malignancies. Engineered TILs, DCs, CAR-T cells, TCR-T cells, and VST therapies have the potential for application to viral malignancies. CAR-T cells target the virally-encoded cell surface antigen through an antibody-based scFv. In contrast, TCR-T cells target a virus-derived peptide on MHC complex. In TIL therapy, tumor is surgically resected and T-cells are expanded from the tumor ex vivo. TILs target viral antigens as well as non-viral proteins. The VST therapy aims to enhance the host immune system’s ability to clear infected cells by using activated virus specific T-cells. While the DCs enable to induce and amplify virus-specific CTLs.
Current antiviral treatments effectively suppress viral replication but fail to eliminate chronic or latent infections. Eradicating viral reservoirs remains a critical therapeutic challenge. Beyond tumors, virus-specific adoptive cell therapies (ACTs) have shown promise in purging viral infections, suggesting a potential role in treating viral malignancies [14]. ACTs are particularly suited to viral malignancies due to the expression of targetable tumor-associated viral antigens exclusively in cancerous cells, providing an unparalleled opportunity to subvert such oncoproteins as tumor-specific targets. Furthermore, the current tantalizing goal is to activate immune cells by targeting viral antigens, rejuvenate antiviral effects and achieve the goal of recognizing and killing virus-related tumor cells.
Tremendous progress has been made in the development of ACTs for viral malignancies, including tumor-infiltrating lymphocyte (TIL), dendritic cell (DC), chimeric antigen receptor (CAR)-T cell, T-cell receptor (TCR)-T cell, and virus-specific T-cell (VST) therapies (Fig. 1b), and these findings require reanalysis and reflection. Our review covers preclinical and clinical ACTs for the ablation of oncovirus infections and associated viral malignancies, highlighting the therapeutic potential of targeting virally encoded antigens.
HPV
High-risk HPV types (HPV16/18) are well-established drivers of various cancer, including cervical carcinoma (CC), head and neck cancer (HNC) and oropharyngeal cancer (OPC) [15]. This oncogenic potential is primarily attributed to viral integration and oncoproteins [16,17,18,19]. The HPV genome encodes early (E1-E7) and late (L1 and L2) proteins during the viral life cycle. When the HPV genome gets integrated, constitutive E6 and E7 expression is observed, which is critical for the transformation and maintenance of malignancy by interfering with cellular homeostasis, inhibiting the immune response and inducing immune escape [20]. The pRb pathway is disrupted by the E7 protein, releasing the E2F transcription factor and leading to cell cycle dysregulation and unrestricted proliferation [21, 22]. E6 promotes p53 degradation, thus inhibiting p53-mediated apoptosis and facilitating an ongoing cell cycle for viral replication [23]. Multiple pathways, including the Wnt/β-catenin, Bak and PI3K/Akt pathways promote cancer progression by interfering with cell proliferation, differentiation, and apoptosis and inducing abnormal gene expression [24].
Even prophylactic vaccines are envisaged to protect immunized individuals against cancer-associated HPV genotypes. For established HPV infection or maintenance in a latent or asymptomatic state in basal cells, where the HPV integrates with the host cell genome and no longer expresses viral L1/2 antigens, the conventional preventive HPV vaccines have been demonstrated to be ineffective [25]. In contrast, therapeutic HPV vaccines focusing on HPV primary oncoproteins, specifically E6 and E7, represent a promising avenue for enhancing clinical outcomes among advanced-stage and recurrent patients without eliciting autoimmune or severe adverse events (Fig. 2a; Table 2). Notwithstanding, it is crucial to acknowledge that the most frequently encountered severe toxicities primarily manifest as hematologic complications, which are anticipated sequelae of lymphocyte-depleting conditioning regimens commonly employed in such therapeutic strategies.

Virally encoded antigens and relevant utilization of oncovirus-specific ACTs in viral malignancies. a HPV-specific ACTs. b HBV-specific ACTs. c HCV-specific ACTs. d CMV-specific ACTs. e EBV-specific ACTs. f HIV-1-specific ACTs.
HPV-specific CAR-T cells
The E7 oncoprotein localizes to the intracellular compartment and consequently cannot be targeted with antibodies or generic CAR-T cells. In the HPV-infected epithelium, E7 proteins are cleaved into short peptide fragments by proteasomes and presented on the cell surface by HLAs, and thus recognized and attacked by T-cells. We presume that CAR-T cells targeting antigen-peptide HLA complexes have higher specificity and lower off-target toxicity. Novelly constructed CAR-T cells targeting the E711 − 19/HLA-A*02:01 complex (E711 − 19:HLA CAR-T) are waited for intensively investigated. Upon delivery using viral vectors, extracellular vesicles or some targeting sequences, the intracellular single-chain antibodies (scFvs) counteracting the E6 and E7 oncoproteins demonstrated antitumor efficacy in vitro and in vivo [26,27,28]. To our knowledge, CAR-T cells targeting the oncovirus-restricted surface antigen have not yet been reported.
HPV-specific TCR-T cells
TCR-T cells that recognize both surface and internal viral antigens demonstrate robust infiltration and persistence in the treatment of solid tumors. As a model for proof-of-principle studies in epithelial cancers, the treatment of HPV-induced cancers using virus-specific TCR-T cells has been established [29]. Epitope E711 − 19-targeted and HLA-A*02:01-restricted TCR transgenic T (E711 − 19 TCR-T) cells can specifically recognize and kill HPV16-positive cancerous cell lines and mediate regression of established human HPV16-positive CC in an in vivo model [30]. These data provide preclinical support for first-in-human, phase I clinical trials of such E711 − 19 TCR-T cells. Robust tumor regression was demonstrated as clinical objective response rate (ORR) in 6/12 metastatic HPV-associated epithelial cancers, including complete remission (CR) of lesions and marked responses even in anti-PD-1 refractory patients [31].
TCR-T cells directed against E692 − 101 of HPV16 (E692 − 101TCR-T) were validated high activity towards HPV16-positive HLA-A*11:01 CC cells in vitro and efficiently repressed tumor growth in a murine model [32]. Another earlier study revealed that the avid E629 − 38TCR-T, recognized an HLA-A*02:01-restricted epitope of HPV16 E6 successfully targeted HPV16-positive epithelial tumor cells and caused tumor regression [33]. In a subsequent phase I/II study, E629 − 38TCR-T cells showed high levels of peripheral blood engraftment 1 month after treatment and induced an ORR in 2/12 patients with chemotherapy-refractory, metastatic HPV16-positive epithelial cancers [34]. Overall, TCR-T therapy based on E6 and E7 oncoproteins of HPV16-positive epithelial cancers are capable of in vivo expansion, long-term persistence, and tumor regression.
HPV-specific TILs
Therapeutic TILs for successful treatment of patients with HPV-positive CC are directed for HPV E6 and/or E7 antigens [35,36,37]. However, that bulk of E6 and E7 specific T-cells reside predominantly in the PD-1-expressing T-cell compartment, are rendered functionally inactive within the TME and display no preferential in vivo expansion [35, 36], suggesting that PD-1 blockade or proper stimulation can be exploited for unleashing diverse antitumor T-cell reactivities. Using a lymphocyte depletion chemotherapy regimen followed by aldesleukin administration and TIL infusion, 9 women with metastatic CC participated in the reported phase I/II clinical trial of single infusion of E6 and E7 targeted TILs (HPV-TILs). Ultimately, 3/9 patients experienced objective tumor responses, whereas 2/9 patients experienced CR 15 and 22 months after treatment [38]. In the subsequent phase II study, 5/18 patients with metastatic CC showed ORR, 2 of whom achieved CR, and 2/11 HPV-positive noncervical cancer cohort also showed ORR. Even more importantly, the magnitude of HPV reactivity and peripheral blood repopulation with HPV-TILs are correlated with the clinical response [39]. These studies indicate that HPV-TIL therapy is feasible and can induce persistent CR in metastatic HPV-positive CC.
HPV-specific DC vaccines
DCs orchestrate adaptive immunity by phagocytosing viral antigens and presenting peptide epitopes, but only a limited number of peptide epitopes are capable of priming specific CTL precursors for a given HLA. More concerningly, the inadequate antigen presentation on mature DCs is frequently observed in HPV-positive tumor bearing individuals [40,41,42]. Although cognate peptide-loaded autologous DCs can stimulate a specific CTL response against HPV16 E786 − 93, such immunogenic peptide does not appear to be processed or presented by HPV16-infected cells [43]. This finding raises controversy regarding the antitumor activity of HPV-specific CTLs and challenges our understanding of CTL epitope mapping.
To broaden clinical applicability, diverse strategies employing genetically modified DCs expressing E6/E7, pulsed with E6/E7 fusion proteins, infected with recombinant adenovirus, or hybridization with tumor cells have facilitated a versatile presentation of all possible CTL epitopes [43,44,45]. To this end, DC-based vaccines can overcome the limitations of peptide epitopes with respect to specific HLA-haplotypes and improve antigen presentation in tumor-bearing individuals. As expected, intramuscular administration of E7-transfected murine DCs substantially decreased tumorigenicity and generated strong immunity against HPV16/18 E7-expressing neoplasms [46]. The enhancement of immune responses by cytokines and immunostimulatory gene therapy should prospectively potentiates antitumor activity, as extensively reviewed elsewhere [44, 47, 48]. However, the immune inhibitory effect on HPV16 E7-expressing DCs has been shown to be mediated by the coexpression of IL12 [49]. In preclinical models, local administration of the tumor lysate-pretreated DC vaccines, containing HPV16 E6/E7 oncoprotein, effectively inhibited recurrence or minimal residual tumors in mice [50].
In a pilot clinical study of 15 CC patients, HPV E7 antigen-loaded autologous DCs (E7-DCs) induced a specific antibody response in 3/11 evaluated patients and a specific cellular immune response in 4/11 patients. Unfortunately, neither histopathological regression nor viral clearance of treated patients was observed, attributed to HLA expression loss [51]. Moreover, even with a sharp decrease in L1 expression and a limited cellular immune response against the L1 antigen in cervical lesions with established HPV16 infection, DCs pulsed with major L1 capsid protein-based HPV16-like particles (VLPs, L1-DCs) can elicit strong specific CTLs and lyse HPV16-infected autologous tumor cells [52]. Evidence also suggests that CTLs induced by VLP vaccination can target cells expressing low L1 protein levels [53, 54]. Collectively, ongoing optimization of DC-based cancer vaccines is essential given their current therapeutic limitations in HPV-related cancers.
