
Abstract
Liver cancer is one of the most common malignant tumors worldwide, it is ranked sixth in incidence and fourth in mortality. According to the distinct origin of malignant tumor cells, liver cancer is mainly divided into hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA). Since most cases are diagnosed at an advanced stage, the prognosis of liver cancer is poor. Tumor growth depends on the dynamic interaction of various cellular components in the tumor microenvironment (TME). As the most abundant components of tumor stroma, cancer-associated fibroblasts (CAFs) have been involved in the progression of liver cancer. The interplay between CAFs and tumor cells, immune cells, or vascular endothelial cells in the TME through direct cell-to-cell contact or indirect paracrine interaction, affects the initiation and development of tumors. Additionally, CAFs are not a homogeneous cell population in liver cancer. Recently, single-cell sequencing technology has been used to help better understand the diversity of CAFs in liver cancer. In this review, we mainly update the knowledge of CAFs both in HCC and CCA, including their cell origins, chemoresistance, tumor stemness induction, tumor immune microenvironment formation, and the role of tumor cells on CAFs. Understanding the context-dependent role of different CAFs subsets provides new strategies for precise liver cancer treatment.
Similar content being viewed by others


Background
Liver cancer causes 841,000 new cases and 782,000 deaths every year, making it the sixth most commonly diagnosed cancer and the fourth leading cause of cancer death worldwide [1]. Primary liver cancer includes HCC (comprising of 75%-85%) and CCA (comprising of 10%-15%) or a mixed form of HCC and CCA. In developing countries, such as China and Eastern Asia, the main risk factor for HCC is hepatitis B virus (HBV) infection, while in developed countries, it is mainly hepatitis C virus (HCV) and alcoholic cirrhosis [2]. Recently, nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) are rising in rank as contributors to HCC development [3]. The presence of these etiologies can lead to and exacerbate the progression of liver cirrhosis, and one-third of patients with cirrhosis will develop HCC during their lifetime [4]. Current treatment strategies for liver cancer include hepatic resection, liver transplantation, and systemic chemotherapy (e.g., sorafenib or Lenvatinib), or a combination of different treatment modalities [5,6,7,8]. For example, a recent clinical study showed that the combined application of molecular-targeted agents (bevacizumab) and immune checkpoint inhibitors (atezolizumab) has been shown to improve overall survival rates relative to sorafenib in HCC [9]. However, despite great therapeutic advances, the overall prognosis of HCC remains poor.
Although tumor cells hold the main role in driving carcinogenesis, an increasing interest has been focused on the TME. TME includes the cellular and the non-cellular components in solid tumors. The cellular compartment is composed of surrounding stromal cells, immune cells, and angiogenic endothelial cells. The non-cellular component contains extracellular matrix (ECM), various growth factors, chemokines, and cytokines [10]. CAFs constitute the main components of tumor stroma that are closely associated with tumor initiation, progression, stemness, chemoresistance, and prognosis [11]. CAFs can directly communicate with tumor cells and other stromal cells in a paracrine manner or remodel the ECM structure to create a microenvironment conducive to tumor cell invasion and metastasis, which indirectly leads to tumor progression. However, increasing evidence has demonstrated that CAFs do not always exert a tumor-supportive role in oncogenesis, they may also play a tumor-suppressive effect that is context-dependent, namely phenotypic heterogeneity and functional diversity. For example, Meflin-positive fibroblasts could form a tumor inhibitory CAFs subpopulation in pancreatic ductal adenocarcinoma (PDAC), which are correlated to favorable outcomes [12]. The diversified cell origin of CAFs may partly contribute to the heterogeneity of CAFs function, and the phenotypic switch of CAFs under the corrupting influence of TME during tumor evolution may be another reason for the heterogeneity. Therefore, understanding the dynamic communication between various cells in the tumor, especially how the reciprocal crosstalk between CAFs and other cells reshapes the TME, is essential for comprehending and treating liver cancer from an evolutionary and holistic perspective. In this review, we discuss the heterogeneous cellular origin of CAFs, their functional malleability in HCC and CCA, respectively, and mainly focus on the latest research progress of CAFs interacting with various cellular components in the TME of the liver cancer.
The characterizations and cellular origin of CAFs in HCC
The unique characteristics of CAFs
Liver normal stroma cells constitute the connective tissues that supply a supportive framework for the liver tissues. Among the stromal components, normal fibroblasts produce a variety of collagens or matrix metalloproteinase (MMP) to maintain the integrity of ECM or remodel the ECM to keep the matrix components in a dynamic equilibrium [13]. When the liver is stimulated by various stimuli in the context of liver cirrhosis or in the process of tumor development, normal fibroblasts or other types of cells can be activated into myofibroblasts or cancer-associated fibroblasts, exhibiting enhanced secretory function and ECM accumulation.
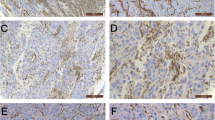
CAFs are easy to isolate from fresh liver cancer tissues and to culture in vitro over several passages with stable phenotype. Different from the normal fibroblasts (NFs), CAFs are identified by spindle-shaped morphology but with multiple branches of cytoplasm, larger indented nuclei, plentiful ribosomes, increased rough endoplasmic reticulum, and a well-developed Golgi apparatus [14, 15]. α-smooth muscle actin (α-SMA) is expressed by multiple CAFs subsets, that is usually be used to identify CAFs in liver cancer [16]. Other reported proteins such as fibroblast activation protein (FAP), vimentin, platelet-derived growth factor (PDGF) receptor (PDGFR)-α and β, and fibroblast-specific protein 1 (FSP-1) can also serve as markers of CAFs [17]. In our previous study, the extracted CAFs were identified by immunofluorescence staining of α-SMA and Vimentin, and the morphological characteristics of CAFs could be observed [18], as shown in Fig. 1A. The distribution of CAFs in HCC tissues can be aggregated, sporadic, and localized along hepatic sinusoids, Fig. 1B showed two different distributions of CAFs in HCC. Moreover, studies have shown that the abundance of CAFs positively correlated with tumor size, and the higher the density of CAFs, the worse the prognosis of HCC patients [19, 20]. Nevertheless, due to the lack of specific lineage biomarkers, it remains a challenging topic in studying CAFs in vivo.

Morphological manifestations of fibroblasts in HCC. A CAFs and NFs extracted from HCC tissue and normal liver tissue were identified by immunofluorescence staining for α-SMA and Vimentin. CAFs exhibited more abundant cytoplasmic content than NFs. B Representative graphs showed two HCC cases with different α-SMA + CAFs distribution densities, with case1 exhibiting more rich CAFs infiltration relative to case2. Peng, H., R. Xue, Z. Ju, J. Qiu, J. Wang, W. Yan, et al., Ann Transl Med, 2020. 8(14): 856
Functionally, activated CAFs exhibit enhanced secretory phenotype, secreting various cytokines including transforming growth factor-β (TGF-β), hepatocyte growth factor (HGF), insulin-like growth factor (IGF), interleukin-6 (IL-6) et al., which can, in turn, induce the proliferation, migration, immune response of liver cancer cells [21]. Apart from this, one of the hallmarks of CAFs is their high capacity for ECM synthesis, including fibronectin as well as different types of collagens [22]. Unbalanced synthesis and degradation of local ECM induce mechanical stiffening of the tissues, which may modulate tumorigenesis and influence the prognosis of patients. In general, CAFs communicate with various cells in the TME by producing various cytokines and ECM proteins to construct a microenvironment suitable for tumor growth and dissemination.
The cellular origin of CAFs in HCC
Emerging studies demonstrate that CAFs can be derived from multiple cell types, including normal resident tissue fibroblasts, hepatic stellate cells (HSCs), mesenchymal stem cells (MSCs), epithelial or endothelial cells [23]. The genetic fate-mapping technique using lecithin retinol acyltransferase-cyclization recombination enzyme (Lrat-Cre) and PDGF receptor beta (PDGFRB)-Cre determine HSCs as the dominant source of myofibroblasts in liver fibrosis [24, 25]. HSCs are commonly residing in the perisinusoidal space and occupy 15% of resident cells in the liver, their unique function is to store vitamin A [26]. Under the stimulation of several cytokines, HSCs can undergo a morphophysiological transformation and acquire a myofibroblast-like phenotype, manifested by increased α-SMA expression and enhanced collagen secretion [27]. TGF-β plays an essential role in all stages of liver disease progression, from inflammation to cirrhosis and liver cancer. During tumorigenesis, TGF-β is a well-known growth factor that can activate HSCs into α-SMA ( +) CAFs [28]. Another research revealed that hepatocyte-derived PDGF-C could also transform HSCs into myofibroblast-like cells to accelerate the progression of HCC [29].
The application of the latest genetic tracing and single-cell RNA sequencing (scRNA-seq) approaches provides more favorable evidence to trace the origin of CAFs. In a recent study, the authors analyzed scRNA-seq data from mouse liver cells and found transcription factor 21 (Tcf21) could be identified as a specific marker that distinguished quiescent HSCs from activated HSCs. By tracing Tcf21 positive cells, they observed these cells mainly marked periportal and pericentral HSCs, Tcf21 positive HSCs were quiescent under steady-state but became activated in the DEN/CCL4-induced state, generating 85% of CAFs in liver tumors [30]. Another recent study using genetic tracing in combination with scRNA-seq analysis demonstrated that liver metastasis-associated CAFs are primarily HSCs derived, as the majority of CAFs showed abundant expression of HSCs signature that included HSC markers [31].
MSCs are multipotent cells, which can self-renew and differentiate into adipocytes, cartilage, bone, and other cells under the appropriate conditions [32]. MSCs can be recruited to the stroma of HCC and play an essential role in HCC initiation, progression [33]. MSCs infiltrating the TME can be transformed into CAFs-like cells after being acclimated by surrounding tumor cells. For example, a recent study investigated that when co-cultured with Huh7 cells, MSCs significantly upregulated the expression of CAFs markers (α-SMA, Vimentin, c-MYC, MMP2, VEGF, IL-6, FGFR1, IL-8, Tenascin-C), thereby acquiring the CAFs-like phenotype and characteristics [34]. Another study revealed after exposure to SK-Hep1, MSCs can also exhibit the properties of CAFs [35]. However, both studies are only limited to cell lines co-cultivation assays in vitro, the in vivo lineage-tracing experiments are required to determine the conversion of MSCs to CAFs in the future.
Additionally, epithelial cells through epithelial-mesenchymal transition (EMT) or endothelial cells undergoing endothelial to mesenchymal transition (EndMT) can acquire mesenchymal properties, which might serve as another source of CAFs [36, 37]. For example, Zeisberg et al. have revealed that TGF-β induced adult mouse hepatocytes to undergo phenotypic and functional switch typical of EMT, which contributed to the population of FSP1-positive fibroblasts in CCL4-induced liver fibrosis [37]. However, this origin may be controversial, another research utilizing triple transgenic mice demonstrated that hepatocytes in vivo neither acquire mesenchymal marker expression nor exhibit a myofibroblasts-like morphology [38]. In some specific contexts, HCC cells may undergo EMT and express markers of CAFs. For instance, hypoxic conditions can induce upregulation of FAP expression in HCC cells [39]. Another study explored that TGF-β promoted α-SMA expression in HCC cells [40]. A recent study using the in vitro EndMT model found that after treatment with TGF-β1 and IL-1β, human fetal liver sinusoidal endothelial cells tend to transition to fibroblast-like cells, while mesenchymal markers were increased, and endothelial markers were decreased [41]. However, these studies are limited to the expression of surface markers of CAFs and lack the exploration of the functional features of CAFs in vivo.
