
Abstract
Incorporation of viral DNA may interfere with the normal sequence of human DNA bases on the genetic level or cause secondary epigenetic changes such as gene promoter methylation or histone acetylation. Colorectal cancer (CRC) is the second leading cause of cancer mortality in the USA. Chromosomal instability (CIN) was established as the key mechanism in cancer development. Later, it was found that CRC results not only from the progressive accumulation of genetic alterations but also from epigenetic changes. JC virus (JCV) is a candidate etiologic factor in sporadic CRC. It may act by stabilizing β-catenin, facilitating its entrance to the cell nucleus, initialing proliferation and cancer development. Diploid CRC cell lines transfected with JCV-containing plasmids developed CIN. This result provides direct experimental evidence for the ability of JCV T-Ag to induce CIN in the genome of colonic epithelial cells. The association of CRC hMLH1 methylation and tumor positivity for JCV was recently documented. JC virus T-Ag DNA sequences were found in 77% of CRCs and are associated with promoter methylation of multiple genes. hMLH1 was methylated in 25 out of 80 CRC patients positive for T-Ag (31%) in comparison with only one out of 11 T-Ag negative cases (9%). Thus, JCV can mediate both CIN and aberrant methylation in CRC. Like other viruses, chronic infection with JCV may induce CRC by different mechanisms which should be further investigated. Thus, gene promoter methylation induced by JCV may be an important process in CRC and the polyp-carcinoma sequence.
Similar content being viewed by others


Introduction
The association between chronic viral infection and human malignancy is well established: papillomaviruses are involved in the pathogenesis of human cervical cancer (Dell and Gaston 2001), Epstein-Barr virus has been linked to Burkitt's lymphoma, nasopharyngeal carcinoma, post-transplant lymphoma, Hodgkin's disease, and gastric carcinoma (Thompson and Kurzrock 2004); hepatitis viruses B and C are associated with hepatocellular carcinoma (Monto and Wright 2001), Simian virus 40 has been linked to mesothelioma (Rizzo et al. 2001), and many more. The mechanisms responsible for the malignant transformation in cases of long-lasting viral infection differ according to the particular virus and cancer and have been extensively studied. An established association between viral infection and cancer may lead the way for development of a vaccine which could prevent both the infection as well as the malignant transformation. Incorporation of viral DNA may interfere with the normal sequence of human DNA bases, in a genetic manner, or cause secondary epigenetic changes such as gene promoter methylation or histone acetylation. Activation of oncogenes or silencing of tumor suppressor genes may be the result of long lasting inflammation. These gene products enhance proliferation and inhibit apoptosis and differentiation, driving the cells towards carcinogenesis.
Colorectal cancer is the second leading cause of cancer mortality in the USA. Chromosomal instability has been established as the key mechanistic component of cancer development (Fearon and Vogelstein 1990). Later, it was found that CRC results not only from the progressive accumulation of genetic alteration, but also from epigenetic changes (Lengauer et al. 1997). Three main pathways have been described: chromosomal instability (CIN), microsatellite instability (MSI), and CpG island methylation phenotype (CIMP; Boland and Goel 2010).
Hypermethylation in CRC
Microsatellite instability is involved in the genesis of about 15% of CRCs (Ionov et al. 1993; Thibodeau et al. 1993a; Aaltonen et al. 1993). The multiple errors in repetitive DNA sequences (microsatellites) result from a failure of the DNA mismatch repair system to edit errors made during DNA replication (Carethers and Boland 2003). This system is inactivated either by hypermethylation of the promoter, which silences gene transcription of hMLH1 (epigenetic phenomenon in sporadic CRC), or because of germ-line mutations (Herman et al. 1998; Lynch et al. 1993). Other genes of the mismatch repair family, such as MSH2, MSH6, and PMS2 may be inactivated by genetic processes, and cause Lynch syndrome. 5′ Cytosine methylation of CpG dinucleotide is the only known epigenetic modification of DNA. DNA methylation is mediated by DNA methyltransferase catalyzing the transfer of a methyl group from methyl donor S-adenosil-methyonine to 5′ cytosine postponed by guanine and forming methylcytosine (Lyko et al. 1999; Robert et al. 2003). The overall frequency of CpG dinucleotides is relatively low in the human genome and accounts for approximately 1% of total DNA bases (Ehrlich et al. 1982). However, up to 60% of genes have a 5′ promoter region with 0.3–2 kb stretches of DNA called “CpG Island” (Bird 1986). The great majority of CpG dinucleotides which are not associated with CpG islands are heavily methylated; however, CpG dinucleotides in CpG islands of promoter regions are usually unmethylated, whether or not the gene is being transcribed (Antequera and Bird 1993).