HBV
HBV is a hepatocyte specific and enveloped DNA virus. The HBV genome contains four partially overlapping open reading frames (ORFs), namely the preS/S region, preC/C region, P region, and X region. According to the different starting codon positions, preS/S can be divided into three different structural domains, including preS1/S, preS2/S, and S, which are responsible for the large protein (LHB), medium protein (MHB), and small protein (SHB) of the envelope protein hepatitis B surface antigen (HBsAg), respectively. Upon entry into hepatocytes, the virus releases its relaxed circular DNA (rcDNA), which is then transported to the nucleus where it forms covalently closed circular DNA (cccDNA). Transcription of cccDNA generates pregenomic RNA (pgRNA), which serves as a template for both viral genome replication and the translation of viral proteins. Newly synthesized pgRNA is encapsidated along with the viral polymerase to form nucleocapsids. Within nucleocapsids, the reverse transcription process generates rcDNA, which can either replenish the cccDNA pool or be enveloped and released as mature virions [55]. This replication cycle perpetuates HBV infection and contributes to its persistence in the host. Owing to the reverse transcription process of HBV, its DNA can be integrated into the chromosome of the targeted cell, resulting in genomic instability, direct insertional mutagenesis and abnormal expression of oncogenes and tumor suppressor genes [56].
Persistent HBV infection predisposes to the initiation and development of hepatocellular carcinoma (HCC) through necro-inflammation and direct carcinogenic effects. The prevalent HBV-related HCC (HBV-HCC) is mostly characterised by HBV-DNA integrations, even in cases serologically negative for HBV antigens [57]. Additionally, the immunosuppressive TME facilitates virus escape and chronic HBV (CHB) progression. The oncogenic process is multifaceted, involving intervention in various signal pathways through microRNAs, compromised immune responses, increased chromosomal alterations, endoplasmic reticulum (ER)-stress toward hepatocellular transformation, epigenetic dysregulation of tumor suppressor genes, and overexpression of fetal liver/hepatic progenitor cell genes [58, 59]. In terms of a plethora of oncogenic factors, prolonged expression of the viral HBV x antigen (HBxAg) and aberrant preS1/S2 envelope proteins dysregulate cell transcription and proliferation, making liver cells sensitive to carcinogenic factors [60,61,62].
Currently available therapies, including prophylactic vaccine and antiviral treatment, effectively control HBV infection or replication but do not achieve clearance for intermediate and advanced HCC. Functional cure, defined by HBsAg loss, does not equate to viral eradication in CHB patients, as residual cccDNA or HBV-DNA integrations encoding HBsAg can lead to disease relapse in HBV carriers [63]. Purging cccDNA in hepatocytes through deamination-induced decay following antiviral therapy is a major therapeutic goal in CHB [55]. ACTs remain pivotal in the management of infection and in the prevention of HBV-HCC relapse (Fig. 2b; Table 3), although exploitation of HBV antigens as tumor-specific targets for ACTs has been criticized due to their inconsistent expression in HCC. Additionally, risk assessment must address two considerations: the adequacy of cellular immunity to achieve durative and complete HBV clearance in CHB patients subjected to prolonged exposure to HBV antigens; assessing whether the robust cytotoxic effects of ACTs might precipitate severe hepatotoxicity and acute liver damage.
HBV-specific CAR-T cells
HBsAg remains positive in both CHB and HBV-HCC with integrated viral genomes. Targeting HBV surface proteins therefore seems promising. SHB and LHB specific CAR-T cells demonstrated recognition of soluble HBsAg and HBsAg-positive hepatocytes, eliciting secretion of IFNγ and IL-2 and selective eliminating of cccDNA-positive cells. More abundance of SHB on the surface of HBV-induced cancer facilitates the ER membrane targeting of HBsAg and steady ER-plasma membrane exchange [14, 64]. Concordantly, SHB-CAR-T cells exhibited faster activation and greater cytokine secretion than LHB-CAR-T cells [65].
In HBV-transgenic mice lacking cccDNA formation, but possessing a functional immune system, large amounts of circulating viral antigens do not inactivate transferred HBsAg-CAR-T cells or lead to uncontrolled immune-mediated damage in vivo. But rather HBsAg-CAR-T cells would recognize all the extracellular and secreted HBsAg proteins (SHB, MHB, and LHB proteins, combined as HBsAg) and demonstrate efficacy against HBV-infected hepatocytes [66]. Reduced HBsAg-CAR-T cell persistence alongside increased viral parameters were observed after initial and sequential transfer into HBV-transgenic mice, suggesting potential causes, such as T-cell exhaustion or overactivation via antigen binding or Fc receptor interaction with the CAR [66, 67]. Notably, through irradiation and tolerization of immunocompetent mice, fully human SHB-CAR-T cell transfer persisted at high numbers and induced a sustained antiviral effect [67]. Accordingly, interactions with the different arms of the endogenous immune system, bystander immune cell activation, and combination therapies are warranted for combating virally induced HCC.
Further preclinical exploration of CAR-T cells as HBV immunotherapy in models with authentic infections harboring episomal HBV cccDNA is warranted. Murine HBsAg-CAR-T cells transferred into HBV-infected humanized liver chimeric mice accumulate in the liver, significantly reducing plasma HBsAg and HBV-DNA levels compared with those in controls [68]. Notably, HBsAg-CAR-T cells did not kill HBV-positive cell lines in cytotoxicity assays, indicating noncytopathic viral clearance. Upon triple knockdown of exhaustion markers (PD-1, Tim-3, and Lag-3), CAR-T cells, which target the preS1 domain of HBsAg and exhibit the tumor-reactive marker CD39+ (preS1-CAR-T), potently inhibit tumor growth and increase IFNγ secretion in a patient-derived xenograft (PDX) mouse model [69].
HBV specific TCR-T cells
HBV-specific TCR-T cells utilize TCR sequences sourced from endogenous T-cells of patients with self-limited HBV infection or are exogenously engineered to recognize HBV antigens presented by infected cells [70, 71]. In some cases, HBV-HCC negative for HBV antigens may contain translationally active HBV-DNA integrations, generating functional T-cell epitopes recognized by and activate HBV-specific T-cells [72].
High-affinity HBsAg- and HBV core antigen (HBcAg)-specific TCRs in resting and activated T-cells from healthy donors and CHB patients can transform these cells into polyfunctional effector cells, which exhibit antiviral efficacy with limited liver injury through direct cytotoxicity [73]. Whilst a single transfer of TCR-T cells into HBV-infected, humanized mice eliminated HBV infection and suppressed virological markers without damaging non-infected cells [73]. Additionally, TCR-T therapy has shown promising results in maintaining memory T-cell function, which is crucial for long-term immune surveillance against HBV reactivation and the occurrence of HBV-HCC. Intriguing, resting T-cells reprogrammed by HBV-specific TCR reduced HBV replication in humanized immunodeficient mice without lysing HBV-infected hepatoma cells and simultaneously have comparable IFNγ levels and lower perforin and granzyme levels [74].
In the first-in-man proof-of-concept clinical trials of TCR-T cells, the HCC-specific antigen HBsAg was targeted by adoptively transferred HBV-specific TCR-T cells in a compassionate setting for HBV-HCC patients with extrahepatic metastasis after liver transplant. Retrovirally transduced TCR-T cells (HBsAg183 − 191-TCR-T), which are designed to target HLA-A*02:01/HBsAg183 − 191 complexes, dramatically reduced HBsAg levels by approximately 10-fold in concomitance with TCR-T cell expansion, albeit with limited survival due to metastatic disease progression [75,76,77]. These findings underscore the potential efficacy of TCR-T cells targeting HBV antigens in inducing sustained immune control over HBV-related tumors.
In both preclinical and clinical settings, multiple infusions of short-lived mRNA-based HBV-specific TCR-T (HBV-TCR-T) cells for HBV-HCC individuals exhibited clinically relevant suppression of HCC and a reduction or stabilization of circulating HBsAg and HBV DNA levels, indicating on-target effects [72, 78,79,80,81]. The transcribed mRNA can be intuitively safe because of the transient self-limiting inflammatory reaction and a dearth of transgene integration into the host genome. However, the results from a phase I trial in a compassionate setting for patients with HCC recurrence post-liver transplant revealed that HBsAg- or HBcAg-directed TCR-T cells engineered by concomitant electroporation of mRNAs encoding specific TCRs have no superior anti-tumor efficacy [80]. From our perspective, the aforementioned status has implications for armoring more robust and drug-resistant TCR-T cells to overcome the immunosuppressive TME.
HBV-specific DC vaccines
DC-based vaccines loaded with HBV-specific antigens represent a promising immunotherapeutic strategy to restore antiviral immunity crucial for controlling CHB and HBV-HCC [82]. DCs loaded with HLA-restricted peptides such as HBcAg18 − 27 and HBsAg335 − 343 have demonstrated efficacy in priming specific CTLs ex vivo and in humanized mice. Stimulation of PBMCs or TILs from CHB patients with these peptide-loaded DCs resulted in significant HBV-specific CTL responses, including IFNγ secretion, CD107 expression upon restimulation, reduction in systemic viral load, and lysis of HBV antigen-expressing hepatocytes [83].
Elegant work has validated the safety and efficacy of antigen-pulsed DCs in a large cohort of CHB patients. DCs derived from CHB patients and pulsed with HBsAg or HBcAg effectively induced CTL responses, reversed immune tolerance in CHB, promoted DC maturation, cytokine production, and enhanced CTL activation [84,85,86]. Since CD14-HBsAg complexes were detected in vitro and in the serum of HBV infected patients. It’s proposed that HBsAg activates DCs through CD14-dependent mechanisms [87], crucial for initiating effective HBV-specific immune responses.
Moreover, DCs loaded with HBV subviral particles (HBVsvp) offer an innovative approach to activate HBV-specific CTLs, bypassing dysfunctional DCs and T-cells in CHB patients, thereby inducing Th1 polarization and strong cytolytic activity [88]. Phase I trials utilizing autologous DCs pulsed with irradiated tumor stem cells (DC-TC) have shown initial safety in patients with cirrhosis and HBV infection, suggesting potential therapeutic benefits [89]. Collectively, these findings underscore the promise of DC-based vaccines as a therapeutic avenue against HBV-associated HCC.
HCV
HCV belongs to the Flaviviridae family and has a single positive-sense RNA (+ ssRNA), which codifies for an icosahedral nucleocapsid composed of C protein and envelope glycoproteins (E1 and E2), as well as non-structural proteins (NS1, NS2, NS3, NS4A/4B, NS5A/NS5B). The molecular mechanisms underlying HCV-HCC primarily revolves around a complex interplay of viral proteins with cellular pathways, leading to dysregulated cellular functions, genomic instability, and tumor transformation [90, 91].
HCV reaches its peak titers several weeks before the onset of detectable humoral or cellular immune responses and the initiation of liver disease. In western countries and Japan, chronic HCV infection is the primary cause of HCC. Wherein the highly variable HCV genomes under the selective pressure of host immune response are major risk factors for HCC development and impede the effectiveness of prophylactic and therapeutic treatments [92,93,94]. With the advent of potent antivirals targeting the viral life cycle: the NS3/4A protease, the NS5A protein and the RNA-dependent RNA polymerase NS5B protein, the incidence of HCV-HCC has substantially decreased [95]. Nevertheless, in cases where the HCV titer remains relatively low during chronic infections, we should armor immune cells and pre-empt T-cell exhaustion or anergy to clear HCV infection. HCV specific ACTs are list in Table 4 (Fig. 2c).
