
Abstract
Cardiovascular complications are the major cause of the marked morbidity and mortality associated with chronic kidney disease (CKD). The classical cardiovascular risk factors such as diabetes and hypertension undoubtedly play a role in the development of cardiovascular disease (CVD) in adult CKD patients; however, CVD is just as prominent in children with CKD who do not have these risk factors. Hence, the CKD-specific pathophysiology of CVD remains incompletely understood. In light of this, studying children with CKD presents a unique opportunity to analyze CKD-associated mechanisms of CVD more specifically and could help to unveil novel therapeutic targets.
Here, we comprehensively review the interaction of the human gut microbiome and the microbial metabolism of nutrients with host immunity and cardiovascular end-organ damage. The human gut microbiome is evolutionary conditioned and modified throughout life by endogenous factors as well as environmental factors. Chronic diseases, such as CKD, cause significant disruption to the composition and function of the gut microbiome and lead to disease-associated dysbiosis. This dysbiosis and the accompanying loss of biochemical homeostasis in the epithelial cells of the colon can be the result of poor diet (e.g., low-fiber intake), medications, and underlying disease. As a result of dysbiosis, bacteria promoting proteolytic fermentation increase and those for saccharolytic fermentation decrease and the integrity of the gut barrier is perturbed (leaky gut). These changes disrupt local metabolite homeostasis in the gut and decrease productions of the beneficial short-chain fatty acids (SCFAs). Moreover, the enhanced proteolytic fermentation generates unhealthy levels of microbially derived toxic metabolites, which further accumulate in the systemic circulation as a consequence of impaired kidney function. We describe possible mechanisms involved in the increased systemic inflammation in CKD that is associated with the combined effect of SCFA deficiency and accumulation of uremic toxins. In the future, a more comprehensive and mechanistic understanding of the gut–kidney–heart interaction, mediated largely by immune dysregulation and inflammation, might allow us to target the gut microbiome more specifically in order to attenuate CKD-associated comorbidities.
Similar content being viewed by others

CKD-associated cardiovascular disease in children and adolescents
The prevalence of chronic kidney disease (CKD) in children continues to increase worldwide. The reported case numbers range between 10.7 and 74.7 per million of the age-related population [1,2,3]. Despite ongoing efforts to improve treatment, mortality is high among patients with CKD, primarily due to cardiovascular diseases (CVD) and progression to end-stage kidney disease (ESKD) [4, 5]. Thus, there is an urgent need to identify extra-renal comorbidities at an early stage and to assess patients’ risk factors in order to effectively modify therapeutical interventions and therefore reduce mortality in children with CKD [6].
The lifespan of pediatric patients with ESKD is 30–40 years below that of healthy children [7]. While CVD is absent in healthy children, cardiovascular events account for 40–45% of deaths in children with ESKD [7,8,9]. The relevance of CVD in CKD has been highlighted in a statement by the American Heart Association, which classifies CKD patients in the highest cardiovascular risk stratification alongside individuals with homozygous familial hypercholesterolemia, diabetes mellitus type 1, heart transplants, or coronary aneurysms due to Kawasaki disease [4, 8, 10].
Adult CKD patients also have a drastic increase in cardiovascular morbidity [5]. In adults, the CVD is mainly driven by the primary courses of CKD: diabetes and arterial hypertension. However, these comorbidities and underlying diseases are usually absent in children with CKD. Thus, in the absence of confounding classical risk factors, cardiovascular mortality is expected to be induced largely by CKD-specific mechanisms.
This hypothesis is underlined by the observation that unlike in the aging population, pediatric patients with CKD rarely show symptomatic atherosclerosis classically associated with a consecutive calcification of the tunica intima of arteries [11]. By contrast, current data suggest the early occurrence of endothelial dysfunction in ESKD with diffuse nonocclusive medial calcification, known as Mönckeberg’s arteriosclerosis [12,13,14]. Medial calcification leads to arterial wall stiffening and thus reduces the compliance and elasticity of arteries resulting in increased systolic pressure and cardiac workload. Subsequently, the development of endothelial dysfunction and vascular calcification results in left ventricular hypertrophy and myocardial fibrosis, which can lead to sudden cardiac death by arrhythmia or cardiac failure [9, 15]. Recently, studies have focused on translating these morphologic findings into clinical surrogate parameters indicating the progression of CVD. In fact, a prospective observational study in more than 700 European children identified key criteria for the development of CVD such as carotid intima-media thickness (cIMT), pulse wave velocity (PWV), left ventricular mass (LVM), and vascular calcification [16,17,18]. Highly significant is the fact that vascular remodeling, myocardial adaptation, and arterial stiffening are clinically detectable in many children at early times when symptoms of CVD may still be absent [19].