Cancer gene genomes simultaneously show global hypomethylation, specific promoter hypermethylation of tumor suppressor genes, and hypomethylation of oncogenes (Gaudet et al. 2003; Eden et al. 2003; Greger et al. 1989). In addition to DNA methylation, common epigenetic changes in cancer include histone modification by acethylation (Jenuwein and Allis 2001).
The methyl groups do not affect base pairing and are therefore not hereditary. DNA methylation inhibits transcription by interfering with transcriptional initiation. Some of transcription factors cannot bind to DNA with methylated CpG Island to start transcription (Prendergast and Ziff 1991). Additionally, methylated DNA has general effect on chromatin. Methyl cytosine binding proteins (MBPs) may recognize and bind to methylated DNA. MBPs are part of complexes containing histone deacetylases that inactivate chromatin configuration around gene and silenced gene expression by exclusion of transcriptional factors (Nan et al. 1998).
Tumors with high-frequency microsatellite instability tend to be diploid to possess a mucinous histology and to have a surrounding lymphoid reaction. They are more prevalent in the proximal colon, have a rapid (2–3 years) progression from adenomatous polyp to cancer, and are insensitive to 5-fluoro-uracil-based chemotherapy. Nevertheless, they are associated with longer survival than stage-matched tumors with microsatellite stability (Lothe et al. 1993; Gryfe et al. 2000; Halling et al. 1999; Thibodeau et al. 1993b; Lukish et al. 1998; Wright et al. 2000). Deng and colleagues demonstrated that, in CRC, regional hypermethylation and global hypomethylation are associated with altered chromatin conformation and histone acetylation, which have a causal correlation with MSI and CIN, respectively (Deng et al. 2006). MSI cancers had closed chromatin conformation and low levels of histone acetylation. Gene silencing is mediated by genetic processes, such as mutation, deletion, or insertion of nucleotides or by epigenetic mechanisms such as methylation of the CpG Island in a gene promoter by the enzyme DNA methyltransferase. Recently, microRNAs (miRNAs) were found to regulate gene transcription. This group of substances may decrease or increase proliferation by silencing oncogenes or tumor suppressor genes. Whether CpG island hypermethylation constitutes a mechanism for miRNA silencing, and its role in the development of cancer remains unexplored. Balaguer and colleagues demonstrated that epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis (Balaguer et al. 2010).
JC virus infection and hMLH1 hypermethylation in CRC
Some oncoviruses, including JC virus, may initiate carcinogenesis by epigenetic modification of different oncogenes. JC virus, a small non-enveloped negatively supercoiled 5,130 bp dsDNA virus, is a member of the polyomavirus family, which includes BK virus, SV40, and, recently described, KI virus, WU virus, and MC virus (White and Khalili 2004; Allander et al. 2007; Gaynor et al. 2007; Feng et al. 2008). JCV was first isolated in 1971 from the brain of a patient (John Cunningham) with Hodgkin's lymphoma who developed progressive multifocal encephalopathy after chemotherapy. Structurally, JCV contains only six genes which encode the T- and t-antigens, three viral capsid proteins (VP1-3), and an agnoprotein involved in viral assembly. The T-Ag takes up over half of the viral genome (Raj et al. 1998). With most humans subclinically infected during childhood, this virus is ubiquitous. Over 70% of adults possess neutralizing antibodies to the virus, some 50% of adults shed JCV DNA in the urine (Kitamura et al. 1994; Rossi et al. 2007). Several studies suggest that JCV establishes latency in B lymphocytes (Monaco et al. 1996; Sabath and Major 2002); however, the viral genome has also been detected in tissue samples from brain, lung, spleen, liver, tonsils, and stomach to colon.