HCV-specific CAR-T cells
The highly variable HCV E2 glycoprotein (HCV-E2) is a major target of the host immune response. Anti-HCV-E2 CARs were designed based on a previously described broadly cross-reactive and cross-neutralising human monoclonal antibody (mAb), directed against conserved HCV-E2 epitopes [96]. The cytotoxic ability of anti-HCV-E2 CARs-grafted T (HCV-E2-CAR-T) cells was evaluated in vitro against HCV-E2-transfected cells as well as hepatocytes infected with HCV. In a proof-of-concept study, retrovirus-transduced HCV-E2-CAR-T were endowed with specific antigen recognition accompanied by degranulation and secretion of proinflammatory and antiviral cytokines [97].
Introducing the HCV NS3 protease (HCV-NS3) between the scFv and hinge domain allowed for protease-regulated CAR circuits, enabling precise control over CAR-T cell activation during cancer therapy. In the absence of HCV-NS3 inhibitor, NS3 displays the proteolytic process, disrupts the CAR structure and prevents the activation signals. Conversely, administering protease inhibitors inhibited NS3 cleavage, preserving CAR integrity and facilitating T-cell activation [98, 99]. The anti-tumor potency and reversibility of drug-regulated CAR-T cells targeting tumor-associated antigens (TAA) were evaluated in solid and hematological tumors [100, 101]. As such, future investigations may explore CARs targeting HCV antigens, potentially leveraging clinically approved HCV-NS3 antiviral protease inhibitors to synergistically combat HCV-HCC.
HCV-specific TCR-T cells
Two specific HCV TCRs that mounted a polyfunctional response to the cognate HLA-A2-restricted NS31073 − 1081 and NS51992 − 2000 peptide, and enabled to eliminate human hepatoma cells with persistent HCV RNA replication [102]. The expanded study revealed that NS3-specific TCR-T cells were prone to induce the antigen-specific cytolysis of target cells, while NS5-specific TCR-T cells favored a non-cytotoxic mechanism [103, 104], mirroring some marked differences in avidity and functional profile between HCV-specific TCR-T in tumor cell lines. High-avidity NS3-specific TCR-T cells rapidly activated apoptotic signaling pathways, causing hepatotoxicity, whereas the low-avidity NS5-specific TCR-T cells promoted the proliferative and metabolic pathways as the extended survival of HCV target cells [104]. At this juncture we surmised that, high-avidity TCR-T cells demonstrate superior antiviral activity, while low-affinity TCR-T cells are considered more suitable for chronic viral infections due to less immune pathology.
HCV-specific DC vaccines
Considering the modest immunogenicity of HCV-E2 glycoprotein, modification of HCV-E2 is warranted to applicably potentiate DC function and elicit a robust protective immune response [105]. Moreover, the effectiveness of the DC vaccines loaded with two selected HCV-E2 peptides have been validated to activate peptide-specific cellular immune activation and induce significant levels of anti-viral cytokines and antibodies [106]. As a proof of concept, vaccination with HCV-NS3-expressing DCs (NS3-DCs) in mice played a predisposing role in T-cell activation, cross-presenting capability of DCs in the draining lymph nodes, and clearance of NS3-positive hepatocytes from the livers [107]. DC vaccines, particularly when reinforced by interactions with other immune cells, hold promise for enhancing protective immunity against HCV [108].
Human CMV
The prevalent human CMV, also known as human herpesvirus 5 (HHV-5), is characterized by a double-stranded linear DNA genome of approximately 235 kb encoding over 200 genes and is implicated in various exoderm-derived malignancies [109,110,111]. A previous consensus has been reached on the coexistence of CMV and immunocompromised hosts, but the mechanism has not been fully elucidated [112]. More concerningly, CMV can increase cellular proliferation, angiogenesis, and immune evasion, thus enabling several hallmarks of cancer. Intriguingly, anti-CMV VST cells accumulate in extremely high numbers and serve as “bystanders” of the tumor. To harness CMV-specific immunity against malignancies, diverse strategies have been devised to redirect VSTs towards eradicating cancerous cells (Fig. 2d; Table 5) [113].
CAR-redirected CMV-specific cytotoxic T-cells (CMV-CAR-T)
The immunodominant CMV antigens, namely the pp65, IE1, and IE2 proteins, evoke physiological CMV-specific T-cells [114]. These T-cells can be isolated and/or reinvigorated using ex vivo CMV-peptide stimulation prior to CAR transduction, followed by in vivo expansion through a CMV vaccine boost [115,116,117]. CD19-CAR-T cell therapy faces limitations such as inadequate engraftment, differentiation, exhaustion, prolonged B cell aplasia, and increased susceptibility to CMV infections [118]. In contrast, CMV-CD19-CAR-T cells integrate anti-CD19 effector functions with potent anti-CMV activity, exhibiting superior proliferation, survival, and in vivo antitumor efficacy compared to conventional CD19-CAR-T cells [115]. Moreover, in a phase I dose-escalation trial of progressive glioblastoma (GBM) patients without prior lymphodepletion, Ahmed et al. evaluated the feasibility and safety of CMV-Her2-CAR-T cells and reported a promising median overall survival (OS) of 11.1 months from the first T-cell infusion and 24.5 months from diagnosis [119]. Overall, compared to generic CAR-T cells, CMV-CAR-T cells have shown superior proliferation, survival, and in vivo antitumor efficacy. They are well tolerated with only minor adverse events (Table 5).
TCR-engineered CMV-specific cytotoxic T-cells (CMV-TCR-T)
To prevent CMV reactivation after haploidentical peripheral blood SCT (PBSCT), a phase I clinical trial assessed the safety and efficacy of CMV-specific T-cells engineered to recognize HLA-restricted peptides from the CMV pp65 protein [120]. Another clinical trial investigated ex vivo expanded CMV-specific T-cells in recurrent GBM patients and reported a median OS of 4.4–13.4 months and a median progression-free survival (PFS) of approximately 8.1 months. However, in vitro analysis did not reveal significant changes in CMV-specific T-cell polyfunctionality [121].
Manufacturing CMV-TCR-T cells appears to be more challenging than CMV-CAR-T cells due to the downregulation of endogenous TCR expression upon forced expression of the artificial TCR. Redirected by the minor histocompatibility antigens (HA), HA-TCR-transferred CMV-specific T (HA-CMV-TCR-T) cells exerted dually potent antileukemic as well as anti-CMV reactivity, showing comparable TCR-specific cytolytic activity to generic TCR-engineered T-cells [122]. Notwithstanding, a follow-up study disclosed that repetitive stimulation skews CMV-TCR-T cells to predominantly express the triggered TCR [123]. In a phase I clinical trial, CMV-TCR-T cells were safely infused into 5/9 patients, but the overall efficacy of this treatment approach was too low to warrant further clinical development [124].
CMV-specific CAR-T cells
At the early stage of the CMV replication cycle, CAR-T (gB-CAR-T) cells directed against glycoprotein B (gB) accessible on the surface of infected cells can mediate antiviral effector functions, such as cytokine production and cytolysis [125]. However, a set of viral antiapoptotic factors directly abrogate T-cell cytotoxicity at later stages of the replication cycle. These gB-CAR-T cells were not tested in vivo because of the low degree of sequence similarity of gB protein between murine and human CMV, thus, recombinant mouse CMV expressing human CMV-gB is obligatory.
CMV-specific DC vaccines
A multiperformance recombinant adenovirus coexpressing the CMV immediate early gene (CMV-IE) enables the selective infection of DCs in vivo (CMV-IE-DCs), which induce T-cell activation to kill cancerous cells and prolong survival in CMV-IE-implanted murine glioma models [126]. Owing to the attenuated ability of CMV-specific T-cells in patients to generate multiple cytokines and chemokines, a pilot trial in which 22 patients with GBM received CMV pp65 RNA-loaded-DCs to augment the polyfunctionality of CMV pp65-specific T-cells revealed that polyfunctional T-cell responses are potential biomarkers for effective antitumor immunotherapy [116]. Encouragingly, three separate clinical trials have demonstrated that DC vaccines targeting the CMV pp65 protein (CMV-pp65-DCs) confer long-term survival benefits to nearly 1/3 of GBM patients, showing no tumor recurrence five years posttreatment [127]. Enhanced insights into tumor etiology and immune principles underscore the unique advantages of virus-targeted DC vaccines in specific tumor immunotherapies.
EBV
EBV belongs to human herpesvirus 4 (HHV-4) of the herpesvirus family and features a 175 kb double-stranded DNA genome encoding over 85 proteins. EBV enters epithelial, B, NK/T cells through a variety of membrane proteins, including gp350, gB, gH, gL and gp42 [128]. EBV has been definitively linked to a variety of lymphoid and epithelial cell malignancies, including B/T/NK cell lymphomas, nasopharyngeal carcinoma (NPC), gastric carcinoma and lung carcinoma due to the immune cell exhaustion and dysregulation [129, 130]. Upon primary infection, the immune-evasive EBV establishes latency and allows the viral genome to persist in the lymphatic system by driving the expansion of infected B cells [131]. EBV-infected B cells selectively express latent viral proteins: EBV nuclear antigen (EBNA) and latent membrane proteins (LMPs). As signaling proteins, LMPs promote the overexpression of some TAAs in B cells and upregulate costimulatory ligands to jointly activate T-cells [132]. As importantly, continuous EBNA1 expression is crucial for maintaining EBV genome replication in EBV-positive tumors, whereas EBNA3 is upregulated in EBV-induced lymphoma and can induce potent anti-EBV-specific CTLs. In addition, compared with those in the control lymphocytes of healthy individuals, EBNA1 and EBNA3 mRNA levels in EBV-induced lymphoma cells are increased by thousands of folds [133]. Accordingly, these latent EBV proteins are viable targets for cellular immunotherapies to clear EBV-infected targets (Fig. 2e; Table 6).
EBV-specific CTLs
EBV-specific CTLs face challenges due to EBV’s variable viral gene expression and multiple evasion mechanisms, complicating epitope selection. During the latent phase, EBV-specific CTLs are often infrequent, relatively immature, and anergic, potentially allowing tumor cells to evade immune surveillance. Previous studies have demonstrated that expanding LMP1/EBNA1-specific CTLs by coculturing with irradiated autologous PBMCs infected with an adenoviral vector encoding EBNA1 and multiple CTL epitopes from LMP1 and LMP2 (AdE1-LMPpoly), followed by reinfusion into EBV-positive recurrent and metastatic NPC patients, effectively controlled tumor progression with a median OS of 17.2 months [134]. However, preparative lymphodepleting chemotherapy prior to administering higher doses of EBV-specific CTLs did not improve clinical outcomes in patients with EBV-associated NPC [135]. In addition, LMP specific CTL was expanded using autologous DCs and EBV-transformed B-lymphoblastoid cell lines transduced with an adenoviral vector expressing LMP, which could induce durable CR in lymphoma patients at a median of 3.1 years after CTL infusion. Within 2 months after CTL infusion, epitope spread can be detected in patients who achieve clinical responses [136]. To enrich BARF1-specific CTLs for NPC treatment, EBV lytic cycle inducers can be used to upregulate the BARF1 oncogene in LCLs to promote more pronounced immunogenic properties [137, 138], suggesting new strategies to bolster EBV-targeting immunotherapy.