In addition to the above possible cell sources, portal fibroblasts (PFs) as a small population of “periductular mesenchymal cells” can also be an origin of CAFs [42]. Lysophostatidic acid (LPA) secreted by HCC can act on PFs to convert them into CAFs, after transdifferentiation, PFs acquired the expression of α-SMA and enhanced the proliferation, migration, and invasion of HCC [31, 43]. This source awaits more research to prove in the future.
The roles of CAFs in HCC progression
Numerous evidence has shown that CAFs are involved in the regulation of tumor cell proliferation, invasion, migration, metastasis, etc. In the treatment of HCC, CAFs promote tumor chemoresistance and recurrence. By secreting various forms of cytokines, growth factors, or extracellular vesicles, CAFs communicate closely with HCC cells either directly or indirectly. Recent data based on spatial proteome profiling of HCC cells and CAFs further supports the interaction between CAFs and HCC cells. For instance, CAFs might release BGN, VCAN, which bind to the receptor TLR2 on the surface of HCC cells in a paracrine way to initiate downstream signal transduction. Alternatively, CD81 expression on CAFs can directly bind to GPC3 on HCC cells, affecting the biological behavior of both [44].
Nevertheless, considering the heterogeneity of CAFs, they might not always play a tumor-promoting role in the TME, on the contrary, they may also exert a tumor-restraining effect in tumor growth. A recent study showed that some specific CAFs subgroups in the TME can exert a mutually antagonistic effect in HCC. By analyzing scRNA-seq data, CAFs in liver metastasis can be divided into three subgroups that are myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs), and PF/mesothelial CAF (PF/mesCAF). myCAFs expressing type I collagen could suppress tumor growth in a manner of mechanical restriction [31]. Another study based on proteomic and scRNA-seq analysis showed that HCC-infiltrating CAFs can be divided into three subtypes [45], the heterogeneous population of CAFs in HCC was summarized in Table 1.
CAFs enhance chemoresistance of HCC cells
Resistance to anti-tumor therapeutics often leads to tumor progression. Sorafenib and Lenvatinib have been administrated for patients with advanced HCC, however, the median overall survival (OS) with either sorafenib or Lenvatinib is only about 13 months [7, 8]. The interaction between CAFs and HCC is one of the potential mechanisms that weaken the sensitivity of tumor cells to chemotherapeutic drugs. Our previous research showed CAFs secreted HGF to upregulate the expression of cell differentiation (CD)-73, and CD73 positive HCC cells were more resistant to the effects of sorafenib and cisplatin [18]. Additionally, a recent study by our team showed that CAFs can induce the Reticulocalbin 1 (RCN1) expression in HCC, and high-expressing RCN1 can attenuate the sensitivity of HCC cells to sorafenib via the IRE1α-XBP1s pathway [48]. Apart from that, chemoresistant HCC cells can also promote CAFs functional enhancement, which in turn provides a more favorable microenvironment for HCC cells to survive. For example, when co-cultured with sorafenib-resistant HCC cells, CAFs can be activated through the B-cell activating factor (BAFF) / NF-κB pathway to further enhance the chemoresistance of HCC [49].
The influence of CAFs on the stemness of HCC cells
Cancer stem cells (CSCs) are categorized by their enhanced self-renewal properties and multilineage differentiation, this population of cells confer resistance to therapy and facilitate the metastasis, recurrence of HCC [50]. In HCC progression, CAFs can provide a favorable tumor niche to support CSCs survival and sustain their stemness in a paracrine manner.
Prior studies have elucidated that HGF, as a mediator in the conditioned media of CAFs, paly a major role in the induction of liver CSCs. Mechanistically, CAFs-derived HGF enhanced stemness through the extracellular signal-regulated kinase (ERK)1/2–FRA1–HEY1 signaling pathways in HCC [51]. In addition to its direct role on stemness maintenance, HGF can also further enhance the stemness capacity of HCC by upregulating the expression of stemness-related molecules. CD73 positive HCC cells have been demonstrated to exert stemness maintaining function by upregulating SOX9 expression and preventing its protein degradation [52]. Our previous study revealed that CAFs could secret the amount of HGF to enhance the sphere-forming capacity of CD73 positive HCC cells via the MET-ERK1/2 pathway [18]. Consistently, Keratin19 has been determined to be a CSCs marker in HCC [53], and it can be induced by HGF from CAFs via a MET-ERK1/2-AP1 and SP1 axis [54]. Another study indicated that CAFs-derived IL-6 promoted stem cell-like properties in HCC cells by enhancing STAT3/Notch signaling [55]. The cluster of differentiation24 (CD24) has previously been identified as another CSCs marker in liver cancer [56]. CD24 positive HCC cells possessed a high capacity of self-renewal, and CAFs-secreted HGF and IL6 enhanced stemness of CD24 positive HCC cells through activated STAT3 pathway [57]. Peri-tumor fibroblasts also produced more IL-6, which induce HCC stemness via IL-6-STAT3-pathway [58]. Moreover, peri-tumor tissue-derived fibroblasts could also recruit CSCs to maintain their stemness via generating a series of cytokines [59].
Besides, other cytokines have recently been reported to regulate HCC stemness. Follistatin-like 1 (FSTL1), a pro-inflammatory factor, which is found to be upregulated during inflammation, is predominantly secreted from the CAFs rather than liver cancer cells. FSTL1 binding to the TLR4 receptor on HCC cells could augment the stemness through deregulated AKT/mTOR/4EBP1 signaling pathways [60]. Additionally, cardiotrophin-like cytokine factor 1 (CLCF1), a cytokine that belongs to the IL-6 superfamily, can be secreted by CAFs to induce the secretion of (C-X-C motif) ligand 6 (CXCL6) and TGF-β in HCC, which subsequently promote tumor cell stemness in an autocrine manner. Furthermore, CXCL6 and TGF-β could activate the CAFs through ERK1/2 signaling to generate more CLCF1 to further sustain the stemness of HCC [61]. However, the liver X receptors (LXRs) can inhibit the expression of key markers in CAFs, thus limiting the differentiation of CAFs. Activation of LXRs can antagonize the effect of TGFβ1-induced CAFs on hepatosphere formation [40].
Previous studies have revealed that FOX members can enhance stem cell-like characteristics in cancers [62]. Luo et al. demonstrated that FOXQ1 was induced by CAFs in co-culture systems in vitro and in vivo, and HCC initiation was promoted by CAFs via FOXQ1/NDRG1 axis. Moreover, the activated FOXQ1/NDRG1 axis could feedback to the recruitment of infiltrating CAFs through the pSTAT6/CCL26 signaling pathway, thus, further enhancing the initiation of HCC [63]. Increasing evidence shows that autophagy is essential in the maintaining of stemness in liver CSCs [64, 65]. Zhao et al. found that the potential mechanism of CAFs to promote HCC stemness may be mediated by autophagy via the mTOR pathway in HCC [66]. The Notch signaling pathway also plays an important role in promoting the self-renewal of liver CSCs [67]. CAFs could maintain CSCs stemness by activating Notch3/LSD1 signaling [68]. Resolvin D1 (RvD1) is an endogenous anti-inflammatory lipid mediator, which targets stromal cells to exert an anti-tumor effect. RvD1 could suppress the cartilage oligomeric matrix protein (COMP) secreted by CAFs, thus, abrogating the promoting effects of CAFs on stemness in HCC via FPR2/ROS/FOXM1 signaling [69].
CAFs shape tumor immune microenvironment in HCC
Tumor-infiltrating lymphocytes (TILs) are a highly heterogeneous population of immune cells that exert pivotal roles in immune evasion and response to immunotherapy [70]. Accumulating previous studies have confirmed the critical roles of the crosstalk between CAFs and TILs in tumorigenesis and progression. In HCC, TILs include innate immune cells and adaptive immune cells, the former contains tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), NK cells, and DC cells, while the latter is composed of T lymphocytes, CD4 + /CD8 + T cells, regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs) [71]. The tumor immune microenvironment (TIME) constituted by these immune cells determines the state of the immune response, and numerous studies have reported that CAFs promote tumor immune escape by influencing the proportion and activity of TIME.
TAMs are abundant infiltration in HCC, these cells can be divided into two subpopulations: M1 macrophages and M2 macrophages. M1 macrophages can be polarized by lipopolysaccharides (LPS), interferon-γ (IFN-γ), or tumor necrosis factor (TNF), to exert pro-inflammatory function, while IL-4, IL-10, and IL-13 lead to the M2 polarization with immunosuppressive function [72]. The research on the function of M1-polarized macrophages is limited in HCC. In the TME of HCC, CAFs tend to promote the M2 polarization of macrophages, possibly mediated by the secretion of IL6 [73]. Moreover, CAFs could generate CXCL12 to induce the secretion of plasminogen activator inhibitor‑1 (PAI‑1) in TAMs, and the up-regulation of PAI-1 in TAMs accelerated the malignant progression of HCC [74]. Endosialin is a member of the C-type lectin-like receptor family and is specifically expressed in cancer cells and tumor stromal cells [75, 76]. Recently, a study demonstrated that Endosialin was mainly expressed in CAFs, endosialin-positive CAFs could recruit the TAMs through interaction with CD68 in TAMs and secrete growth arrest-specific protein 6 (GAS6) to mediate the M2 polarization to promote the HCC progression [77].
Similar to TAMs, TANs also exhibit a dual role in HCC, they can be either anti-tumorigenic (N1) or pro-tumorigenic (N2) [78]. In HCC, CAFs could induce chemotaxis of neutrophils through the stromal cell-derived factor (SDF)-1a/CXCR4 pathway and promote PDL1 expression in neutrophils, the recruited neutrophils exerted immunosuppressive function by inhibiting the T cell immunity via the IL6-STAT3-PDL1 signaling pathway [79]. As mentioned above, in addition to sustain the stemness of HCC, CLCF1 produced by CAFs could also enhance TANs infiltration and polarization through increasing the secretion of CXCL6 and TGF-β in HCC cells [61].
NK cells as members of the innate immune system, which initiate anti-tumoral cytotoxic in various solid tumors. HCC patients often displayed reduced numbers of NK cells in the peripheral compared with healthy subjects and exhibited poor capacity to kill tumor cells [80]. CAFs tend to inhibit the activation and cytotoxic activity of NK cells in the HCC microenvironment. For example, CAFs secreted prostaglandin E2 (PGE2) and indoleamine 2,3-dioxygenase (IDO) could deactivate the NK cells and attenuate their cytotoxic activity, thereby forming an unresponsive niche for HCC progression [81].