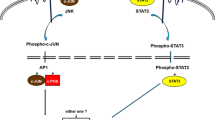
JC virus is a candidate etiologic factor for sporadic CRC (Niv et al. 2004). It may act by stabilizing β-catenin, facilitating its entrance to the cell nucleus and starting proliferation and cancer formation. Diploid CRC cell lines transfected with JCV-containing plasmids developed CIN (Niv et al. 2004). This result provided direct experimental evidence for the ability of T-Ag to induce CIN in the genome of colonic epithelial cells. The association of hMLH1 promoter methylation and tumor positivity for JC virus in CRC was recently documented (Goel et al. 2006). JC virus T-Ag DNA sequences found in 77% of CRCs were associated with promoter methylation of multiple genes (Fig. 1). hMLH1 was methylated in 25 out of 80 CRC patients positive for T-Ag (31%) in comparison with only one out of 11 negative cases (9%). The latter patient was microsatellite-stable. This observation is somewhat unexpected since, in this case, suppression of DNA repair by silencing hMLH1 gene should be sufficient to cause genomic instability and uncontrolled proliferation. We confirmed this observation in a small sample of Israeli patients (Niv et al. 2010). In this paper, we looked at both JCV T-Ag in the CRC tissue and hMLH1 promoter methylation in different ethnic groups in Israel with different incidences of CRC. We found 83.3% positivity of the tumors for JCV T-Ag, which is very similar to that in previous publications (Laghi et al. 1999; Ricciardiello et al. 2003). Unexpectedly, we demonstrated hMLH1 promoter methylation only in JCV T-Ag-positive tumors.

Working hypothesis: JC virus T-Ag initiates proliferation through WNT signaling or hypermethylation of hMLH1
Nosho et al. (2009), in large study that included 766 patients with sporadic colorectal cancer, investigated the correlation between expression of T-Ag in tumor tissue and different molecular alterations (MSI, CIMP, CIN, loss of heterozygosity of different loci, BRAF and KRAS mutation, p53, b-catenin, cyclin D1, and cyclooxygenase-2 expression). In this study, JCV T-Ag was demonstrated in 35% of tumors, but, in contrast to results of Goel and colleagues, expression of T-Ag was inversely associated with MSI and CIMP. In multivariate logistic regression analysis, JCV T-Ag expression was independently associated with p53 expression and CIN. In other study, Nosho et al. (2008) investigated the association between JCV T-Ag expression and methylator phenotype in 50 cases of colorectal adenoma. T-Ag expression was demonstrated in 16 (32%) of cases, and the proportion of positive cases increased progressively from unmethylated cases to CIMP-low and CIMP-high.
Possible mechanisms of epigenetic processes induced by chronic viral infection in cancer
An association between methylation and exposure to carcinogens such as viruses has been observed before but has not led to mechanistic insights into the process (Boland et al. 2005; Shen et al. 2002). Human papillomavirus chronic infection is associated with methylation of several genes involved in carcinogenesis such as APC, CDKN2A, PTEN, and others (Whiteside et al. 2008). Similarly, in nasopharyngeal carcinoma associated with Epstein-Barr virus (EBV), hypermethylation of certain cellular promoters is attributed to the up-regulation of DNA methyltransferase1 (DNMT1) by the viral oncoprotein LMP1 via JNK/AP1-signaling (Niller and Wolf 2009). The primary molecular abnormality in EBV-associated gastric cancer is global CpG Island methylation in the promoter region of many cancer-related genes (Fukayama 2010). Viral latent membrane protein 2A up-regulates cellular DNMT1 through phosphorylation of STAT3 and hypermethylation and silencing of the tumor suppressor gene, PTEN. Increased DNMT1 also caused methylation of E-cadherin, hMLH1, and THBS-1 genes, and was associated with poor prognosis (Etoh et al. 2004). Studying adenovirus type 12 oncogenesis in the hamster model, Doerfler et al. demonstrated significant alteration in DNA methylation patterns that affected the transcription of cellular genes (Doerfler 2009). Foreign DNA may cause extensive methylation in cellular genome segments even at loci remote from the site of insertion. In addition, down-regulation of microRNA by viral component can induce aberrant DNA methylation by targeting DNA methyltransferase 1, as demonstrated in hepatitis B virus-related hepatocellular carcinoma, in this case, miR-152 (Huang et al. 2010).
Conclusion
JC virus can mediate both CIN and aberrant methylation in CRC. Like other viruses, chronic infection with JCV may induce CRC by different mechanisms that should be further specified. Gene promoter methylation induced by JCV may be an important process in CRC and the polyp-carcinoma sequence. Further research in this field is needed, looking at a possible association between HNPCC tumors and JCV, the presence of JCV in normal mucosa of cancer-free patients, association between JCV with prognosis, and survival in CRC cases of CIN, CIMP, and MSI pathways.