Initial preparation of EBV-specific CTLs involved stimulating PBMCs with autologous EBV-transformed LCLs, followed by transduction with E1-deficient adenovirus. These E1-transgenic CTLs released oncolytic adenovirus at tumor sites, leading to tumor regression upon exposure to HLA-matched, EBV-infected cells [139]. Recent research highlights the potential of ectopically expressed LMP1 in tumor B cells to prime autologous CD4+ T-cells (LMP1-CD4+ T) against a wide array of endogenous tumor antigens, including TAAs and neoantigens, suggesting efficient treatment for B cell malignancies [132]. These groundbreaking studies underscore the necessity of reevaluating conventional paradigms in both viral and tumor immunity.
EBV-specific DC vaccines
Although autologous DCs transduced with an adenovirus encoding truncated LMP1 (ΔLMP1) and full-length LMP2 (Ad-ΔLMP1-LMP2-DCs) enable to activate LMP1/2-specific T-cells in vitro, no increase in the frequency of peripheral LMP1/2-specific T- cells was detected in advanced NPC patients. Meanwhile, they induced delayed-type hypersensitivity responses but did not result in significant toxicity [140]. Considering its limited efficacy, future research should prioritize the administration of more potent DC vaccines to patients with lower tumor burdens. In a pilot study of 29 subjects, intradermal injection of LMP2-DCs using an adenovirus expressing LMP2 (Ad-LMP2) achieved a five-year survival rate of 94.4% in NPC responders, indicating enhanced responses to LMP2 peptide pools [141].
EBV specific CAR-T cells
Compared to CTL treatment regimens, the development of EBV-specific CAR-T is somewhat slower. CAR-T cells engineered with the scFv specific to the extracellular domain of LMP1 (LMP1-CAR-T) were activated in co-culture with LMP1-overexpressing NPC cells, leading to production of IFNɣ and IL-2. Intra-tumoral injection of LMP-CAR-T cells in a xenograft mouse model reduced tumor growth [142]. Moreover, a clinical trial is currently underway (NCT02980315) to evaluate LMP1-CAR-T cells for treating EBV-associated malignant tumors.
The lytic envelope gp350 is prominently expressed on the surface of cells during EBV lytic reactivation and persists in subsets of latently infected cells. A proof-of-concept preclinical study revealed that gp350-targeting CAR-T cells (gp350-CAR-T) exerted cytotoxic effects against EBV-positive tumor cells and hindered EBV-associated lymphoproliferation and lymphomagenesis in a fully humanized mouse model [143, 144]. However, using TCR alpha chain (TRAC) locus-knock-in, off-the-shelf edited gp350-CAR-T cells showed limited efficacy against lymphoma due to weak and variable gp350 expression [145], highlighting EBV’s immune evasion mechanisms that can affect CAR-T cell characteristics and efficacy.
EBV-specific TCR-T cells
The TCR specific to LMP1 (LMP1-TCR) provoked high levels of cytokine secretion and cytolytic activity, displaying explosive ex vivo proliferation upon antigen activation, and inhibited tumor growth in a xenogeneic model [146]. Ongoing efforts aim to generate more robust EBV TCRs by incorporating a CD28 domain preceding CD3, which augments antigen-specific IFNγ production without compromising the cytotoxic response [147]. Clinical trials investigating LMP2-specific TCR-T cells are ongoing (NCT04509726, NCT03925896). Despite promising results, the effectiveness of adoptive immunotherapy for EBV-associated cancers remains constrained by limited targetable EBV antigens and their suboptimal immunogenicity.
HIV-1
HIV-1 latent reservoirs are established days after infection and persist through clonal expansion of infected cells. Individuals living with HIV-1 face heightened risks of developing T-cell lymphoma and B-cell non-Hodgkin’s lymphoma (B-NHL), predominantly diffuse large B-cell lymphoma (DLBCL) and Burkitt’s lymphoma [148]. Pathogenesis studies highlight multifaceted mechanisms encompassing oncogenic proteins, immune system dysregulation, genetic predisposition, and other factors. Despite antiretroviral therapy (ART) effectively suppressing active viral replication, it fails to eliminate integrated latent viruses, necessitating lifelong treatment. Strategies to target HIV-1 latent reservoirs and associated lymphomas propose cytolytic immunotherapies as adjunctive to ART (Table 7; Fig. 2f).
HIV-1-specific CAR-T
The huge success of CAR-T therapy for B cell leukemias is rooted in pioneering preclinical and clinical study of HIV-1 infection. Furthermore, CAR-T cell has been recommended for the clinical therapy for HIV-1-positive lymphoma patients [149, 150]. In a randomised phase II clinical trial, first-generation CAR-T cells using CD4 ectodomain (CD4-CAR-T) to target the HIV-1 gp120 expressed on the surface of HIV-1-infected cells, noted a trend toward viral-load rebound and long-term engraftment in patients [151]. Given that CD4 and CCR5 are primary coreceptors of HIV-1 infection, CD4-CAR-T cells are susceptible to HIV-1 infection. For that reason, broadly neutralizing antibodies (bNAbs) against HIV-1 are engineered in the CAR construct (bNAb-derived CAR-T) cells showed a higher neutralizing capacity for different HIV-1 strains and circumvented HIV-1 infection [152]. Furthermore, bNAb-derived CAR-T cells with the deletion of CCR5 exhibited superior viral replication control compared to counterparts lacking this modification [153]. Nevertheless, emergence of resistant viral variants through spontaneous mutations poses a challenge to sustained efficacy, necessitating ongoing refinement. Innovative approaches like DuoCAR-T cells, targeting multiple binding sites on gp120 and the extracellular region of gp41, exhibited promising efficacy in eliminating HIV-1 in preclinical models of humanized mice with intrasplenic infection, presenting a multifaceted strategy against globally prevalent HIV-1 strains [154, 155].
Other HIV-1-specific ACTs
Anti-HIV-1 TCR-T cells manifested robust, antigen-specific polyfunctional cytokine profiles upon encountering antigens, but ineffectively controlled HIV-1. Conversely, CAR-T cells demonstrated accelerated recognition and elimination of HIV-1-infected targets relative to TCR-T cells, attributed to their ability to activate Caspase 3 and induce apoptosis in HIV-1-infected cells [156]. Therefore, it is hypothesized that the speed of target recognition and killing determines the efficacy of engineered T-cell therapies for infectious HIV-1.
Therapeutic DC-based vaccines pulsed with heat inactivated autologous HIV-1 (HIV-1-DC) have shown feasibility, safety, and well-tolerated outcomes in clinical settings [157]. Additionally, DCs electroporated with mRNA encoding Tat, Rev, and Nef (DC-TRN) significantly modulated NK cell and HIV-1-specific T-cell responses, leading to substantial reductions in plasma HIV-1 viral loads following interruption of antiretroviral therapy [158, 159]. These findings underscore the potential of DC-based approaches in augmenting immune responses crucial for controlling HIV-1 infection and HIV-1-defined cancers.
Other oncoviruses in viral malignancies
KSHV is etiologically linked to Kaposi’s sarcoma and primary effusion lymphoma, where both latency and lytic reactivation phases contribute to the pathogenesis of KSHV-associated malignancies [10, 160]. Notably, KSHV’s immune evasion strategies, mediated by genes like KSHV K3 and K5 encoding membrane-tethered E3 ubiquitin ligases, interfere with MHC expression, thereby evading immune surveillance by T and NK cells. This evasion mechanism could be exploited in the development of off-the-shelf allogeneic CAR-T cells. Incorporating K3 or K5 into CAR constructs has been shown to decrease the recognition and cytotoxicity against allogeneic T-cells in both culture and animal models [161].
Akin to MCPV, the elevated prevalence and viral load of polyomavirus JC (JCPV) within tumor tissues strongly suggest an active role in tumorigenesis rather than a bystander effect [162,163,164]. Furthermore, some lymphomas are characterized as virus-associated cancers due to the high incidence of viruses such as HIV-1, EBV, KSHV, HCV, HBV, and others, all of which exert pathogenic effects [90, 165]. Cooperative interactions between different oncoviruses represent an additional contributory mechanism in viral malignancies [165].
Conclusions and perspective
Preclinical and clinical studies have sought to utilize a flood of innovation ACTs for the prophylaxis and treatment of virus infection in both refractory and advanced malignancies [63, 166,167,168]. Capitalizing on the etiological link between viral malignancies and oncoviruses, we have summarized the relevant literature on the use of virus-specific ACTs to avoid or ablate viral malignances, and this information may also provide guidance for the selection of effective oncovirus-encoded antigens (Fig. 2a-f). This therapeutic approach is often combined with vaccinations, immune checkpoint inhibitors, systemic aldesleukin, virotherapy, and support by organ transplantation. Notably, virus-specific ACT mediated antitumor effects were observed even in heavily pretreated patients. These immune antitumor effects may be even more clinically evident when used as a first-line treatment at the early stage of virus infection, since an intense immunosuppressive TME that is typically encountered in refractory cancer patients may not be present.