Tumor-infiltrating DCs are essential in the activation of naïve T cells and initiate the adaptive immune response against tumors in the TIME. Moreover, there exists a population of regulatory DCs (rDC), which promote T cell anergy and induce Treg differentiation, thus contributing to immunotolerance in HCC [82]. CAFs are capable of attracting normal DCs into the tumor site through secreting SDF-1α and educating them to acquire tolerogenic characteristics, which resemble rDC. These CAFs-educated rDC cells highly expressed IDO, showing strong immunosuppression of T cell response and facilitating the proliferation of CD4 + CD25 + Foxp3 + Treg cells via IL-6-mediated STAT3 activation [83]. However, whether CAFs can directly recruit and activate infiltrating Treg cells has not been studied, which deserves further investigation in the future.
MDSCs are a highly heterogeneous population composed of two cell subsets: monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs), which are similar to monocytes and neutrophils in phenotypes and morphologies, respectively [84]. MDSCs possess potent immunosuppressive effects. PMN-MDSCs are mainly enriched in TIME and generate large amounts of reactive oxygen species (ROS) to mediate T cell tolerance. However, M-MDSCs accumulate in peripheral blood expressing high levels of iNOS but releasing low ROS to suppress the immune response [85]. Emerging studies have shown that CAFs could promote the generation of MDSCs through paracrine manner. In HCC, CAFs recruit monocytes into TIME by SDF-1a/CXCR4 pathway, then induce the monocytes to differentiate into CD14 + HLA-DR-/low MDSCs, which depended on IL-6/STAT3 manner. These educated MDSCs could impair T cells function to block the anti-tumor immune response [86]. Another research also confirmed that CAFs could produce higher levels of chemokines and cytokines, such as macrophage colony stimulating factor (M-CSF), monocyte chemotactic protein-1(MCP-1), and TGFβ1, to recruit the MDSCs into the TIME, however, the specific molecular mechanism needs to be further studied [87]. Additionally, activated HSCs could also promote the accumulation of MDSCs in HCC by releasing cyclooxygenase-2 (COX-2) and PGE2 [88]. The interaction between CAFs and TIL in liver cancer was summarized in Table 2
CAFs facilitate HCC angiogenesis
HCC is a hypervascularized tumor, and neo-angiogenesis leads to tumor cells dissemination, invasion, disease recurrence, and metastasis. Besides, tumor angiogenesis is a dynamic process that is mediated by pro-angiogenesis factors secretion by the tumor cells and stromal cells in the TME. For instance, CAFs secreted vascular endothelial growth factor (VEGF) in the surrounding tumor sites to promote the proliferation and angiogenesis of human umbilical vein endothelial cells (HUVECs) via enhancer of zeste homolog-2 (EZH2) /vasohibin 1 (VASH1) pathway [94]. In addition to VEGF, the placental growth factor (PlGF) is another angiogenic factor that can promote angiogenesis in HCC [95]. A recent study has shown that CD90 positive CAFs have a strong correlation with PIGF expression in HCC tissues, and highly expressed of CD90 and PIGF in CAFs were related to angiogenesis-related markers in vascular endothelial cells, such as CD31, CD34, and CD105, thus to facilitate the angiogenesis of HCC [96].
Tumor vasculogenic mimicry (VM) is an alternative way that tumor cells establish the blood supply in the absence of HUVECs. In the mouse xenografts model, implantation of CAFs with tumor cells could significantly enhance the VM formation in HCC tissues when compared with implanting tumor cells alone. Mechanistically, CAFs-derived TGF-β and SDF1 facilitated VM formation by inducing the expression of endothelial cells markers and the ECM remodeling-associated genes, such as VE-cadherin, MMP2, and laminin5γ2 in tumor cells [97].
However, in addition to the positive effect on angiogenesis, specific CAFs subsets can also play a opposite role in tumor blood vessels. According to a recent study, prolargin was solely expressed and secreted by a subset of CAFs deriving from portal fibroblasts, which bound and antagonized several pro-angiogenic growth factors, such as FGF1, FGF2, HGF, and TGF-β1, to inhibit the angiogenesis of HCC and was positively correlated with good clinical outcome in HCC patients [45]. SPARCL1 is a secreted protein and plays a tumor suppressor role in several tumors [98, 99]. Another recent scRNA-seq-based analysis found that SPARCL1-positive fibroblasts, which were located in the large blood vessels in the stromal niche of liver tumor, representing a group of vessels associated fibroblasts, could maintain the self-stabilization of blood vessels, thereby reducing tumor cell invasion and is associated with a favorable prognosis for the HCC patients [41]. In general, the above studies reveal that the effect of CAFs on tumor blood vessels is partly attributed to their cell origin or spatial localization. However, little is known about the function of tumor suppressive CAFs, which requires more research on the in vivo spatial transcriptome in the future.
Activation of CAFs in HCC
A large number of documents have shown that there exists a bi-directional communication between tumor cells and stromal cells, that is, CAFs can not only influence the initiation and progression of the tumor, but tumor cells or other cells in the TME can also stimulate the activation of CAFs, thereby forming the feedback loop further accelerates the deterioration of the tumor.
HCC cells can act on the precursor cells of CAFs, such as HSCs or other progenitors, to activate them in the manner of paracrine or exosomes. TGFβ is a well-researched inflammatory factor that can educate HSCs into myofibroblast-like cells, as mentioned above, HCC-derived TGFβ and CXCL6 could activate CAFs to enhance their secretory function [61]. TGFβ can also stimulate HCC cells to produce more connective tissue growth factor (CTGF), and TGFβ-dependent CTGF secretion can drive tumorigenesis with high stroma infiltration [100]. Mechanistically, CTGF as a matricellular protein related to fibrosis can be secreted by HCC cells to induce adjacent HSCs activation in the TME and this tumor-promoting effect of HSCs can be abolished by anti-CLGF neutralizing antibody [101]. Consistently, hepatocyte-derived PDGF-C promoted the conversion of HSCs into myofibroblasts-like cells by binding to the PDGF receptor located on HSCs [29]. Tissue inhibitor of matrix metalloproteinases (TIMPs) inhibits MMP proteolytic activity and mediates the remodeling of ECM. A study has shown that TIMPs expression in HCC can induce the liver fibroblasts into CAFs and then protect HCC cells from apoptosis via SDF-1/CXCR4/PI3K/AKT signaling pathways [102]. Extracellular sulfatase 2 (SULF2) is a member of sulfatase family genes, when co-cultured with HSCs, SULF2 could be secreted by the HCC cells to induce the differentiation of HSCs into CAFs via the TGFβ1/SMAD3 signaling pathway [103]. Another study has reported upon co-cultured with HCC cells, HSCs produced more HGF and stimulated STMN1 expression via MET pathway in HCC cells, subsequently, STMN1 enhanced PDGF homodimeric protein expression, which might facilitate HSCs activation to acquire CAFs phenotype [104]. More recently, research showed that endoplasmic reticulum (ER) stress also mediated the mutual communication between HSCs and HCC. In the context of HCC, unfolded proteins accumulated and activated ER stress. Inositol requiring enzyme 1α (IREα), as a three-transmembrane protein, can sense the presence of misfolded proteins under ER stress [105, 106]. HCC cells activated IREα in HSCs, thereby leading to HSCs activation in vitro 2D and 3D co-culture systems [107]. Periostin is a matricellular protein involved in collagen deposition, which contributes to the development of various tumors [108]. Periostin can stimulate HSCs activation in an autocrine integrin-FAK-STAT3-periostin circuit and enhance HCC cells proliferation via the ERK pathway in a paracrine manner [109]. Additionally, the sox 9/inhibin subunit beta B (INHBB) axis also plays a critical role between HSCs and HCC cells. Sox9 positive HCC cells induce INHBB expression and activin B secretion, and accordingly promote the activation of surrounding HSCs, ultimately favoring HCC metastasis [110]. In addition to HSCs, PFs can also be activated and converted into myofibroblasts-like phenotypes under the stimulation of LPA secreted by HCC cells [43].
Emerging evidence indicates that CAFs exhibit a senescence-associated secretory phenotype (SASP), that is able to secrete more pro-inflammatory cytokines in HCC. Bone morphogenetic protein 4 (BMP4) is a member of the TGF-β superfamily and is involved in organogenesis in the liver [111]. A study has found that BMP4 was highly expressed in CAFs compared to NFs and produced several SASP factors to enhance tumor invasiveness. Moreover, BMP4 expressed by HCC cells could be acted as an exogenous stimulating factor to exacerbate the activation of CAFs in the HCC microenvironment [112]. Similarly, deoxycholic acid (DCA) can cause the senescence of HSCs and induce the production of SASP factors. Furthermore, the surgical specimens of HCC patients also confirmed that the senescent HSCs were located in the stroma surrounding HCC, revealing the role of bile acid in HSCs activation in TME [113]. Gluconeogenic enzyme fructose 1,6-bisphosphatase 1 (FBP1) as a metabolic tumor suppressor in HCC, can trigger HSCs activation and senescence by releasing HMGB1 after being deleted in hepatocytes, showing a SASP [114].
Exosomes are a subtype of extracellular vesicles, which contain various biological substances that can be secreted from one cell and transferred to another, acting as a carrier for signal transmission [115]. Tumor cells can also promote the activation of HSCs in the form of exosomes [116]. A prior study has shown that exosomal miRNA-21 secreted by HCC cells promoted HSCs activation via PDK1/AKT signaling pathway [117], meanwhile, miRNA-21 could induce the progression of liver cirrhosis to liver cancer by promoting HSCs activation and collagen deposition via the TGF-β signaling pathway [118]. Exosomal miR-1247-3p secreted by high-metastatic HCC cells regulated fibroblasts activation via B4GALT3-β1-integrin-NF-κB axis in lung pre-metastatic niche from liver cancer, and the activated CAFs enhanced the secretion of pro-inflammatory cytokines, thereby promoting the stemness, EMT, chemoresistance, and tumorigenicity of HCC cells [119].
In addition to the above-mentioned stimulatory cytokines, mechanical factors such as matrix stiffness are also involved in CAFs activation. Matrix stiffness can promote malignancies by shaping the biological characteristics of tumor cells via directly regulating their growth and motility [120]. A recent study showed that stiffness induced HSCs activation through the CD36-AKT-E2F3 mechano-signaling pathway, which in turn promoted FGF2 transcription and secretion to promote HCC growth and distant metastasis [121]. Another research also provided the evidence that stiffness promoted HSCs activation relied on p300 nuclear accumulation, which was mechanistically mediated by RHOA-AKT pathway [122]. It is worth noting that both TGF-β-mediated and stiffness-mediated HSCs activation require p300, however, the transcription targets of the two are different [123].