References
Aaltonen LA, Peltomaki P, Leach FS et al (1993) Clues to the pathogenesis of familial colorectal cancer. Science 260:812–816
Allander T, Andreasson K, Gupta S, Bjerkner A, Bogdanovic G, Persson MA, Dalianis T, Ramqvist T, Andersson B (2007) Identification of a third human polyomavirus. J Virol 81(8):4130–4136
Antequera F, Bird A (1993) Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci USA 90(24):11995–11999
Balaguer F, Link A, Lozano JJ, Cuatrecasas M, Nagasaka T, Boland CR, Goel A (2010) Epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis. Cancer Res 70:6609–6618
Bird AP (1986) CpG-rich islands and the function of DNA methylation. Nature 321:209–213, Review
Boland CR, Goel A (2010) Microsatellite instability in colorectal cancer. Gastroenterology 138:2073–2087
Boland CR, Luciani MG, Gasche C, Goel A (2005) Infection, inflammation and gastrointestinal cancer. Gut 54:1321–1331
Carethers JM, Boland CR (2003) Neoplasia of the gastrointestinal tract. In: Yamada T, Alpers DH, Kaplowitz N, Laine L, Owyang C (eds) Powell DW. Lippincott-Raven, Philadelphia, pp 557–583
Dell G, Gaston K (2001) Human papillomaviruses and their role in cervical cancer. Cell Mol Life Sci 58:1923–1942
Deng G, Nguyen A, Tanaka H, Matsuzaki K, Bell I, Metha KR, Terdiman JP, Waldman FM, Kakar S, Gum J, Crawley S, Sleisenger MH, Kim YS (2006) Regional hypermethylation and global hypomethylation are associated with altered chromatin conformation and histone acetylation in colorectal cancer. Int J Cancer 118:2999–3005
Doerfler W (2009) Epigenetic mechanisms in human adenovirus type 12 oncogenesis. Sem Cancer Biol 19:136–143
Eden A, Gaudet F, Waghmare A, Jaenisch R (2003) Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300(5618):455
Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10(8):2709–2721
Etoh T, Kanai Y, Ushijima S, Nakanishi Y, Sasako M, Kitano S, Hirohshi S (2004) Increased DNA methyltransferase 1 protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am J Pathol 164:689–699
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61:759–767
Feng H, Shuda M, Chang Y, Moore PS (2008) Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319(5866):1096–1100
Fukayama M (2010) Epstein-Barr virus and gastric carcinoma. Pathol Int 60:337–350
Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R (2003) Induction of tumors in mice by genomic hypomethylation. Science 300:489–492
Gaynor AM, Nissen MD, Whiley DM, Mackay IM, Lambert SB, Wu G, Brennan DC, Storch GA, Sloots TP, Wang D (2007) Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog 3(5):e64
Goel A, Li MS, Nagasaka T, Shin SK, Fuerst F, Ricciardiello L, Wasserman L, Boland CR (2006) Association of JC virus T antigen with the methylator phenotype in sporadic colorectal cancers. Gastroenterology 130:1950–1961
Greger V, Passarge E, Höpping W, Messmer E, Horsthemke B (1989) Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet 83(2):155–158
Gryfe R, Kim H, Hsieh ETK et al (2000) Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 342:69–77
Halling KC, French AJ, McDonnell SK et al (1999) Microsatellite instability and 8p allelic imbalance in stage B2 and C colorectal cancers. J Natl Cancer Inst 91:1295–1303
Herman JG, Umar A, Polyak K et al (1998) Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 95:6870–6875
Huang J, Wang Y, Guo Y, Sun S (2010) Down-regulated microRNA-152 induces aberrant DNA methylation in hepatitis B virus-related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology 52:60–70
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M (1993) Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 363:558–561
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293(5532):1074–1080, Review
Kitamura T, Kunitake T, Guo J et al (1994) Transmission of the human polyomavirus JC virus occurs both within the family and outside the family. J Clin Microbiol 32:2359–2363
Laghi L, Randolph AE, Chauhan DP et al (1999) JC virus DNA is present in the mucosa of the human colon and in colorectal cancers. Proc Natl Acad Sci USA 96:7484–7489
Lengauer C, Kinzler KW, Vogelstein B (1997) DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci USA 94:2545–2550
Lothe RA, Peltomaki P, Meling GI et al (1993) Genomic instability in colorectal cancer: relationship to clinicopathological variables and family history. Cancer Res 53:5849–5852