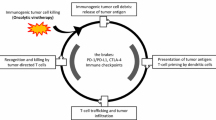
The bottleneck of ACTs for treating viral malignancies lies in several factors, including inadequate expansion and persistence of adoptive cells, MHC downregulation, suppressive TME, and targetable viral antigen level. Traditional strategies for solid tumor treatment aimed at increasing the trafficking, infiltration, and persistence of highly active adoptive cells are also applicable to the treatment of viral malignancies. Specially T-cells can be engineered with costimulatory signaling, immune checkpoint inhibitors, CMV/EBV TCR coexpression, and tissue homing ligands, which have demonstrated several advantages over the prototype, including enhanced expansion, persistence, antiviral activities. One major challenge is effectiveness of this therapy in recognizing and targeting infected cells. This is largely dependent on the ability of the transferred T-cells to interact with MHC molecules presenting viral antigens on the surface of infected cells. Another crucial issue is the TME within the infected tissue, which can create an inhibitory environment that hinders the function of the transferred cells. This may include factors such as immune suppressive cells, cytokines, and a lack of adequate nutrients for T-cells to proliferate and function optimally. Furthermore, the availability and selection of appropriate viral antigens for targeting also play a significant role in ACT success. In addition, an important direction for future research involves targeting multiple highly conserved sites of more than one viral antigen and utilizing a variety of therapeutic targets to overcome the viral escape mechanisms. Identifying highly immunogenic and conserved antigens that elicit a strong T-cell response is essential for effectively clearing the viral infection. Overall, overcoming these obstacles requires a comprehensive understanding of the immune response to viral infections and the development of strategies to optimize the function of adoptive cells in the context of the complex TME. Furthermore, investigating the optimal timing for intervening in the progression from viral infection to chronic inflammation to cancer development is crucial. Early intervention strategies to prevent or delay the carcinogenic process represent a significant area for further exploration and discussion in the field of cellular immunotherapy for viral infections. In general, ACTs can target viral antigens and tumor-specific markers, and provide potent immune responses against viral infections and their associated malignancies. More importantly, we need to determine which specific scenarios can be administrated by certain form of ACTs.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- ACTs:
-
Adoptive cell therapies
- alloSCT:
-
Allogeneic after stem cell transplantation
- ART:
-
Antiretroviral therapy
- B-NHL:
-
B-cell non-Hodgkin’s lymphoma
- CAR:
-
Chimeric antigen receptor
- CHB:
-
Chronic HBV
- CMV-IE:
-
CMV immediate early gene
- CR:
-
Complete remission
- cccDNA:
-
Covalently closed circular DNA
- CMV:
-
Cytomegalovirus
- DC:
-
Dendritic cell
- DLBCL:
-
Diffuse large B-cell lymphoma
- ER:
-
Endoplasmic reticulum
- EBV:
-
Epstein-Barr virus
- EBNA:
-
Epstein-Barr virus nuclear antigens
- GBM:
-
Glioblastoma
- gB:
-
Glycoprotein B
- HBcAg:
-
HBV core antigen
- HBVsvp:
-
HBV subviral particles
- HBxAg:
-
HBV x antige
- HNC:
-
Head and neck cancer
- HBsAg:
-
Hepatitis B surface antigen
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HCC:
-
Hepatocellular carcinoma
- HA:
-
Histocompatibility antigens
- HHV-4:
-
Human herpesvirus 4
- HHV-5:
-
Human herpesvirus 5
- HIV-1:
-
Human immunodeficiency virus type 1
- HPV:
-
Human papilloma virus
- hTERT:
-
Human telomerase reverse transcriptase
- IARC:
-
International Agency for Research on Cancer
- KSHV:
-
Kaposi’s sarcoma-associated herpesvirus
- LMPs:
-
Latent membrane proteins
- LCLs:
-
Lymphoblastic cell lines
- MHC:
-
Major histocompatibility complex
- MCPV:
-
Merkel cell polyomavirus
- NPC:
-
Nasopharyngeal carcinoma
- NK:
-
Natural killer
- ORR:
-
Objective response rate
- ORFs:
-
Open reading frames
- OPC:
-
Oropharyngeal cancer
- OS:
-
Overall survival
- PDX:
-
Patient-derived xenograft
- PBSCT:
-
Peripheral blood SCT
- JCPV:
-
Polyomavirus JC
- pgRNA:
-
Pregenomic RNA
- PFS:
-
Progression-free survival
- rcDNA:
-
Relaxed circular DNA
- +ssRNA:
-
Single positive-sense RNA
- scFvs:
-
Single-chain antibodies
- TCR:
-
T cell receptor
- TRAC:
-
TCR alpha chain
- TAA:
-
Tumor associated antigen
- TIL:
-
Tumor-infiltrating lymphocyte
- VST:
-
Virus-specific T cell
References
Tayyar R, Ho D. Herpes Simplex Virus and Varicella Zoster Virus infections in Cancer patients. Viruses. 2023;15(2).
Jankovic M, Knezevic T, Tomic A, Milicevic O, Jovanovic T, Djunic I et al. Human cytomegalovirus oncoprotection across diverse populations, Tumor histologies, and Age groups: the relevance for prospective Vaccinal Therapy. Int J Mol Sci. 2024;25(7).
Guyon J, Haidar Ahmad S, El Baba R, Le Quang M, Bikfalvi A, Daubon T et al. Generation of glioblastoma in mice engrafted with human cytomegalovirus-infected astrocytes. Cancer Gene Ther. 2024.
El Baba R, Pasquereau S, Haidar Ahmad S, Monnien F, Abad M, Bibeau F, et al. EZH2-Myc driven glioblastoma elicited by cytomegalovirus infection of human astrocytes. Oncogene. 2023;42(24):2031–45.
Nehme Z, Pasquereau S, Haidar Ahmad S, El Baba R, Herbein G. Polyploid giant cancer cells, EZH2 and myc upregulation in mammary epithelial cells infected with high-risk human cytomegalovirus. EBioMedicine. 2022;80:104056.
Krenzlin H, Behera P, Lorenz V, Passaro C, Zdioruk M, Nowicki MO, et al. Cytomegalovirus promotes murine glioblastoma growth via pericyte recruitment and angiogenesis. J Clin Investig. 2019;129(4):1671–83.
Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60.
Jasinski-Bergner S, Mandelboim O, Seliger B. Molecular mechanisms of human herpes viruses inferring with host immune surveillance. J Immunother Cancer. 2020;8(2).
Kaiserlian D, Dubois B. Dendritic cells and viral immunity: friends or foes? Semin Immunol. 2001;13(5):303–10.
Galati L, Chiantore MV, Marinaro M, Di Bonito P. Human oncogenic viruses: characteristics and Prevention Strategies-lessons learned from human papillomaviruses. Viruses. 2024;16(3).
Qi J, Ding C, Jiang X, Gao Y. Advances in developing CAR T-Cell therapy for HIV Cure. Front Immunol. 2020;11:361.
Ferreira DA, Tayyar Y, Idris A, McMillan NAJ. A hit-and-run affair - A possible link for cancer progression in virally driven cancers. Biochim et Biophys acta Reviews cancer. 2021;1875(1):188476.
Contreras A, Sánchez SA, Rodríguez-Medina C, Botero JE. The role and impact of viruses on cancer development. Periodontology 2000. 2024.
Seif M, Einsele H, Löffler J. CAR T cells beyond Cancer: hope for Immunomodulatory Therapy of Infectious diseases. Front Immunol. 2019;10:2711.
Schlecht NF, Kulaga S, Robitaille J, Ferreira S, Santos M, Miyamura RA, et al. Persistent human papillomavirus infection as a predictor of cervical intraepithelial neoplasia. JAMA. 2001;286(24):3106–14.
Ferber MJ, Montoya DP, Yu C, Aderca I, McGee A, Thorland EC, et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene. 2003;22(24):3813–20.
Xu S, Shi C, Zhou R, Han Y, Li N, Qu C, et al. Mapping the landscape of HPV integration and characterising virus and host genome interactions in HPV-positive oropharyngeal squamous cell carcinoma. Clin Translational Med. 2024;14(1):e1556.
Tian R, Huang Z, Li L, Yuan J, Zhang Q, Meng L, et al. HPV integration generates a cellular super-enhancer which functions as ecDNA to regulate genome-wide transcription. Nucleic Acids Res. 2023;51(9):4237–51.
Yu L, Majerciak V, Lobanov A, Mirza S, Band V, Liu H, et al. HPV oncogenes expressed from only one of multiple integrated HPV DNA copies drive clonal cell expansion in cervical cancer. mBio. 2024;15(5):e0072924.
Steinbach A, Riemer AB. Immune evasion mechanisms of human papillomavirus: an update. Int J Cancer. 2018;142(2):224–9.
Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018;25(1):114–32.
Boulet G, Horvath C, Vanden Broeck D, Sahebali S, Bogers J. Human papillomavirus: E6 and E7 oncogenes. Int J Biochem Cell Biol. 2007;39(11):2006–11.
Wang JCK, Baddock HT, Mafi A, Foe IT, Bratkowski M, Lin TY, et al. Structure of the p53 degradation complex from HPV16. Nat Commun. 2024;15(1):1842.
Skelin J, Luk HY, Butorac D, Boon SS, Tomaić V. The effects of HPV oncoproteins on host communication networks: therapeutic connotations. J Med Virol. 2023;95(12):e29315.
Yang A, Farmer E, Wu TC, Hung CF. Perspectives for therapeutic HPV vaccine development. J Biomed Sci. 2016;23(1):75.
Paolini F, Amici C, Carosi M, Bonomo C, Di Bonito P, Venuti A, et al. Intrabodies targeting human papillomavirus 16 E6 and E7 oncoproteins for therapy of established HPV-associated tumors. J Experimental Clin cancer Research: CR. 2021;40(1):37.
Ferrantelli F, Arenaccio C, Manfredi F, Olivetta E, Chiozzini C, Leone P, et al. The intracellular delivery of Anti-HPV16 E7 scFvs through Engineered Extracellular vesicles inhibits the proliferation of HPV-Infected cells. Int J Nanomed. 2019;14:8755–68.
Accardi L, Paolini F, Mandarino A, Percario Z, Di Bonito P, Di Carlo V, et al. In vivo antitumor effect of an intracellular single-chain antibody fragment against the E7 oncoprotein of human papillomavirus 16. Int J Cancer. 2014;134(11):2742–7.
Norberg SM, Hinrichs CS. Engineered T cell therapy for viral and non-viral epithelial cancers. Cancer Cell. 2023;41(1):58–69.
Jin BY, Campbell TE, Draper LM, Stevanović S, Weissbrich B, Yu Z et al. Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model. JCI Insight. 2018;3(8).
Nagarsheth NB, Norberg SM, Sinkoe AL, Adhikary S, Meyer TJ, Lack JB, et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med. 2021;27(3):419–25.
Jiang J, Xia M, Zhang L, Chen X, Zhao Y, Zeng C, et al. Rapid generation of genetically engineered T cells for the treatment of virus-related cancers. Cancer Sci. 2022;113(11):3686–97.
Draper LM, Kwong ML, Gros A, Stevanović S, Tran E, Kerkar S, et al. Targeting of HPV-16 + epithelial Cancer cells by TCR Gene Engineered T Cells Directed against E6. Clin cancer Research: Official J Am Association Cancer Res. 2015;21(19):4431–9.
Doran SL, Stevanović S, Adhikary S, Gartner JJ, Jia L, Kwong MLM, et al. T-Cell receptor gene therapy for Human Papillomavirus-Associated Epithelial cancers: a first-in-Human, phase I/II study. J Clin Oncology: Official J Am Soc Clin Oncol. 2019;37(30):2759–68.
Stevanović S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Sci (New York NY). 2017;356(6334):200–5.
de Vos PJ, Heusinkveld M, Ramwadhdoebe TH, Löwik MJ, van der Hulst JM, Goedemans R, et al. An unexpectedly large polyclonal repertoire of HPV-specific T cells is poised for action in patients with cervical cancer. Cancer Res. 2010;70(7):2707–17.