The characterizations and cellular origin of CAFs in CCA
The unique features of CAFs in CCA
CCA is the second most frequent primary malignancy of the biliary system in liver cancer [124]. According to the anatomical location, CCA can be subdivided into three distinct subtypes: intrahepatic CCA (ICC), perihilar CCA (PCC), and distal CCA (DCC) [125]. Although CCA is a relatively rare malignant tumor; however, its incidence has increased in the last decade due to a rise in ICC [125]. The three subtypes have different clinical characteristics, therapeutic strategies, and prognosis. Considering the features of intratumor heterogeneity, one subtype can also be divided into different subgroups. A recent scRNA-seq analysis-based study revealed the intratumoral diversity of ICC cells. The malignant cells can be classified by four subclusters: subcluster 0 malignant cells highly expressed markers related to the EMT process, subcluster 1 showed enrichment in cell-cycle and hypoxia-dominant signature, subcluster 2 exhibited high expression of immune-related genes and subcluster 3 malignant cells highly expressed SPINK1, which was closely associated with poor prognosis in ICC [47]. Unlike the histological features of HCC, the most prominent hallmark of CCA is the abundant desmoplastic stroma infiltration within the tumor, in which the presence of CAFs is responsible for the dense stroma of CCA [126]. By using immunohistochemical staining for α-SMA, the degree of CAFs infiltration in different CCA samples can be determined [127]. Other markers, such as FSP-1, PDGFR, can also be used to identify CAFs in CCA, among which positive FSP-1 has the highest expression rate in CAFs, reaching 84.5% [128]. Interestingly, however, unlike other solid tumors, such as breast and pancreatic cancer, where abundant stroma is associated with poor patient outcomes [129, 130], in ICC, patients with high proportion of stroma area exhibit a better disease-free survival, which indicates that desmoplastic stroma seems to exert a protective effect [131].
The cellular origin of CAFs in CCA
The origin of CAFs in CCA is still not very clear, and previous studies suggested that it may be derived from activated HSCs or PFs [132, 133]. Recently, a study used Lrat-Cre-driven lox-stop-lox-TdTomato (TdTom) system to label HSCs in two ICC murine models, they found that in the context of ICC, 85%–95% of Col1a1-GFP + CAFs and 85%–93% of α-SMA + CAFs came from HSCs. Subsequently, they confirmed that HSCs-originated CAFs represented the subclusters with the most ligand-receptor interactions with tumor cells via scRNA-seq analysis of murine as well as human CCA samples [46].
Hence, given the unique properties of CCA, comprehending the activation status of CAFs in the stroma, specific CAFs subtypes, and the interaction of CAFs with surrounding cells provides new insights into the malignant progression and treatment of CCA.
The roles of CAFs in CCA progression
Previous studies have shown that CAFs could produce various factors in the TME to promote the progression of CCA. For instance, SDF-1 secreted by CAFs binds to CXCR4 on the surface of CCA cells and mediates the invasion of CCA through the ERK1/2 and AKT pathways [134]. HGF could also be released by CAFs to stimulate CCA cells invasion in vitro assay [135]. In addition, some ECM components secreted by CAFs, such as periostin, tenascin-C, were involved in tumor migration and invasion [136, 137], and a variety of matrix metalloproteases (MMPs), including MMP1, MMP2, and MMP9, could also be produced by CAFs, which influenced tumor development via mediating ECM remodeling [138,139,140]. It is worth noting that CAFs in CCA are also heterogeneous, and different subtypes of CAFs can affect tumor development through distinct mechanisms. According to a recent study based on scRNA-seq analysis, CAFs in ICC can be categorized into three subpopulations, inflammatory and growth factor-enriched CAFs (iCAFs), myofibroblastic CAFs (myCAFs), and mesothelial CAFs (mesCAFs). iCAFs mediated ICC growth through the HGF-MET axis, while myCAFs promoted ICC progression by producing hyaluronan synthase 2 rather than type I collagen [46]. Another scRNA-seq analysis employed a negative selection strategy to enrich fibroblasts, dividing ICC-infiltrating CAFs into 5 subclusters, apart from myCAF and iCAF, vascular CAFs (vCAFs), antigen-presenting CAFs (apCAFs), EMT-like CAFs (eCAFs), and lipid metabolism-related fibroblasts are also classified [47], the molecular characteristics of each subgroup were summarized in Table 1.
CAFs enhance chemoresistance of CCA cells
As mentioned above, CD90-positive CAFs secreted PIGF to promote angiogenesis in HCC. Likewise, PIGF is mainly expressed in stromal cells and is associated with poor prognosis in CCA. In vivo studies have shown that by blocking PIGF production in CAFs, the stiffness of the tumor could be weakened, thereby improving the hypoxic status, and enhancing the blood supply of the tumor, which was more conducive to the application of chemotherapeutic drugs [141]. The results suggested that by antagonizing some cytokines secreted by CAFs, its chemoresistance effect could be attenuated. Tyrosine kinase inhibitors (TKI), such as erlotinib, are administrated to treat CCA, however, they did not provide significant improvement of survival in clinical trials in CCA [142]. A recent study found that erlotinib-resistant CCA cells highly expressed insulin receptor (IR) and insulin-like growth factor (IGF) 1 receptor (IGF1R), and In vivo tumor formation model constructed from these resistant cells showed rich CAFs infiltration. Meanwhile, CAFs-secreted IGF2 stimulated IR/IGF1R signaling activation in resistant cells, which in turn promoted CAFs proliferation and activation for CCA-CAF interaction [143]. Additionally, in a 3D co-culture model, CAFs have a significant impact on reducing the sensitivity of CCA cells to chemotherapeutic drugs, including gemcitabine, cisplatin, and 5-fluorouracil (5-FU), but the insufficient effect on erlotinib [144]. The previous study has demonstrated that IL-6 secreted by CAFs could inhibit autophagy in CCA cells to stimulate CCA progression [145]. Moreover, IL-6 released by CAFs could educate neighboring CCA cells, rendering them less sensitive to chemotherapeutics via inhibiting the autophagy stress-response to the drug [146]. MiR-206 acted as a suppressor factor in liver cancer and played a suppressive role in the activation of HSCs, the crosstalk of CAFs with CCA cells reduced MiR-206 expression, which induced the conversion of NFs into CAFs and enhanced their secretion of IL6. When overexpressed MiR-206, the mutual interplay between CAFs and CCA was attenuated, and the resistance to gemcitabine was also been blockage [147].
The role of CAFs on stemness in CCA
In most solid tumors, CSCs occupy only a small proportion, however, in CCA, CSCs account for up to 30% of the tumor bulk, indicating that CCA is a CSCs-based tumor [148]. Several CSCs surface markers have been identified in CCA, such as CD133, epithelial cell adhesion molecule (EpCAM), CD44, CD13, and CD90 [149,150,151,152,153]. The supportive ‘CSCs niche’ formed by the interaction of the abundant seeds ‘tumor cells and fertile soil ‘CAFs’ can maintain the proliferation and self-renewal of CSCs. According to the scRNA-seq data analysis mentioned above, one of the CAFs subsets, CD146-positive vascular CAFs (vCAFs), could secrete IL6 and significantly enhance the stemness ability of CCA [47]
In addition to direct regulation of CSCs, CAFs can also indirectly sustain tumor stemness by influencing the immune microenvironment. A previous study has shown that a subset of CAFs, FAP + CAFs, could recruit MDSCs into the TME by secreting CCL2 to exert immunosuppressive function [154]. Moreover, accumulating evidence has demonstrated that MDSCs could promote cancer stemness in a paracrine fashion [155]. Recently, a study showed that CAFs may indirectly regulate tumor stemness by educating MDSCs in the TME. By using an orthotopic ICC model, co-injection of CAFs and ICC cells in the livers of nude mice significantly induced the stemness of cancer, which could be attenuated by depletion of CAFs or MDSCs. Mechanistically, IL-6 and IL-33 secreted by CAFs stimulated the hyperactivated 5-lipoxygenase (5-LO) metabolism in CD33 + MDSCs, contributing to a large accumulation of downstream metabolite leukotriene B4 (LTB4). Abundant LTB4 acted on its receptor Leukotriene B4 receptor type 2 (BLT2) in ICC cells to promote ICC stemness via activation of PI3K/Akt-mTORC1 signaling [90]. This study also revealed that in addition to cytokines secreted by multiple cells that can enhance tumor stemness, amino acid metabolism is also involved in the regulation of tumor stemness.
CAFs modulate immune responses in CCA
TILs in CCA also own their specific infiltration characteristics. According to the latest scRNA-seq data analysis of CCA, T cells and NK cells in the CCA were divided into 8 distinct subsets, and it was found that CD8 + T cells were in an exhausted status, and CD4 + Foxp3 + Treg cells were enriched in the TME while exhibiting a highly immunosuppressive feature [47]. CAFs also modulate the function of immune cells to drive an immunosuppressive microenvironment. Several studies have identified the interaction between CAFs and MDSCs. As mentioned above, FAP + CAFs could enhance MDSCs recruitment by secreting CCL2, and this tumor-promoting effect relied on MDSCs but independently of their immunosuppressive function [89]. After recruitment of MDSCs into the TME, CAFs set out to further educate MDSCs to enhance their stemness capacity via 5-LO/LTB4-BLT2 signaling [90]. Caveolin-1 (CAV1) has previously been reported to play a major role in cellular senescence, but its effect on tumors depends on the cancer type [156,157,158]. In ICC, CAV1 was highly expressed in CAFs, and CAV1 + CAFs were associated with poor prognosis in ICC patients. Immunohistochemical analysis showed that CAV1 + CAFs also correlated with infiltration of Foxp3 + TILs, which suggested that CAV1 + CAFs might recruit Tregs into the TME to mediate the prognosis of ICC [93]. Co-culturing of CAFs and CCA cells up-regulated CXCL5 expression in CCA cells, and secreted CXCL5 could promote the migration and invasion of CCA. Furthermore, CXCL5 had a direct recruitment effect on TANs in HCC [159]. Similarly, CD66 + TANs were positively correlated with CXCL5 in CCA tissues, and CXCL5 secreted by stromal cells mediated TANs chemotaxis through activation of PI3K-AKT and ERK1/2 pathways [91, 92].
CAFs regulate angiogenesis in CCA
Unlike HCC, CCA is a hypovascular tumor, and the dense stroma constructed by abundant CAFs induces the collapse of tumor blood vessels to form a hypoxic microenvironment. In addition to structural factors, CAFs can also secrete cytokines to mediate the angiogenesis of CCA. The PDGF family has been reported to promote the HSCs migration and proliferation, among which PDGF-D secreted by CCA cells promoted CAFs recruitment via PDGFRβ and Rho GTPase and JNK activation in CCA [160]. Hypoxia-induced PDGF secretion from CCA cells could bind to the receptors PDGFRβ expressed on the CAFs, thereby stimulating CAFs to produce VEGF-A and VEGF-C. In CCA, CAFs and lymphatic endothelial cells were spatially adjacent to each other, moreover, the lymphatic vessels provided an important dissemination pathway for the metastasis of CCA cells. Ultimately, VEGF secreted by CAFs promoted the chemotaxis and assembly of lymphatic endothelial cells to form the appropriate lymphatic vasculature that supported CCA invasion [161]. PlGF, a member of the VEGF family, which has been documented to enhance VEGF-driven angiogenesis by binding to VEGFR1 and Nrp1 on endothelial cells [162], was recently demonstrated to be produced by CAFs to deteriorate the hypoxic state in CCA through enhancing the stiffness of the stroma, compressing the tumor vessels [141]. In addition, it was worth noting that, unlike VEGF, PIGF was only up-regulated in pathological conditions such as hypoxia, and undetectable in healthy conditions [163]. Therefore, PIGF could be targeted to selectively inhibit the angiogenesis in pathological conditions without interfering normal blood vessel growth.