Lukish JR, Muro K, DeNobile J et al (1998) Prognostic significance of DNA replication errors in young patients with colorectal cancer. Ann Surg 227:51–56
Lyko F, Ramsahoye BH, Kashevsky H, Tudor M, Mastrangelo MA, Orr-Weaver TL, Jaenisch R (1999) Mammalian (cytosine-5) methyltransferases cause genomic DNA methylation and lethality in Drosophila. Nat Genet 23(3):363–366
Lynch HT, Smyrk TC, Watson P et al (1993) Genetics, natural history, tumour spectrum, and pathology of hereditary non-polyposis colorectal cancer: an updated review. Gastroenterology 104:1535–1549
Monaco MC, Atwood WJ, Gravell M, Tornatore CS, Major EO (1996) JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J Virol 70:7004–7012
Monto A, Wright TL (2001) The epidemiology and prevention of hepatocellular carcinoma. Semin Oncol 28:441–449
Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393(6683):386–389
Niller HH, Wolf H, Minarovits J. Epigenetic dysregulation of the host cell genome in Epstein-Barr virus-associated neoplasia. Semin Cancer Biol 2009;19:158–164
Niv Y, Goel A, Boland CR (2004) JC virus and colorectal cancer: a possible trigger in the chromosomal instability pathway. Curr Opin Gastroenterol 21:85–89
Niv Y, Vilkin A, Brenner B, Kendel Y, Morgenstern S, Levi Z (2010) hMLH1 promoter methylation and JC virus T antigen presence in the tumor tissue of colorectal cancer Israeli patients of different ethnic groups. Eur J Gastroenterol Hepatol 22:938–941
Nosho K, Yamamoto H, Takahashi T, Mikami M, Hizaki K, Maehata T, Taniguchi H, Yamaoka S, Adachi Y, Itoh F, Imai K, Shinomura Y (2008) Correlation of laterally spreading type and JC virus with methylator phenotype status in colorectal adenoma. Hum Pathol 39(5):767–775
Nosho K, Shima K, Kure S, Irahara N, Baba Y, Chen L, Kirkner GJ, Fuchs CS, Ogino S (2009) JC virus T-antigen in colorectal cancer is associated with p53 expression and chromosomal instability, independent of CpG island methylator phenotype. Neoplasia 11(1):87–95
Prendergast GC, Ziff EB (1991) Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science 251(4990):186–189
Raj GV, Gallia GL, Chang CF, Khalili K (1998) T-antigen-dependent transcriptional initiation and its role in the regulation of human neurotropic JC virus late gene expression. J Gen Virol 79:2147–2155
Ricciardiello L, Baglioni M, Giovannini C et al (2003) Induction of chromosomal instability in colonic cells by the human polyoma JC virus. Cancer Res 63:7256–7262
Rizzo P, Bocchetta M, Powers A et al (2001) SV40 and the pathogenesis of mesothelioma. Semin Cancer Biol 11:63–71
Robert MF, Morin S, Beaulieu N, Gauthier F, Chute IC, Barsalou A, MacLeod AR (2003) DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat Genet 33(1):61–65
Rossi A, Delbue S, Mazziotti R, Valli M, Borghi E, Mancuso R, Calvo MG, Ferrante P (2007) Presence, quantitation and characterization of JC virus in the urine of Italian immunocompetent subjects. J Med Virol 79:408–412
Sabath BF, Major EO (2002) Traffic of JC virus from sites of initial infection to the brain: The path to progressive multifocal leukoencephalopathy. J Infect Dis (Suppl2): S180–186
Shen L et al (2002) DNA methylation and environmental exposures in human hepatocellular carcinoma. J Natl Cancer Inst 94:755–761
Thibodeau SN, Bren G, Schaid D (1993) Microsatellite instability in cancer of the proximal colon. Science 260:816–819
Thompson MP, Kurzrock R (2004) Epstein-Barr virus and cancer. Clin Cancer Res 10:803–821
White MK, Khalili K (2004) Polyomaviruses and human cancer: molecular mechanisms underlying patterns of tumorigenesis. Virology 324:1–16
Whiteside MA, Siegel EM, Unger ER (2008) Human papillomavirus and molecular considerations for cancer risk. Cancer 113(10 suppl):2981–2994
Wright CM, Dent OF, Barker M et al (2000) Prognostic significance of extensive microsatellite instability in sporadic clinicopathological stage C colorectal cancer. Br J Surg 87:1197–1202
Conflict of interest
The authors have no conflicts of interest or any financial relationship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Vilkin, A., Niv, Y. Association between hMLH1 hypermethylation and JC virus (JCV) infection in human colorectal cancer (CRC). Clin Epigenet 2, 1–5 (2011). https://doi.org/10.1007/s13148-010-0013-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13148-010-0013-3