Santegoets SJ, Welters MJP, Schrikkema DS, Freriks MR, Kok H, Weissbrich B, et al. The common HLA class I-restricted tumor-infiltrating T cell response in HPV16-induced cancer. Cancer Immunol Immunotherapy: CII. 2023;72(6):1553–65.
Stevanović S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncology: Official J Am Soc Clin Oncol. 2015;33(14):1543–50.
Stevanović S, Helman SR, Wunderlich JR, Langhan MM, Doran SL, Kwong MLM, et al. A phase II study of Tumor-infiltrating lymphocyte therapy for human papillomavirus-associated epithelial cancers. Clin cancer Research: Official J Am Association Cancer Res. 2019;25(5):1486–93.
Silva AJD, Moura IA, Gama M, Leal LRS, Pinho SS, Espinoza BCF et al. Advancing immunotherapies for HPV-Related cancers: exploring Novel vaccine strategies and the influence of Tumor Microenvironment. Vaccines. 2023;11(8).
Li X, Wang H, Lai W, Liao J, Mo W, Huang K et al. Prevention and treatment of HPV-related cancer through a mRNA vaccine expressing APC-targeting antigen. Immunology. 2024.
Wang Z, Wang Q, Tao Y, Chen J, Yuan Z, Wang P. Characterization of immune microenvironment in patients with HPV-positive and negative head and neck cancer. Sci data. 2023;10(1):694.
Bauer M, Wagner H, Lipford GB. HPV type 16 protein E7 HLA-A2 binding peptides are immunogenic but not processed and presented. Immunol Lett. 2000;71(1):55–9.
Zeng M, Zhang W, Li Y, Yu L. Harnessing adenovirus in cancer immunotherapy: evoking cellular immunity and targeting delivery in cell-specific manner. Biomark Res. 2024;12(1):36.
Nonn M, Schinz M, Zumbach K, Pawlita M, Schneider A, Dürst M, et al. Dendritic cell-based tumor vaccine for cervical cancer I: in vitro stimulation with recombinant protein-pulsed dendritic cells induces specific T cells to HPV16 E7 or HPV18 E7. J Cancer Res Clin Oncol. 2003;129(9):511–20.
Wang TL, Ling M, Shih IM, Pham T, Pai SI, Lu Z, et al. Intramuscular administration of E7-transfected dendritic cells generates the most potent E7-specific anti-tumor immunity. Gene Ther. 2000;7(9):726–33.
Chang EY, Chen CH, Ji H, Wang TL, Hung K, Lee BP, et al. Antigen-specific cancer immunotherapy using a GM-CSF secreting allogeneic tumor cell-based vaccine. Int J Cancer. 2000;86(5):725–30.
Mikysková R, Indrová M, Símová J, Jandlová T, Bieblová J, Jinoch P, et al. Treatment of minimal residual disease after surgery or chemotherapy in mice carrying HPV16-associated tumours: Cytokine and gene therapy with IL-2 and GM-CSF. Int J Oncol. 2004;24(1):161–7.
Mackova J, Kutinova L, Hainz P, Krystofova J, Sroller V, Otahal P, et al. Adjuvant effect of dendritic cells transduced with recombinant vaccinia virus expressing HPV16-E7 is inhibited by co-expression of IL12. Int J Oncol. 2004;24(6):1581–8.
Reinis M, Indrová M, Mendoza L, Mikysková R, Bieblová J, Bubeník J, et al. HPV16-associated tumours: therapy of surgical minimal residual disease with dendritic cell-based vaccines. Int J Oncol. 2004;25(4):1165–70.
Ferrara A, Nonn M, Sehr P, Schreckenberger C, Pawlita M, Dürst M, et al. Dendritic cell-based tumor vaccine for cervical cancer II: results of a clinical pilot study in 15 individual patients. J Cancer Res Clin Oncol. 2003;129(9):521–30.
Bellone S, El-Sahwi K, Cocco E, Casagrande F, Cargnelutti M, Palmieri M, et al. Human papillomavirus type 16 (HPV-16) virus-like particle L1-specific CD8 + cytotoxic T lymphocytes (CTLs) are equally effective as E7-specific CD8 + CTLs in killing autologous HPV-16-positive tumor cells in cervical cancer patients: implications for L1 dendritic cell-based therapeutic vaccines. J Virol. 2009;83(13):6779–89.
De la Paz G, Monroy-García A, Mora-García Mde L, Peña CG, Hernández-Montes J, Weiss-Steider B, et al. An HPV 16 L1-based chimeric human papilloma virus-like particles containing a string of epitopes produced in plants is able to elicit humoral and cytotoxic T-cell activity in mice. Virol J. 2009;6:2.
Pinto LA, Edwards J, Castle PE, Harro CD, Lowy DR, Schiller JT, et al. Cellular immune responses to human papillomavirus (HPV)-16 L1 in healthy volunteers immunized with recombinant HPV-16 L1 virus-like particles. J Infect Dis. 2003;188(2):327–38.
Xia Y, Stadler D, Lucifora J, Reisinger F, Webb D, Hösel M, et al. Interferon-γ and Tumor Necrosis Factor-α produced by T cells reduce the HBV persistence form, cccDNA, without Cytolysis. Gastroenterology. 2016;150(1):194–205.
Zhao LH, Liu X, Yan HX, Li WY, Zeng X, Yang Y, et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun. 2016;7:12992.
Qian Z, Liang J, Huang R, Song W, Ying J, Bi X, et al. HBV integrations reshaping genomic structures promote hepatocellular carcinoma. Gut. 2024;73(7):1169–82.
Jiang Y, Han QJ, Zhang J. Hepatocellular carcinoma: mechanisms of progression and immunotherapy. World J Gastroenterol. 2019;25(25):3151–67.
Lapierre P, Janelle V, Langlois MP, Tarrab E, Charpentier T, Lamarre A. Expression of viral Antigen by the liver leads to Chronic Infection through the Generation of Regulatory T Cells. Cell Mol Gastroenterol Hepatol. 2015;1(3):325–e411.
Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64(1 Suppl):S84–101.
Ringelhan M, Protzer U. Oncogenic potential of hepatitis B virus encoded proteins. Curr Opin Virol. 2015;14:109–15.
Lin X, Zuo S, Luo R, Li Y, Yu G, Zou Y, et al. HBX-induced miR-5188 impairs FOXO1 to stimulate β-catenin nuclear translocation and promotes tumor stemness in hepatocellular carcinoma. Theranostics. 2019;9(25):7583–98.
Petersen J, Thompson AJ, Levrero M. Aiming for cure in HBV and HDV infection. J Hepatol. 2016;65(4):835–48.
Gorelick FS, Shugrue C. Exiting the endoplasmic reticulum. Mol Cell Endocrinol. 2001;177(1–2):13–8.
Bohne F, Chmielewski M, Ebert G, Wiegmann K, Kürschner T, Schulze A, et al. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology. 2008;134(1):239–47.
Krebs K, Böttinger N, Huang LR, Chmielewski M, Arzberger S, Gasteiger G, et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145(2):456–65.
Festag MM, Festag J, Fräßle SP, Asen T, Sacherl J, Schreiber S, et al. Evaluation of a fully human, Hepatitis B Virus-Specific Chimeric Antigen Receptor in an Immunocompetent Mouse Model. Mol Therapy: J Am Soc Gene Therapy. 2019;27(5):947–59.
Kruse RL, Shum T, Tashiro H, Barzi M, Yi Z, Whitten-Bauer C, et al. HBsAg-redirected T cells exhibit antiviral activity in HBV-infected human liver chimeric mice. Cytotherapy. 2018;20(5):697–705.
Zou F, Tan J, Liu T, Liu B, Tang Y, Zhang H, et al. The CD39(+) HBV surface protein-targeted CAR-T and personalized tumor-reactive CD8(+) T cells exhibit potent anti-HCC activity. Mol Therapy: J Am Soc Gene Therapy. 2021;29(5):1794–807.
Banu N, Chia A, Ho ZZ, Garcia AT, Paravasivam K, Grotenbreg GM, et al. Building and optimizing a virus-specific T cell receptor library for targeted immunotherapy in viral infections. Sci Rep. 2014;4:4166.
Gehring AJ, Xue SA, Ho ZZ, Teoh D, Ruedl C, Chia A, et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J Hepatol. 2011;55(1):103–10.
Tan AT, Yang N, Lee Krishnamoorthy T, Oei V, Chua A, Zhao X, et al. Use of expression profiles of HBV-DNA Integrated into Genomes of Hepatocellular Carcinoma Cells to select T cells for Immunotherapy. Gastroenterology. 2019;156(6):1862–e769.
Wisskirchen K, Kah J, Malo A, Asen T, Volz T, Allweiss L, et al. T cell receptor grafting allows virological control of Hepatitis B virus infection. J Clin Investig. 2019;129(7):2932–45.
Koh S, Kah J, Tham CYL, Yang N, Ceccarello E, Chia A, et al. Nonlytic lymphocytes Engineered to Express Virus-Specific T-Cell receptors limit HBV infection by activating APOBEC3. Gastroenterology. 2018;155(1):180–e936.
Bertoletti A, Brunetto M, Maini MK, Bonino F, Qasim W, Stauss H. T cell receptor-therapy in HBV-related hepatocellularcarcinoma. Oncoimmunology. 2015;4(6):e1008354.
Koh S, Tan AT, Li L, Bertoletti A. Targeted Therapy of Hepatitis B Virus-Related Hepatocellular Carcinoma: Present and Future. Diseases (Basel, Switzerland). 2016;4(1).
Qasim W, Brunetto M, Gehring AJ, Xue SA, Schurich A, Khakpoor A, et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J Hepatol. 2015;62(2):486–91.
Meng F, Zhao J, Tan AT, Hu W, Wang SY, Jin J, et al. Immunotherapy of HBV-related advanced hepatocellular carcinoma with short-term HBV-specific TCR expressed T cells: results of dose escalation, phase I trial. Hep Intl. 2021;15(6):1402–12.
Tan AT, Bertoletti A. HBV-HCC treatment with mRNA electroporated HBV-TCR T cells. Immunotherapy Adv. 2022;2(1):ltab026.
Yang F, Zheng X, Koh S, Lu J, Cheng J, Li P, et al. Messenger RNA electroporated hepatitis B virus (HBV) antigen-specific T cell receptor (TCR) redirected T cell therapy is well-tolerated in patients with recurrent HBV-related hepatocellular carcinoma post-liver transplantation: results from a phase I trial. Hep Intl. 2023;17(4):850–9.
Tan AT, Meng F, Jin J, Zhang JY, Wang SY, Shi L, et al. Immunological alterations after immunotherapy with short lived HBV-TCR T cells associates with long-term treatment response in HBV-HCC. Hepatol Commun. 2022;6(4):841–54.