Activation of CAFs in CCA
Similarly, the mutual communications between CCA cells and CAFs also make CCA cells secrete some cytokines to promote the activation and function enhancement of CAFs to further aggravate tumor progression. This vicious cycle could be initiated by CAFs-generated heparin-binding epidermal growth factor (HB-EGF), which bound to epidermal growth factor receptor (EGFR) on CCA cells to promote CCA cells invasion through (ERK)1/2 and STAT3 signaling pathways. Activation of EGFR in CCA cells could produce TGF-β, which facilitated CAFs activation and HB-EGF production [164]. Previous studies reported that the interaction of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor fibroblast growth factor-inducible 14 (Fn14) could lead to the progression of liver fibrosis by modulating myofibroblast proliferation [165]. In CCA, TWEAK overexpression promoted the proliferation of collagen-producing CAFs, which expressed Fn14 in a substantial proportion of CCA patients. Accordingly, the TWEAK/Fn14 signaling axis may serve as an early driver of tumor niche development to promote tumor growth in CCA [166]. CCA-derived exosomes also mediate CAFs activation, the expression level of miR-34c was decreased in exosomes derived from CCA, and downregulation of miR-34c could induce CAFs activation by targeting the Wnt signaling pathway in CCA [167]. Zinc Finger E-Box Binding Homeobox 1 (ZEB1) as a transcription factor was associated with poor prognosis in ICC and ZEB1 expression in ICC cells induced tumor EMT and stemness phenotype. Besides, ZEB1 was involved in the interplay between CCA cells and CAFs by regulating the expression of HGF and IL6 to promote CCA progression. In vitro studies showed that CTGF in the supernatants of ZEB1-overexpressing CCA cells promoted the proliferation of myofibroblasts. Mechanistically, ZEB1 upregulated CTGF expression by directly binding to the promoter of CTGF. Furthermore, ZEB1 was also expressed on CAFs and contributed to the activation of stromal CAFs [168]. PIGF secreted by ICC cells promoted the activation of CAFs to acquire a myofibroblast-like phenotype via AKT/NF-kB pathway, then activated CAFs could, in turn, promote ICC cells invasion [141]. Programmed cell death ligand 1 (PD-L1) as a known immune checkpoint molecule could induce immune tolerance in the TME, however, a recent study found that PDL1 expressed by HSCs was required for the transformation of HSCs into myofibroblasts but was independent of the immunosuppressive function of PDL1. Mechanistically, the PD-L1 extracellular domain bound to TGF-β receptors II (TβRII) of HSCs to protect TβRII from lysosomal degradation, and the cytoplasmic domain of PD-L1 protected TGF-β receptors I (TβRI) mRNA from degradation by the RNA exosome complex in HSCs [169].
Conclusions
In recent years, accumulating studies have emphasized the non-negligible role of CAFs as the main component of tumor stroma in the TME of liver cancer. In this review, we mainly summarized the cellular origin of CAFs and several aspects that CAFs implicated in the progression of liver cancer: chemotherapy resistance, tumor stemness maintenance, induction of immunosuppressive microenvironment, tumor angiogenesis, and activation of CAFs by liver cancer cells (Fig. 2), which were summarized in Table 3. Using modern scRNA-seq analysis and gene lineage tracing techniques have identified HSCs as the major cellular source of CAFs and also revealed the heterogeneity of CAFs within liver cancer. However, it should be noted that the use of scRNA-seq analysis to identify specific subsets of CAFs is based on the transcriptional expression levels of signature genes, and this analysis does not totally reflect the protein expression profiling. Therefore, it is the necessity to explore the distinct function of CAFs subpopulations, including the use of 3D cultures, organoids that more closely mimic the in vivo microenvironment, and transgenic mouse models and patient-derived xenografts models.

The origin and the role of CAFs in liver cancer. Schematic illustration of potential cellular origins of CAFs, including epithelial cells, endothelial cells, tumor cells, HSCs, portal fibroblasts, and MSCs. The upper part of the picture shows the the influence exerted by CAFs in the TME, including chemoresistance, stemness induction, tumor immune, angiogenesis, and aggressiveness
Based on the biological research of CAFs in liver cancer, currently, many novel therapeutic strategies targeting CAFs have been explored and developed, such as targeting the precursors of CAFs, mainly by inhibiting the activation of HSCs. Alternatively, blocking the tumor-promoting factors that CAFs-produced and the CAFs-mediated signaling pathways, such as developing inhibitors against IL6, CTGF, TGF-β, and SMAD, p-STAT3 signaling, etc. In addition, the application of nano-delivery systems makes it possible to precisely target and eliminate tumor-promoting CAFs and tumor cells. However, due to the lack of tumor clinical trials and considering the existence of tumor-suppressive CAFs, more efforts should be devoted to ensuring the clinical safety and translation of various nanoparticle formulations.
In conclusion, based on the in-depth research on the diverse functions of CAFs, the more precise elimination of tumor-promoting CAFs subsets or systemic combination therapy could become an effective strategy for the liver cancer treatment.
Availability of data and materials
Not applicable.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- CCA:
-
Cholangiocarcinoma
- TME:
-
Tumor microenvironment
- CAFs:
-
Cancer-associated fibroblasts
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- NAFLD:
-
Nonalcoholic fatty liver disease
- NASH:
-
Nonalcoholic steatohepatitis
- ECM:
-
Extracellular matrix
- PDAC:
-
Pancreatic ductal adenocarcinoma
- MMP:
-
Matrix metalloproteinase
- α-SMA:
-
α-Smooth muscle actin
- FAP:
-
Fibroblast activation protein
- PDGF:
-
Platelet-derived growth factor
- FSP-1:
-
Fibroblast-specific protein 1
- TGF-β:
-
Transforming growth factor-β
- HGF:
-
Hepatocyte growth factor
- IGF:
-
Insulin-like growth factor
- IL-6:
-
Interleukin-6
- HSCs:
-
Hepatic stellate cells
- MSCs:
-
Mesenchymal stem cells
- scRNA-seq:
-
Single-cell RNA sequencing
- Lrat-Cre:
-
Lecithin retinol acyltransferase-cyclization recombination enzyme
- Tcf21:
-
Transcription factor 21
- EMT:
-
Epithelial-mesenchymal transition
- EndMT:
-
Endothelial to mesenchymal transition
- PFs:
-
Portal fibroblasts
- LPA:
-
Lysophostatidic acid
- RCN1:
-
Reticulocalbin 1
- BAFF:
-
B-cell activating factor
- CSCs:
-
Cancer stem cells
- FSTL1:
-
Follistatin-like 1
- CLCF1:
-
Cardiotrophin-like cytokine factor 1
- ERK:
-
Extracellular signal-regulated kinase
- LXRs:
-
Liver X receptors
- RvD1:
-
Resolvin D1
- TILs:
-
Tumor-infiltrating lymphocytes
- TAMs:
-
Tumor-associated macrophages
- TANs:
-
Tumor-associated neutrophils
- Tregs:
-
Regulatory T cells
- MDSCs:
-
Myeloid-derived suppressor cells
- TIME:
-
Tumor immune microenvironment
- LPS:
-
Lipopolysaccharides
- IFN-γ:
-
Interferon-γ
- TNF:
-
Tumor necrosis factor
- CXCL12:
-
C‑X‑C motif chemokine ligand 12
- PAI‑1:
-
Plasminogen activator inhibitor‑1
- GAS6:
-
Growth arrest-specific protein 6
- PGE2:
-
Prostaglandin E2
- IDO:
-
Indoleamine 2,3-dioxygenase
- ROS:
-
Reactive oxygen species
- SDF:
-
Stromal cell-derived factor
- MCP-1 :
-
Monocyte chemotactic protein-1
- COX-2 :
-
Cyclooxygenase-2
- VEGF :
-
Vascular endothelial growth factor
- HUVECs:
-
Human umbilical vein endothelial cells
- PlGF:
-
Placental growth factor
- VM:
-
Vasculogenic Mimicry
- CTGF:
-
Connective tissue growth factor
- TIMPs :
-
Matrix metalloproteinases
- SULF2:
-
Sulfatase 2
- ER :
-
Endoplasmic reticulum
- IREα:
-
Inositol requiring enzyme 1α
- SASP:
-
Senescence associated secretory phenotype
- BMP4 :
-
Bone morphogenetic protein 4
- EVs :
-
Extracellular vesicles
- ICC:
-
Intrahepatic CCA
- PCC :
-
Perihilar CCA
- DCC :
-
Distal CCA
- TKI :
-
Tyrosine kinase inhibitors
- IGF2 :
-
Insulin-like growth factor 2
- EGFR :
-
Epidermal growth factor receptor
- ZEB1 :
-
Zinc Finger E-Box Binding Homeobox 1
- PD-L1 :
-
Programmed cell death ligand 1
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Medavaram S, Zhang Y. Emerging therapies in advanced hepatocellular carcinoma. Exp Hematol Oncol. 2018;7:17.
Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18(4):223–38.
Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, Allen C, et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the Global, Regional, and National level: results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017;3(12):1683–91.
Omata M, Cheng AL, Kokudo N, Kudo M, Lee JM, Jia J, et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol Int. 2017;11(4):317–70.
Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Bozzetti F, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med. 1996;334(11):693–9.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90.
Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–73.
Llovet JM, Castet F, Heikenwalder M, Maini MK, Mazzaferro V, Pinato DJ, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19(3):151–72.
EbioMedicine TT. Microenvironment: a druggable target for metastatic disease? EBioMedicine. 2018;31:1–2.
Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591–6.
Mizutani Y, Kobayashi H, Iida T, Asai N, Masamune A, Hara A, et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019;79(20):5367–81.
Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76(1):69–125.
De Wever O, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008;123(10):2229–38.
Lazard D, Sastre X, Frid MG, Glukhova MA, Thiery JP, Koteliansky VE. Expression of smooth muscle-specific proteins in myoepithelium and stromal myofibroblasts of normal and malignant human breast tissue. Proc Natl Acad Sci U S A. 1993;90(3):999–1003.
Sukowati CH, Anfuso B, Croce LS, Tiribelli C. The role of multipotent cancer associated fibroblasts in hepatocarcinogenesis. BMC Cancer. 2015;15:188.
Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99(Pt B):186–96.
Peng H, Xue R, Ju Z, Qiu J, Wang J, Yan W, et al. Cancer-associated fibroblasts enhance the chemoresistance of CD73(+) hepatocellular carcinoma cancer cells via HGF-Met-ERK1/2 pathway. Ann Transl Med. 2020;8(14):856.