Sun K, Wang L, Zhang Y. Dendritic cell as therapeutic vaccines against tumors and its role in therapy for hepatocellular carcinoma. Cell Mol Immunol. 2006;3(3):197–203.
Martinet J, Leroy V, Dufeu-Duchesne T, Larrat S, Richard MJ, Zoulim F, et al. Plasmacytoid dendritic cells induce efficient stimulation of antiviral immunity in the context of chronic hepatitis B virus infection. Hepatology (Baltimore MD). 2012;56(5):1706–18.
Chen W, Shi M, Shi F, Mao Y, Tang Z, Zhang B, et al. HBcAg-pulsed dendritic cell vaccine induces Th1 polarization and production of hepatitis B virus-specific cytotoxic T lymphocytes. Hepatol Research: Official J Japan Soc Hepatol. 2009;39(4):355–65.
Akbar SM, Furukawa S, Horiike N, Abe M, Hiasa Y, Onji M. Safety and immunogenicity of hepatitis B surface antigen-pulsed dendritic cells in patients with chronic hepatitis B. J Viral Hepatitis. 2011;18(6):408–14.
Duan XZ, He HX, Zhuang H. Restoration in vitro of impaired T-cell responses in patients with chronic hepatitis B by autologous dendritic cells loaded with hepatitis B virus proteins (R2). J Gastroenterol Hepatol. 2006;21(6):970–6.
van Montfoort N, van der Aa E, van den Bosch A, Brouwers H, Vanwolleghem T, Janssen HLA, et al. Hepatitis B Virus Surface Antigen activates myeloid dendritic cells via a Soluble CD14-Dependent mechanism. J Virol. 2016;90(14):6187–99.
Farag MMS, Suef RA, Al-Toukhy GM, Selim MA, Elbahnasawy MA, El Sharkawy N, et al. HBVsvp-Pulsed dendritic cell immunotherapy induces Th1 polarization and Hepatitis B Virus-Specific cytotoxic T lymphocytes production. Infect drug Resist. 2020;13:2699–709.
Wang X, Bayer ME, Chen X, Fredrickson C, Cornforth AN, Liang G, et al. Phase I trial of active specific immunotherapy with autologous dendritic cells pulsed with autologous irradiated tumor stem cells in hepatitis B-positive patients with hepatocellular carcinoma. J Surg Oncol. 2015;111(7):862–7.
Zhang Y, Guo W, Zhan Z, Bai O. Carcinogenic mechanisms of virus-associated lymphoma. Front Immunol. 2024;15:1361009.
Machida K, Cheng KT, Sung VM, Shimodaira S, Lindsay KL, Levine AM, et al. Hepatitis C virus induces a mutator phenotype: enhanced mutations of immunoglobulin and protooncogenes. Proc Natl Acad Sci USA. 2004;101(12):4262–7.
Dai X, Guo Y, Hu Y, Bao X, Zhu X, Fu Q, et al. Immunotherapy for targeting cancer stem cells in hepatocellular carcinoma. Theranostics. 2021;11(7):3489–501.
Curtis MR, Epstein RL, Pei P, Linas BP, Ciaranello AL. Cost-effectiveness of strategies for treatment timing for perinatally acquired Hepatitis C Virus. JAMA Pediatr. 2024.
Bull RA, Leung P, Gaudieri S, Deshpande P, Cameron B, Walker M, et al. Transmitted/Founder viruses rapidly escape from CD8 + T cell responses in Acute Hepatitis C virus infection. J Virol. 2015;89(10):5478–90.
Manns MP, Buti M, Gane E, Pawlotsky JM, Razavi H, Terrault N, et al. Hepatitis C virus infection. Nat Reviews Disease Primers. 2017;3:17006.
Sautto G, Tarr AW, Mancini N, Clementi M. Structural and antigenic definition of hepatitis C virus E2 glycoprotein epitopes targeted by monoclonal antibodies. Clin Dev Immunol. 2013;2013:450963.
Sautto GA, Wisskirchen K, Clementi N, Castelli M, Diotti RA, Graf J, et al. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut. 2016;65(3):512–23.
Li HS, Wong NM, Tague E, Ngo JT, Khalil AS, Wong WW. High-performance multiplex drug-gated CAR circuits. Cancer Cell. 2022;40(11):1294–e3054.
Zhang J, Du B, Liu M. Let’s turn the CAR-T cells ON and OFF precisely. Cancer Cell. 2022;40(11):1264–6.
Cao W, Geng ZZ, Wang N, Pan Q, Guo S, Xu S et al. A Reversible Chemogenetic Switch for Chimeric Antigen Receptor T Cells. Angewandte Chemie (International ed in English). 2022;61(10):e202109550.
Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ, Vilches-Moure JG, et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell. 2022;185(10):1745–e6322.
Pasetto A, Frelin L, Aleman S, Holmström F, Brass A, Ahlén G, et al. TCR-redirected human T cells inhibit hepatitis C virus replication: hepatotoxic potential is linked to antigen specificity and functional avidity. J Immunol (Baltimore Md: 1950). 2012;189(9):4510–9.
Silva DN, Chrobok M, Rovesti G, Healy K, Wagner AK, Maravelia P, et al. Process development for adoptive cell therapy in Academia: A Pipeline for Clinical-Scale Manufacturing of multiple TCR-T cell products. Front Immunol. 2022;13:896242.
Balasiddaiah A, Davanian H, Aleman S, Pasetto A, Frelin L, Sällberg M et al. Hepatitis C virus-specific T cell receptor mRNA-Engineered Human T cells: impact of Antigen specificity on Functional Properties. J Virol. 2017;91(9).
Haga Y, Meyer K, Sung MMH, Reagan EK, Weissman D, Ray R. Hepatitis C virus modified sE2(F442NYT) as an antigen in candidate vaccine facilitates human immune cell activation. J Virol. 2024;98(1):e0180923.
Malaina I, Martinez L, Salcines-Cuevas D, Teran-Navarro H, Ocejo-Vinyals JG, Gonzalez-Lopez E, et al. Testing a vaccine candidate against Hepatitis C virus designed by combinatorial optimization. Sci Rep. 2023;13(1):21746.
Mekonnen ZA, Masavuli MG, Yu W, Gummow J, Whelan DM, Al-Delfi Z, et al. Enhanced T cell responses Induced by a necrotic dendritic cell vaccine, expressing HCV NS3. Front Microbiol. 2020;11:559105.
Han CL, Yan YC, Yan LJ, Meng GX, Yang CC, Liu H, et al. Efficacy and security of tumor vaccines for hepatocellular carcinoma: a systemic review and meta-analysis of the last 2 decades. J Cancer Res Clin Oncol. 2023;149(4):1425–41.
Jih J, Liu YT, Liu W, Zhou ZH. The incredible bulk: human cytomegalovirus tegument architectures uncovered by AI-empowered cryo-EM. Sci Adv. 2024;10(8):eadj1640.
Teo WH, Chen HP, Huang JC, Chan YJ. Human cytomegalovirus infection enhances cell proliferation, migration and upregulation of EMT markers in colorectal cancer-derived stem cell-like cells. Int J Oncol. 2017;51(5):1415–26.
Adelman JW, Rosas-Rogers S, Schumacher ML, Mokry RL, Terhune SS, Ebert AD. Human cytomegalovirus induces significant structural and functional changes in terminally differentiated human cortical neurons. mBio. 2023;14(6):e0225123.
van den Pol AN. Viral infections in the developing and mature brain. Trends Neurosci. 2006;29(7):398–406.
Britsch I, van Wijngaarden AP, Helfrich W. Applications of anti-cytomegalovirus T cells for Cancer (Immuno)Therapy. Cancers. 2023;15(15).
Daei Sorkhabi A, Sarkesh A, Saeedi H, Marofi F, Ghaebi M, Silvestris N, et al. The basis and advances in clinical application of cytomegalovirus-specific cytotoxic T cell immunotherapy for Glioblastoma Multiforme. Front Oncol. 2022;12:818447.
Wang X, Urak R, Walter M, Guan M, Han T, Vyas V et al. Large-scale manufacturing and characterization of CMV-CD19CAR T cells. J Immunother Cancer. 2022;10(1).
Reap EA, Suryadevara CM, Batich KA, Sanchez-Perez L, Archer GE, Schmittling RJ, et al. Dendritic cells enhance polyfunctionality of Adoptively Transferred T cells that target cytomegalovirus in Glioblastoma. Cancer Res. 2018;78(1):256–64.
Caruana I, Weber G, Ballard BC, Wood MS, Savoldo B, Dotti G. K562-Derived whole-cell vaccine enhances antitumor responses of CAR-Redirected virus-specific cytotoxic T lymphocytes in vivo. Clin cancer Research: Official J Am Association Cancer Res. 2015;21(13):2952–62.
Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. 2018;131(1):121–30.
Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: a phase 1 dose-escalation Trial. JAMA Oncol. 2017;3(8):1094–101.
Ma C, Chen P, Du J, Wang L, Lu N, Sun J et al. Adoptive transfer of CMV-specific TCR-T cells for the treatment of CMV infection after haploidentical hematopoietic stem cell transplantation. J Immunother Cancer. 2024;12(1).
Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014;74(13):3466–76.
Heemskerk MH, Hoogeboom M, Hagedoorn R, Kester MG, Willemze R, Falkenburg JH. Reprogramming of virus-specific T cells into leukemia-reactive T cells using T cell receptor gene transfer. J Exp Med. 2004;199(7):885–94.
van Loenen MM, Hagedoorn RS, Kester MG, Hoogeboom M, Willemze R, Falkenburg JH, et al. Kinetic preservation of dual specificity of coprogrammed minor histocompatibility antigen-reactive virus-specific T cells. Cancer Res. 2009;69(5):2034–41.
van Balen P, Jedema I, van Loenen MM, de Boer R, van Egmond HM, Hagedoorn RS, et al. HA-1H T-Cell receptor gene transfer to redirect Virus-Specific T Cells for Treatment of Hematological Malignancies after allogeneic stem cell transplantation: a phase 1 clinical study. Front Immunol. 2020;11:1804.
Full F, Lehner M, Thonn V, Goetz G, Scholz B, Kaufmann KB, et al. T cells engineered with a cytomegalovirus-specific chimeric immunoreceptor. J Virol. 2010;84(8):4083–8.
Kim JW, Kane JR, Panek WK, Young JS, Rashidi A, Yu D, et al. A dendritic cell-targeted Adenoviral Vector facilitates adaptive Immune Response Against Human Glioma Antigen (CMV-IE) and prolongs survival in a human glioma Tumor Model. Neurotherapeutics: J Am Soc Experimental Neurother. 2018;15(4):1127–38.
Batich KA, Mitchell DA, Healy P, Herndon JE 2nd, Sampson JH. Once, twice, Three Times a finding: reproducibility of dendritic cell vaccine trials targeting cytomegalovirus in Glioblastoma. Clin cancer Research: Official J Am Association Cancer Res. 2020;26(20):5297–303.