Fang M, Yuan J, Chen M, Sun Z, Liu L, Cheng G, et al. The heterogenic tumor microenvironment of hepatocellular carcinoma and prognostic analysis based on tumor neo-vessels, macrophages and alpha-SMA. Oncol Lett. 2018;15(4):4805–12.
Jia CC, Wang TT, Liu W, Fu BS, Hua X, Wang GY, et al. Cancer-associated fibroblasts from hepatocellular carcinoma promote malignant cell proliferation by HGF secretion. PLoS ONE. 2013;8(5):e63243.
Khan GJ, Sun L, Khan S, Yuan S, Nongyue H. Versatility of cancer associated fibroblasts: commendable targets for anti-tumor therapy. Curr Drug Targets. 2018;19(13):1573–88.
Glentis A, Oertle P, Mariani P, Chikina A, El Marjou F, Attieh Y, et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat Commun. 2017;8(1):924.
Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev. 2021;101(1):147–76.
Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823.
Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, Griggs DW, Prinsen MJ, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19(12):1617–24.
Senoo H, Mezaki Y, Fujiwara M. The stellate cell system (vitamin A-storing cell system). Anat Sci Int. 2017;92(4):387–455.
Yavuz BG, Pestana RC, Abugabal YI, Krishnan S, Chen J, Hassan MM, et al. Origin and role of hepatic myofibroblasts in hepatocellular carcinoma. Oncotarget. 2020;11(13):1186–201.
Fabregat I, Moreno-Caceres J, Sanchez A, Dooley S, Dewidar B, Giannelli G, et al. TGF-beta signalling and liver disease. FEBS J. 2016;283(12):2219–32.
Wright JH, Johnson MM, Shimizu-Albergine M, Bauer RL, Hayes BJ, Surapisitchat J, et al. Paracrine activation of hepatic stellate cells in platelet-derived growth factor C transgenic mice: evidence for stromal induction of hepatocellular carcinoma. Int J Cancer. 2014;134(4):778–88.
Wang SS, Tang XT, Lin M, Yuan J, Peng YJ, Yin X, et al. Perivenous stellate cells are the main source of myofibroblasts and cancer-associated fibroblasts formed after chronic liver injuries. Hepatology. 2021;74(3):1578–94.
Bhattacharjee S, Hamberger F, Ravichandra A, Miller M, Nair A, Affo S, et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest. 2021;131(11):e146987.
Ding DC, Shyu WC, Lin SZ. Mesenchymal stem cells. Cell Transplant. 2011;20(1):5–14.
Yin Z, Jiang K, Li R, Dong C, Wang L. Multipotent mesenchymal stromal cells play critical roles in hepatocellular carcinoma initiation, progression and therapy. Mol Cancer. 2018;17(1):178.
Salah RA, Nasr MA, El-Derby AM, AbdElkodous M, Mohamed RH, El-Ekiaby N, et al. Hepatocellular carcinoma cell line-microenvironment induced cancer-associated phenotype, genotype and functionality in mesenchymal stem cells. Life Sci. 2022;288:120168.
Bhattacharya SD, Mi Z, Talbot LJ, Guo H, Kuo PC. Human mesenchymal stem cell and epithelial hepatic carcinoma cell lines in admixture: concurrent stimulation of cancer-associated fibroblasts and epithelial-to-mesenchymal transition markers. Surgery. 2012;152(3):449–54.
Yeon JH, Jeong HE, Seo H, Cho S, Kim K, Na D, et al. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. 2018;76:146–53.
Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282(32):23337–47.
Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, Penz-Osterreicher M, et al. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51(3):1027–36.
Zou B, Liu X, Zhang B, Gong Y, Cai C, Li P, et al. The expression of FAP in hepatocellular carcinoma cells is induced by hypoxia and correlates with poor clinical outcomes. J Cancer. 2018;9(18):3278–86.
Moren A, Bellomo C, Tsubakihara Y, Kardassis D, Mikulits W, Heldin CH, et al. LXRalpha limits TGFbeta-dependent hepatocellular carcinoma associated fibroblast differentiation. Oncogenesis. 2019;8(6):36.
Wang H, Feng C, Lu M, Zhang B, Xu Y, Zeng Q, et al. Integrative single-cell transcriptome analysis reveals a subpopulation of fibroblasts associated with favorable prognosis of liver cancer patients. Transl Oncol. 2021;14(1):100981.
Wells RG. The portal fibroblast: not just a poor man’s stellate cell. Gastroenterology. 2014;147(1):41–7.
Mazzocca A, Dituri F, Lupo L, Quaranta M, Antonaci S, Giannelli G. Tumor-secreted lysophostatidic acid accelerates hepatocellular carcinoma progression by promoting differentiation of peritumoral fibroblasts in myofibroblasts. Hepatology. 2011;54(3):920–30.
Huang P, Kong Q, Gao W, Chu B, Li H, Mao Y, et al. Spatial proteome profiling by immunohistochemistry-based laser capture microdissection and data-independent acquisition proteomics. Anal Chim Acta. 2020;1127:140–8.
Chiavarina B, Ronca R, Otaka Y, Sutton RB, Yokobori T, Rezzola S, Chiodelli P, Souche R, Pourquier D, Maraver A, Faa G, Khellaf L, Turtoi E, Oyama T, Gofflot S, Bellahcène A, Detry O, Delvenne P, Castronovo V, Nishiyama M, Turtoi A. Fibroblast-derived prolargin is a tumor suppressor in hepatocellular carcinoma. Oncogene. 2022;41(10):1410–20.
Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell. 2021;39(6):883.
Zhang M, Yang H, Wan L, Wang Z, Wang H, Ge C, et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol. 2020;73(5):1118–30.
Wang JW, Ma L, Liang Y, Yang XJ, Wei S, Peng H, et al. RCN1 induces sorafenib resistance and malignancy in hepatocellular carcinoma by activating c-MYC signaling via the IRE1alpha-XBP1s pathway. Cell Death Discov. 2021;7(1):298.
Gao L, Morine Y, Yamada S, Saito Y, Ikemoto T, Tokuda K, Miyazaki K, Okikawa S, Takasu C, Shimada M. The BAFF/NFκB axis is crucial to interactions between sorafenib-resistant HCC cells and cancer-associated fibroblasts. Cancer Sci. 2021;112(9):3545–54.
Liu YC, Yeh CT, Lin KH. Cancer stem cell functions in hepatocellular carcinoma and comprehensive therapeutic strategies. Cells. 2020;9(6):1331.
Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 2016;15(6):1175–89.
Ma XL, Hu B, Tang WG, Xie SH, Ren N, Guo L, et al. CD73 sustained cancer-stem-cell traits by promoting SOX9 expression and stability in hepatocellular carcinoma. J Hematol Oncol. 2020;13(1):11.
Kawai T, Yasuchika K, Ishii T, Katayama H, Yoshitoshi EY, Ogiso S, et al. Keratin 19, a cancer stem cell marker in human hepatocellular carcinoma. Clin Cancer Res. 2015;21(13):3081–91.
Rhee H, Kim HY, Choi JH, Woo HG, Yoo JE, Nahm JH, et al. Keratin 19 expression in hepatocellular carcinoma is regulated by fibroblast-derived HGF via a MET-ERK1/2-AP1 and SP1 Axis. Cancer Res. 2018;78(7):1619–31.
Xiong S, Wang R, Chen Q, Luo J, Wang J, Zhao Z, et al. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Cancer Res. 2018;8(2):302–16.
Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123(5):1911–8.
Li Y, Wang R, Xiong S, Wang X, Zhao Z, Bai S, et al. Cancer-associated fibroblasts promote the stemness of CD24(+) liver cells via paracrine signaling. J Mol Med (Berl). 2019;97(2):243–55.
Zhao Z, Xiong S, Wang R, Li Y, Wang X, Wang Y, et al. Peri-tumor fibroblasts promote tumorigenesis and metastasis of hepatocellular carcinoma via Interleukin6/STAT3 signaling pathway. Cancer Manag Res. 2019;11:2889–901.
Jiang J, Ye F, Yang X, Zong C, Gao L, Yang Y, et al. Peri-tumor associated fibroblasts promote intrahepatic metastasis of hepatocellular carcinoma by recruiting cancer stem cells. Cancer Lett. 2017;404:19–28.
Loh JJ, Li TW, Zhou L, Wong TL, Liu X, Ma VWS, et al. FSTL1 secreted by activated fibroblasts promotes hepatocellular carcinoma metastasis and stemness. Cancer Res. 2021;81(22):5692–705.
Song M, He J, Pan QZ, Yang J, Zhao J, Zhang YJ, et al. Cancer-associated fibroblast-mediated cellular crosstalk supports hepatocellular carcinoma progression. Hepatology. 2021;73(5):1717–35.
Benayoun BA, Caburet S, Veitia RA. Forkhead transcription factors: key players in health and disease. Trends Genet. 2011;27(6):224–32.
Luo Q, Wang CQ, Yang LY, Gao XM, Sun HT, Zhang Y, et al. FOXQ1/NDRG1 axis exacerbates hepatocellular carcinoma initiation via enhancing crosstalk between fibroblasts and tumor cells. Cancer Lett. 2018;417:21–34.
Song YJ, Zhang SS, Guo XL, Sun K, Han ZP, Li R, et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013;339(1):70–81.
Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST, et al. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol Cell. 2017;68(2):281-292 35.
Zhao Z, Bai S, Wang R, Xiong S, Li Y, Wang X, et al. Cancer-associated fibroblasts endow stem-like qualities to liver cancer cells by modulating autophagy. Cancer Manag Res. 2019;11:5737–44.
Liu C, Liu L, Chen X, Cheng J, Zhang H, Shen J, et al. Sox9 regulates self-renewal and tumorigenicity by promoting symmetrical cell division of cancer stem cells in hepatocellular carcinoma. Hepatology. 2016;64(1):117–29.
Liu C, Liu L, Chen X, Cheng J, Zhang H, Zhang C, et al. LSD1 stimulates cancer-associated fibroblasts to drive Notch3-dependent self-renewal of liver cancer stem-like cells. Cancer Res. 2018;78(4):938–49.
Sun L, Wang Y, Wang L, Yao B, Chen T, Li Q, et al. Resolvin D1 prevents epithelial-mesenchymal transition and reduces the stemness features of hepatocellular carcinoma by inhibiting paracrine of cancer-associated fibroblast-derived COMP. J Exp Clin Cancer Res. 2019;38(1):170.
Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–95.
Hao X, Sun G, Zhang Y, Kong X, Rong D, Song J, et al. Targeting immune cells in the tumor microenvironment of HCC: new opportunities and challenges. Front Cell Dev Biol. 2021;9:775462.
Huang Y, Ge W, Zhou J, Gao B, Qian X, Wang W. The role of tumor associated macrophages in hepatocellular carcinoma. J Cancer. 2021;12(5):1284–94.
Yin Z, Ma T, Lin Y, Lu X, Zhang C, Chen S, et al. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J Cell Biochem. 2018;119(11):9419–32.
Chen S, Morine Y, Tokuda K, Yamada S, Saito Y, Nishi M, et al. Cancer-associated fibroblast-induced M2-polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor-1 pathway. Int J Oncol. 2021;59(2):59.
Christian S, Ahorn H, Koehler A, Eisenhaber F, Rodi HP, Garin-Chesa P, et al. Molecular cloning and characterization of endosialin, a C-type lectin-like cell surface receptor of tumor endothelium. J Biol Chem. 2001;276(10):7408–14.
O’Shannessy DJ, Dai H, Mitchell M, Huntsman S, Brantley S, Fenstermacher D, et al. Endosialin and associated protein expression in soft tissue sarcomas: a potential target for anti-endosialin therapeutic strategies. Sarcoma. 2016;2016:5213628.
Yang F, Wei Y, Han D, Li Y, Shi S, Jiao D, et al. Interaction with CD68 and regulation of GAS6 expression by endosialin in fibroblasts drives recruitment and polarization of macrophages in hepatocellular carcinoma. Cancer Res. 2020;80(18):3892–905.
Arvanitakis K, Mitroulis I, Germanidis G. Tumor-associated neutrophils in hepatocellular carcinoma pathogenesis, prognosis, and therapy. Cancers. 2021;13(12):2899.
Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9(4):422.
Bozward AG, Warricker F, Oo YH, Khakoo SI. Natural killer cells and regulatory T cells cross talk in hepatocellular carcinoma: exploring therapeutic options for the next decade. Front Immunol. 2021;12:643310.
Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012;318(2):154–61.
Han Y, Chen Z, Yang Y, Jiang Z, Gu Y, Liu Y, et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology. 2014;59(2):567–79.
Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. 2016;5(2):e198.
Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13(10):739–52.
Wang YA-O, Zhang T, Sun M, Ji X, Xie M, Huang W, et al. Therapeutic values of myeloid-derived suppressor cells in hepatocellular carcinoma: facts and hopes. Cancers. 2021;13(20):5127.
Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene. 2017;36(8):1090–101.
Zhang H, He G, Kong Y, Chen Y, Wang B, Sun X, et al. Tumour-activated liver stromal cells regulate myeloid-derived suppressor cells accumulation in the liver. Clin Exp Immunol. 2017;188(1):96–108.
Li J, Li H, Yu Y, Liu Y, Liu Y, Ma Q, et al. Mannan-binding lectin suppresses growth of hepatocellular carcinoma by regulating hepatic stellate cell activation via the ERK/COX-2/PGE(2) pathway. Oncoimmunology. 2018;8(2):e1527650.
Lin Y, Li B, Yang X, Cai Q, Liu W, Tian M, et al. Fibroblastic FAP promotes intrahepatic cholangiocarcinoma growth via MDSCs recruitment. Neoplasia. 2019;21(12):1133–42.
Lin Y, Cai Q, Chen Y, Shi T, Liu W, Mao L, et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology. 2022;75(1):28–42.
Zhou SL, Dai Z, Zhou ZJ, Chen Q, Wang Z, Xiao YS, et al. CXCL5 contributes to tumor metastasis and recurrence of intrahepatic cholangiocarcinoma by recruiting infiltrative intratumoral neutrophils. Carcinogenesis. 2014;35(3):597–605.
Okabe H, Beppu T, Ueda M, Hayashi H, Ishiko T, Masuda T, Otao R, Horlad H, Mima K, Miyake K, Iwatsuki M, Baba Y, Takamori H, Jono H, Shinriki S, Ando Y, Baba H. Identification of CXCL5/ENA-78 as a factor involved in the interaction between cholangiocarcinoma cells and cancer-associated fibroblasts. Int J Cancer. 2012;131(10):2234–41.
Lan C, Kitano Y, Yamashita YI, Yamao T, Kajiyama K, Yoshizumi T, et al. Cancer-associated fibroblast senescence and its relation with tumour-infiltrating lymphocytes and PD-L1 expressions in intrahepatic cholangiocarcinoma. Br J Cancer. 2022;126(2):219–27.
Huang B, Huang M, Li Q. Cancer-associated fibroblasts promote angiogenesis of hepatocellular carcinoma by VEGF-Mediated EZH2/VASH1 Pathway. Technol Cancer Res Treat. 2019;18:1533033819879905.
Ho, M.C., H. Chen Cn Fau - Lee, F.-J. Lee H Fau - Hsieh, C.-T. Hsieh Fj Fau - Shun, C.-L. Shun Ct Fau - Chang, Y.-T. Chang Cl Fau - Lai, et al. Placenta growth factor not vascular endothelial growth factor A or C can predict the early recurrence after radical resection of hepatocellular carcinoma. Cancer Lett, 2007. 250(2): 237–49.
Liu Z, Chen M, Zhao R, Huang Y, Liu F, Li B, et al. CAF-induced placental growth factor facilitates neoangiogenesis in hepatocellular carcinoma. Acta Biochim Biophys Sin (Shanghai). 2020;52(1):18–25.
Yang J, Lu Y, Lin YY, Zheng ZY, Fang JH, He S, et al. Vascular mimicry formation is promoted by paracrine TGF-β and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett. 2016;383(1):18–27.
Hu H, Zhang H, Ge W, Liu X, Loera S, Chu P, et al. Secreted protein acidic and rich in cysteines-like 1 suppresses aggressiveness and predicts better survival in colorectal cancers. Clin Cancer Res. 2012;18(19):5438–48.
Hurley PJ, Marchionni L, Simons BW, Ross AE, Peskoe SB, Miller RM, Erho N, Vergara IA, Ghadessi M, Huang Z, Gurel B, Park BH, Davicioni E, Jenkins RB, Platz EA, Berman DM, Schaeffer EM. Secreted protein, acidic and rich in cysteine-like 1 (SPARCL1) is down regulated in aggressive prostate cancers and is prognostic for poor clinical outcome. Proc Natl Acad Sci U S A. 2012;109(37):14977–82.
Mazzocca A, Fransvea E, Dituri F, Lupo L, Antonaci S, Giannelli G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology. 2010;51(2):523–34.
Makino Y, Hikita H, Kodama T, Shigekawa M, Yamada R, Sakamori R, et al. CTGF mediates tumor-stroma interactions between hepatoma cells and hepatic stellate cells to accelerate HCC progression. Cancer Res. 2018;78(17):4902–14.
Song T, Dou C, Jia Y, Tu K, Zheng X. TIMP-1 activated carcinoma-associated fibroblasts inhibit tumor apoptosis by activating SDF1/CXCR4 signaling in hepatocellular carcinoma. Oncotarget. 2015;6(14):12061–79.
Wang C, Shang C, Gai X, Song T, Han S, Liu Q, et al. Sulfatase 2-induced cancer-associated fibroblasts promote hepatocellular carcinoma progression via inhibition of apoptosis and induction of epithelial-to-mesenchymal transition. Front Cell Dev Biol. 2021;9:631931.
Zhang R, Gao X, Zuo J, Hu B, Yang J, Zhao J, et al. STMN1 upregulation mediates hepatocellular carcinoma and hepatic stellate cell crosstalk to aggravate cancer by triggering the MET pathway. Cancer Sci. 2020;111(2):406–17.
Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569(1–2):29–63.
Acosta-Alvear D, Karagoz GE, Frohlich F, Li H, Walther TC, Walter P. The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife. 2018;7:e43036.
Pavlović N, Calitz C, Thanapirom K, Mazza G, Rombouts K, Gerwins P, et al. Inhibiting IRE1α-endonuclease activity decreases tumor burden in a mouse model for hepatocellular carcinoma. Elife. 2020;9:e55865.
Ruan K, Bao S, Ouyang G. The multifaceted role of periostin in tumorigenesis. Cell Mol Life Sci. 2009;66(14):2219–30.
Xiao H, Zhang Y, Li Z, Liu B, Cui D, Liu F, et al. Periostin deficiency reduces diethylnitrosamine-induced liver cancer in mice by decreasing hepatic stellate cell activation and cancer cell proliferation. J Pathol. 2021;255(2):212–23.
Chen Y, Qian B, Sun X, Kang Z, Huang Z, Ding Z, et al. Sox9/INHBB axis-mediated crosstalk between the hepatoma and hepatic stellate cells promotes the metastasis of hepatocellular carcinoma. Cancer Lett. 2021;499:243–54.
Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol. 2016;12(4):203–21.
Mano Y, Yoshio S, Shoji H, Tomonari S, Aoki Y, Aoyanagi N, et al. Bone morphogenetic protein 4 provides cancer-supportive phenotypes to liver fibroblasts in patients with hepatocellular carcinoma. J Gastroenterol. 2019;54(11):1007–18.
Nguyen PT, Kanno K, Pham QT, Kikuchi Y, Kakimoto M, Kobayashi T, et al. Senescent hepatic stellate cells caused by deoxycholic acid modulates malignant behavior of hepatocellular carcinoma. J Cancer Res Clin Oncol. 2020;146(12):3255–68.
Li F, Huangyang P, Burrows M, Guo K, Riscal R, Godfrey J, Lee KE, Lin N, Lee P, Blair IA, Keith B, Li B, Simon MC. FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat Cell Biol. 2020;22(6):728–39.
Gurunathan S, Kang MH, Jeyaraj M, Qasim M, Kim JH. Review of the isolation, characterization, biological function, and multifarious therapeutic approaches of exosomes. Cells. 2019;8(4):307.
Li J, Yan Y, Ang L, Li X, Liu C, Sun B, et al. Extracellular vesicles-derived OncomiRs mediate communication between cancer cells and cancer-associated hepatic stellate cells in hepatocellular carcinoma microenvironment. Carcinogenesis. 2020;41(2):223–34.
Zhou Y, Ren H, Dai B, Li J, Shang L, Huang J, et al. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J Exp Clin Cancer Res. 2018;37(1):324.
Lai C, Yeh K, Lin CY, Hsieh YW, Lai HH, Chen JR, et al. MicroRNA-21 plays multiple oncometabolic roles in the process of NAFLD-related hepatocellular carcinoma via PI3K/AKT, TGF-β, and STAT3 signaling. Cancers. 2021;13(5):940.
Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9(1):191.
Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, et al. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60(1):24–34.
Liu Z, Mo H, Liu R, Niu Y, Chen T, Xu Q, et al. Matrix stiffness modulates hepatic stellate cell activation into tumor-promoting myofibroblasts via E2F3-dependent signaling and regulates malignant progression. Cell Death Dis. 2021;12(12):1134.
Dou C, Liu Z, Tu K, Zhang H, Chen C, Yaqoob U, et al. P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology. 2018;154(8):2209–21.
Wang Y, Tu KA-O, Liu D, Guo L, Chen Y, Li Q, et al. p300 Acetyltransferase is a cytoplasm-to-nucleus shuttle for SMAD2/3 and TAZ nuclear transport in transforming growth factor β-stimulated hepatic stellate cells. Hepatology. 2019;70(4):1409–23.