Damania B, Kenney SC, Raab-Traub N. Epstein-Barr virus: Biology and clinical disease. Cell. 2022;185(20):3652–70.
Lenoci D, Resteghini C, Serafini MS, Pistore F, Canevari S, Ma B, et al. Tumor molecular landscape of Epstein-Barr virus (EBV) related nasopharyngeal carcinoma in EBV-endemic and non-endemic areas: implications for improving treatment modalities. Translational Research: J Lab Clin Med. 2024;265:1–16.
Chen C, Wang C, Pang R, Liu H, Yin W, Chen J, et al. Comprehensive single-cell transcriptomic and proteomic analysis reveals NK cell exhaustion and unique tumor cell evolutionary trajectory in non-keratinizing nasopharyngeal carcinoma. J Translational Med. 2023;21(1):278.
Sausen DG, Bhutta MS, Gallo ES, Dahari H, Borenstein R. Stress-Induced Epstein-Barr Virus Reactivation. Biomolecules. 2021;11(9).
Choi IK, Wang Z, Ke Q, Hong M, Paul DW Jr., Fernandes SM, et al. Mechanism of EBV inducing anti-tumour immunity and its therapeutic use. Nature. 2021;590(7844):157–62.
Wojtak K, Perales-Puchalt A, Weiner DB. Novel synthetic DNA immunogens targeting latent expressed antigens of Epstein-Barr Virus Elicit Potent Cellular responses and inhibit Tumor Growth. Vaccines. 2019;7(2).
Smith C, Tsang J, Beagley L, Chua D, Lee V, Li V, et al. Effective treatment of metastatic forms of Epstein-Barr virus-associated nasopharyngeal carcinoma with a novel adenovirus-based adoptive immunotherapy. Cancer Res. 2012;72(5):1116–25.
Secondino S, Zecca M, Licitra L, Gurrado A, Schiavetto I, Bossi P, et al. T-cell therapy for EBV-associated nasopharyngeal carcinoma: preparative lymphodepleting chemotherapy does not improve clinical results. Annals Oncology: Official J Eur Soc Med Oncol. 2012;23(2):435–41.
Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncology: Official J Am Soc Clin Oncol. 2014;32(8):798–808.
Faè DA, Martorelli D, Mastorci K, Muraro E, Dal Col J, Franchin G, et al. Broadening specificity and enhancing cytotoxicity of Adoptive T Cells for Nasopharyngeal Carcinoma Immunotherapy. Cancer Immunol Res. 2016;4(5):431–40.
Martorelli D, Houali K, Caggiari L, Vaccher E, Barzan L, Franchin G, et al. Spontaneous T cell responses to Epstein-Barr virus-encoded BARF1 protein and derived peptides in patients with nasopharyngeal carcinoma: bases for improved immunotherapy. Int J Cancer. 2008;123(5):1100–7.
Yotnda P, Savoldo B, Charlet-Berguerand N, Rooney C, Brenner M. Targeted delivery of adenoviral vectors by cytotoxic T cells. Blood. 2004;104(8):2272–80.
Chia WK, Wang WW, Teo M, Tai WM, Lim WT, Tan EH, et al. A phase II study evaluating the safety and efficacy of an adenovirus-∆LMP1-LMP2 transduced dendritic cell vaccine in patients with advanced metastatic nasopharyngeal carcinoma. Annals Oncology: Official J Eur Soc Med Oncol. 2012;23(4):997–1005.
Zeng Y, Si YF, Lan GP, Wang Z, Zhou L, Tang MZ, et al. LMP2-DC Vaccine elicits specific EBV-LMP2 response to effectively improve immunotherapy in patients with nasopharyngeal Cancer. Biomed Environ Sci: BES. 2020;33(11):849–56.
Tang X, Zhou Y, Li W, Tang Q, Chen R, Zhu J, et al. T cells expressing a LMP1-specific chimeric antigen receptor mediate antitumor effects against LMP1-positive nasopharyngeal carcinoma cells in vitro and in vivo. J Biomedical Res. 2014;28(6):468–75.
Slabik C, Kalbarczyk M, Danisch S, Zeidler R, Klawonn F, Volk V, et al. CAR-T cells targeting Epstein-Barr Virus gp350 validated in a Humanized Mouse Model of EBV infection and lymphoproliferative disease. Mol Therapy Oncolytics. 2020;18:504–24.
Zhang X, Wang T, Zhu X, Lu Y, Li M, Huang Z, et al. GMP development and preclinical validation of CAR-T cells targeting a lytic EBV antigen for therapy of EBV-associated malignancies. Front Immunol. 2023;14:1103695.
Braun T, Pruene A, Darguzyte M, Vom Stein AF, Nguyen PH, Wagner DL, et al. Non-viral TRAC-knocked-in CD19(KI)CAR-T and gp350(KI)CAR-T cells tested against Burkitt lymphomas with type 1 or 2 EBV infection: in vivo cellular dynamics and potency. Front Immunol. 2023;14:1086433.
Cho HI, Kim UH, Shin AR, Won JN, Lee HJ, Sohn HJ, et al. A novel Epstein-Barr virus-latent membrane protein-1-specific T-cell receptor for TCR gene therapy. Br J Cancer. 2018;118(4):534–45.
Schaft N, Lankiewicz B, Drexhage J, Berrevoets C, Moss DJ, Levitsky V, et al. T cell re-targeting to EBV antigens following TCR gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFNgamma production. Int Immunol. 2006;18(4):591–601.
Hübel K. The changing Landscape of Lymphoma Associated with HIV infection. Curr Oncol Rep. 2020;22(11):111.
Hattenhauer ST, Mispelbaum R, Hentrich M, Boesecke C, Monin MB. Enabling CAR T-cell therapies for HIV-positive lymphoma patients - a call for action. HIV Med. 2023;24(9):957–64.
Uldrick TS, Ison G, Rudek MA, Noy A, Schwartz K, Bruinooge S, et al. Modernizing clinical Trial Eligibility Criteria: recommendations of the American Society of Clinical Oncology-friends of Cancer Research HIV Working Group. J Clin Oncology: Official J Am Soc Clin Oncol. 2017;35(33):3774–80.
Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Therapy: J Am Soc Gene Therapy. 2002;5(6):788–97.
Liu B, Zhang W, Xia B, Jing S, Du Y, Zou F et al. Broadly neutralizing antibody-derived CAR T cells reduce viral reservoir in individuals infected with HIV-1. J Clin Investig. 2021;131(19).
Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, et al. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol Therapy: J Am Soc Gene Therapy. 2017;25(3):570–9.
Anthony-Gonda K, Bardhi A, Ray A, Flerin N, Li M, Chen W et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Science translational medicine. 2019;11(504).
Anthony-Gonda K, Ray A, Su H, Wang Y, Xiong Y, Lee D et al. In vivo killing of primary HIV-infected cells by peripheral-injected early memory-enriched anti-HIV duoCAR T cells. JCI Insight. 2022;7(21).
Zhou Y, Jadlowsky J, Baiduc C, Klattenhoff AW, Chen Z, Bennett AD, et al. Chimeric antigen receptors enable superior control of HIV replication by rapidly killing infected cells. PLoS Pathog. 2023;19(12):e1011853.
Laeremans T, den Roover S, Lungu C, D’Haese S, Gruters RA, Allard SD, et al. Autologous dendritic cell vaccination against HIV-1 induces changes in natural killer cell phenotype and functionality. NPJ Vaccines. 2023;8(1):29.
García F, Climent N, Guardo AC, Gil C, León A, Autran B, et al. A dendritic cell-based vaccine elicits T cell responses associated with control of HIV-1 replication. Sci Transl Med. 2013;5(166):166ra2.
Allard SD, De Keersmaecker B, de Goede AL, Verschuren EJ, Koetsveld J, Reedijk ML, et al. A phase I/IIa immunotherapy trial of HIV-1-infected patients with Tat, Rev and Nef expressing dendritic cells followed by treatment interruption. Clin Immunol (Orlando Fla). 2012;142(3):252–68.
Izumiya Y, Algalil A, Espera JM, Miura H, Izumiya C, Inagaki T, et al. Kaposi’s sarcoma-associated herpesvirus terminal repeat regulates inducible lytic gene promoters. J Virol. 2024;98(2):e0138623.
Wang X, Cabrera FG, Sharp KL, Spencer DM, Foster AE, Bayle JH. Engineering Tolerance toward Allogeneic CAR-T cells by regulation of MHC surface expression with human herpes Virus-8 proteins. Mol Therapy: J Am Soc Gene Therapy. 2021;29(2):718–33.
Vilkin A, Niv Y. Association between hMLH1 hypermethylation and JC virus (JCV) infection in human colorectal cancer (CRC). Clin Epigenetics. 2011;2(1):1–5.
Goel A, Li MS, Nagasaka T, Shin SK, Fuerst F, Ricciardiello L, et al. Association of JC virus T-antigen expression with the methylator phenotype in sporadic colorectal cancers. Gastroenterology. 2006;130(7):1950–61.
Wong MK, Yee C. Polyomavirus-positive Merkel cell carcinoma: the beginning of the beginning. J Clin Investig. 2024;134(8).
Verdu-Bou M, Tapia G, Hernandez-Rodriguez A, Navarro JT. Clinical and therapeutic implications of Epstein-Barr Virus in HIV-Related Lymphomas. Cancers. 2021;13:21.
Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–7.
Carrillo MA, Zhen A, Mu W, Rezek V, Martin H, Peterson CW, et al. Stem cell-derived CAR T cells show greater persistence, trafficking, and viral control compared to ex vivo transduced CAR T cells. Mol Therapy: J Am Soc Gene Therapy. 2024;32(4):1000–15.
Morte-Romea E, Pesini C, Pellejero-Sagastizábal G, Letona-Giménez S, Martínez-Lostao L, Aranda SL, et al. CAR immunotherapy for the treatment of infectious diseases: a systematic review. Front Immunol. 2024;15:1289303.
Acknowledgements
Not applicable.
Funding
This study was supported by Chinese National Major Project for New Drug Innovation (2019ZX09201002003). National Natural Science Foundation of China (82030076, 82070161, 81970151, 81670162 and 81870134). Shenzhen Science and Technology Foundation (JCYJ20190808163601776, JCYJ20200109113810154). Shenzhen Key Laboratory Foundation (ZDSYS20200811143757022). Sanming Project of Medicine in Shenzhen (SZSM202111004).
Author information
Authors and Affiliations
Contributions
W.Z. and L.Y. designed and directed the study. W.Z. and M.Z. wrote the manuscript. W.Z., Y.L. and W.Z. critically reviewed the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors critically reviewed and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, W., Zeng, M., Li, Y. et al. Leveraging oncovirus-derived antigen against the viral malignancies in adoptive cell therapies. Biomark Res 12, 71 (2024). https://doi.org/10.1186/s40364-024-00617-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-024-00617-6