Bertuccio P, Malvezzi M, Carioli G, Hashim D, Boffetta P, El-Serag HB, et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J Hepatol. 2019;71(1):104–14.
Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17(9):557–88.
Fabris L, Perugorria MJ, Mertens J, Bjorkstrom NK, Cramer T, Lleo A, et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int. 2019;39(Suppl 1):63–78.
Massani M, Stecca T, Fabris L, Caratozzolo E, Ruffolo C, Furlanetto A, et al. Isolation and characterization of biliary epithelial and stromal cells from resected human cholangiocarcinoma: a novel in vitro model to study tumor-stroma interactions. Oncol Rep. 2013;30(3):1143–8.
Zhang XF, Dong M, Pan YH, Chen JN, Huang XQ, Jin Y. Expression pattern of cancer-associated fibroblast and its clinical relevance in intrahepatic cholangiocarcinoma. Hum Pathol. 2017;65((1532–8392 (Electronic))):92–100.
Kramer CJH, Vangangelt KMH, van Pelt GW, Dekker TJA, Tollenaar R, Mesker WE. The prognostic value of tumour-stroma ratio in primary breast cancer with special attention to triple-negative tumours: a review. Breast Cancer Res Treat. 2019;173(1):55–64.
Erkan M, Michalski CW, Rieder S, Reiser-Erkan C, Abiatari I, Kolb A, Giese NA, Esposito I, Friess H, Kleeff J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2008;6(10):1155–61.
Guedj N, Blaise L, Cauchy F, Albuquerque M, Soubrane O, Paradis V. Prognostic value of desmoplastic stroma in intrahepatic cholangiocarcinoma. Mod Pathol. 2021;34(2):408–16.
Sirica AE, Campbell DJ, Dumur CI. Cancer-associated fibroblasts in intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2011;27(3):276–84.
Dranoff JA, Wells RG. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology. 2010;51(4):1438–44.
Gentilini A, Rombouts K, Galastri S, Caligiuri A, Mingarelli E, Mello T, Marra F, Mantero S, Roncalli M, Invernizzi P, Pinzani M. Role of the stromal-derived factor-1 (SDF-1)-CXCR4 axis in the interaction between hepatic stellate cells and cholangiocarcinoma. J Hepatol. 2012;57(4):813–20.
Menakongka A, Suthiphongchai T. Involvement of PI3K and ERK1/2 pathways in hepatocyte growth factor-induced cholangiocarcinoma cell invasion. World J Gastroenterol. 2010;16(6):713–22.
Aishima S, Taguchi K, Terashi T, Matsuura S, Shimada M, Tsuneyoshi M. Tenascin expression at the invasive front is associated with poor prognosis in intrahepatic cholangiocarcinoma. Mod Pathol. 2003;16(10):1019–27.
Utispan K, Thuwajit P, Abiko Y, Charngkaew K, Paupairoj A, Chau-in S, Thuwajit C. Gene expression profiling of cholangiocarcinoma-derived fibroblast reveals alterations related to tumor progression and indicates periostin as a poor prognostic marker. Mol Cancer, 2010. 9(13).
Terada T, Okada Y, Nakanuma Y. Expression of immunoreactive matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in human normal livers and primary liver tumors. Hepatology. 1996;23(6):1341–4.
Prakobwong S, Yongvanit P, Hiraku Y, Pairojkul C, Sithithaworn P, Pinlaor P, et al. Involvement of MMP-9 in peribiliary fibrosis and cholangiocarcinogenesis via Rac1-dependent DNA damage in a hamster model. Int J Cancer. 2010;127(11):2576–87.
Sirica AE, Dumur CI, Campbell DJ, Almenara JA, Ogunwobi OO, Dewitt JL. Intrahepatic cholangiocarcinoma progression: prognostic factors and basic mechanisms. Clin Gastroenterol Hepatol. 2009;7(11 Suppl):S68-78.
Aoki S, Inoue K, Klein S, Halvorsen S, Chen J, Matsui A, et al. Placental growth factor promotes tumour desmoplasia and treatment resistance in intrahepatic cholangiocarcinoma. Gut. 2022;71(1):185–93.
Pellat A, Vaquero J, Fouassier L. Role of ErbB/HER family of receptor tyrosine kinases in cholangiocyte biology. Hepatology. 2018;67(2):762–73.
Vaquero J, Lobe C, Tahraoui S, Claperon A, Mergey M, Merabtene F, et al. The IGF2/IR/IGF1R pathway in tumor cells and myofibroblasts mediates resistance to EGFR inhibition in cholangiocarcinoma. Clin Cancer Res. 2018;24(17):4282–96.
Boonsri B, Yacqub-Usman K, Thintharua P, Myint KZ, Sae-Lao T, Collier P, et al. Effect of combining EGFR tyrosine kinase inhibitors and cytotoxic agents on cholangiocarcinoma cells. Cancer Res Treat. 2021;53(2):457–70.
Thongchot S, Vidoni C, Ferraresi A, Loilome W, Yongvanit P, Namwat N, et al. Dihydroartemisinin induces apoptosis and autophagy-dependent cell death in cholangiocarcinoma through a DAPK1-BECLIN1 pathway. Mol Carcinog. 2018;57(12):1735–50.
Thongchot S, Vidoni C, Ferraresi A, Loilome W, Khuntikeo N, Sangkhamanon S, Titapun A, Isidoro C, Namwat N. Cancer-associated fibroblast-derived IL-6 determines unfavorable prognosis in cholangiocarcinoma by affecting autophagy-associated chemoresponse. Cancers. 2012;13(9):2134.
Yang R, Wang D, Han S, Gu Y, Li Z, Deng L, et al. MiR-206 suppresses the deterioration of intrahepatic cholangiocarcinoma and promotes sensitivity to chemotherapy by inhibiting interactions with stromal CAFs. Int J Biol Sci. 2022;18(1):43–64.
Cardinale V, Renzi A, Carpino G, Torrice A, Bragazzi MC, Giuliante F, et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am J Pathol. 2015;185(6):1724–39.
Leelawat K, Thongtawee T, Narong S, Subwongcharoen S, Treepongkaruna SA. Strong expression of CD133 is associated with increased cholangiocarcinoma progression. World J Gastroenterol. 2011;17(9):1192–8.
Sulpice L, Rayar M, Turlin B, Boucher E, Bellaud P, Desille M, et al. Epithelial cell adhesion molecule is a prognosis marker for intrahepatic cholangiocarcinoma. J Surg Res. 2014;192(1):117–23.
Gu MJ, Jang BI. Clinicopathologic significance of Sox2, CD44 and CD44v6 expression in intrahepatic cholangiocarcinoma. Pathol Oncol Res. 2014;20(3):655–60.
Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM, Akita H, Takiuchi D, Hatano H, Nagano H, Barnard GF, Doki Y, Mori M. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010;120(9):3326–39.
Sukowati CH, Anfuso B, Torre G, Francalanci P, Croce LS, Tiribelli C. The expression of CD90/Thy-1 in hepatocellular carcinoma: an in vivo and in vitro study. PLoS ONE. 2013;8(10):e76830.
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP Promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76(14):4124–35.
Sica A, Porta C, Amadori A, Pastò A. Tumor-associated myeloid cells as guiding forces of cancer cell stemness. Cancer Immunol Immunother. 2017;66(8):1025–36.
Zou H, Stoppani E, Volonte D, Galbiati F. Caveolin-1, cellular senescence and age-related diseases. Mech Ageing Dev. 2011;132(11–12):533–42.
Volonte D, Vyas AR, Chen C, Dacic S, Stabile LP, Kurland BF, et al. Caveolin-1 promotes the tumor suppressor properties of oncogene-induced cellular senescence. J Biol Chem. 2018;293(5):1794–809.
Yamao T, Yamashita YI, Yamamura K, Nakao Y, Tsukamoto M, Nakagawa S, et al. Cellular senescence, represented by expression of Caveolin-1, in cancer-associated fibroblasts promotes tumor invasion in pancreatic cancer. Ann Surg Oncol. 2019;26(5):1552–9.
Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, et al. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology. 2012;56(6):2242–54.
Cadamuro M, Nardo G, Indraccolo S, Dall’olmo L, Sambado L, Moserle L, et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology. 2013;58(3):1042–53.
Cadamuro M, Brivio S, Mertens J, Vismara M, Moncsek A, Milani C, et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J Hepatol. 2019;70(4):700–9.
Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016;17(10):611–25.
Green CJ, Lichtlen P, Huynh NT, Yanovsky M, Laderoute KR, Schaffner W, Murphy BJ. Placenta growth factor gene expression is induced by hypoxia in fibroblasts: a central role for metal transcription factor-1. Cancer Res. 2001;61(6):2696–703.
Clapéron A, Mergey M, Aoudjehane L, Ho-Bouldoires TH, Wendum D, Prignon A, Merabtene F, Firrincieli D, Desbois-Mouthon C, Scatton O, Conti F, Housset C, Fouassier L. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology. 2013;58(6):2001–11.
Wilhelm A, Shepherd EL, Amatucci A, Munir M, Reynolds G, Humphreys E, et al. Interaction of TWEAK with Fn14 leads to the progression of fibrotic liver disease by directly modulating hepatic stellate cell proliferation. J Pathol. 2016;239(1):109–21.
Dwyer BJ, Jarman EJ, Gogoi-Tiwari J, Ferreira-Gonzalez S, Boulter L, Guest RV, et al. TWEAK/Fn14 signalling promotes cholangiocarcinoma niche formation and progression. J Hepatol. 2021;74(4):860–72.
Qin X, Lu M, Li G, Zhou Y, Liu Z. Downregulation of tumor-derived exosomal miR-34c induces cancer-associated fibroblast activation to promote cholangiocarcinoma progress. Cancer Cell Int. 2021;21(1):373.
Lobe C, Vallette M, Arbelaiz A, Gonzalez-Sanchez E, Izquierdo L, Pellat A, et al. Zinc finger E-box binding homeobox 1 promotes cholangiocarcinoma progression through tumor dedifferentiation and tumor-stroma paracrine signaling. Hepatology. 2021;74(6):3194–212.
Sun L, Wang Y, Wang X, Navarro-Corcuera A, Ilyas S, Jalan-Sakrikar N, et al. PD-L1 promotes myofibroblastic activation of hepatic stellate cells by distinct mechanisms selective for TGF-beta receptor I versus II. Cell Rep. 2022;38(6):110349.
Acknowledgements
Thanks to Yewei Zhang for the writing guidance and editorial revision of this article.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81872255, 62141109), the Leading-edge Technology Programme of Jiangsu Natural Science Foundation: BK20212021.
Author information
Authors and Affiliations
Contributions
Hao Peng and Erwei Zhu wrote the manuscript, and Yewei Zhang edited and critical revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Peng, H., Zhu, E. & Zhang, Y. Advances of cancer-associated fibroblasts in liver cancer. Biomark Res 10, 59 (2022). https://doi.org/10.1186/s40364-022-00406-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-022-00406-z