The hypothesis of arteriosclerosis being an inflammatory disease was first postulated by Ross et al. in 1999 [20]. More recently, approaches using anti-inflammatory therapy directed against prominent cytokines, namely canakinumab against interleukin 1 beta, in the secondary prevention of arteriosclerosis have further highlighted the pivotal role of inflammation in the pathophysiology of cardiovascular complications [21]. In fact, plasma concentrations of pro-inflammatory cytokines are consistently elevated in CKD and oxidative stress is linked to inflammation by activating the nuclear factor “kappa-light-chain-enhancer” of activated B-cells (NF-κB) and inducing pro-inflammatory cytokines [22].
As it became clear that chronic inflammation constitutes one of the main risk factors of CVD [20], increasing attention has been paid to the intestinal microbiome, its metabolism, and its interaction with host inflammatory status. Although data is limited on the inflammatory status of pediatric patients with CKD, results from studies with adult patients and animal experiments highlight the interaction of the gut microbiome, kidneys, and immune system as a crucial contributor to CVD pathology [23,24,25,26,27,28,29,30,31].
In this review, we provide an overview of the mechanisms involved in CKD-associated alterations of the gut microbiome, the microbially produced metabolites, and their relation to the development of CVD. Particularly, we focus on bacterial and host metabolism of amino acids, such as tryptophan (TRP) and tyrosin (TYR), as well as the influence of short-chain fatty acids (SCFAs). While of limited epidemiological importance, research in pediatric CKD patients enables the investigation of CVD mechanisms specific to kidney disease independent of other risk factors and therefore can extend our pathophysiological understanding of CVD in CKD. Furthermore, we discuss novel therapeutic approaches based on the understanding of the microbiome and its metabolites that could help lower the enormous burden of cardiovascular morbidity and mortality in patients suffering from CKD.
The intestinal microbiome and microbiome–host interactions in health and CKD
The microbiome is defined not only by a community of microorganisms living in a defined environment (microbiota), but also by the whole spectrum of molecules produced by the microorganisms (including nucleic acids and metabolites) and the surrounding environment [32].
In 2007, the Human Microbiome Project was launched to improve the understanding of the enormous diversity of microbial flora. Most strikingly, it refuted the assumption that humans share a large core of microbial taxa, sprinkled with a few lineages that make each individual unique. Quite the contrary, interindividual differences are substantially greater than previously expected [33,34,35,36,37]. This observation was consolidated by the European Union project METAgenomics of the Human Intestinal Tract (MetaHIT). This study showed that a microbial species found with up to 10% abundance in one individual can be as rare as one cell in 1000 in another participant within the cohort. In summary, these findings indicate an extraordinarily complex and dynamic consortium of bacteria residing in the human gut which consistently responds to internal and external stimuli [38,39,40]. Despite substantial differences in the abundance of microbial species in the human gut microbiota, a larger overlap of genes encoding for metabolic functions exists [41]. Taken together, healthy microbiomes may differ significantly in species abundance, but the metabolic potential of the gut microbiome from different individuals share common overlapping features.
Bacteria–host interaction: how the gut microbiome is shaped throughout life
During the human lifetime, the intestinal flora undergoes dynamic changes with the most substantial alterations occurring during childhood [42, 43]. Recently, it has been shown that meconium before birth is indeed sterile; however, contrary to a widespread assumption, newborns are not sterile at birth. In fact, the vaginal flora evolves throughout pregnancy to provide the newborn with beneficial microbes, such as Lactobacillus and Prevotella which are transmitted to the child during birth [44,45,46]. These maternally provided microbial communities occupy niches and protect newborns from the colonization of pathogens. Interestingly, while children born through vaginal delivery share a significant proportion of 16S rRNA sequences with their biological mother for up to 2 years, the gut residing in the gut of children born via cesarean section (C-section) is more similar to the hospital environment and the mother’s skin. This may contribute to the 64–82% higher risk to sustain skin infections with methicillin-resistant Staphylococcus aureus in the case of neonates delivered via C-section compared to vaginal birth. Although not proven yet because of the high risk of confounding factors, the altered composition of gut microbiota that results from C-section births may be associated with an increased risk of obesity, atopic diseases like asthma, and Crohn’s disease in later life [47,48,49,50,51,52].
Despite the mode of birth, breastfeeding is among the most influential perinatal factors shaping the intestinal microbiome composition, partly by providing both a source of beneficial commensals, including Staphylococci, Streptococci, lactic acid bacteria, and Bifidobacteria [53], and human milk oligosaccharides as an energy source for beneficial gut bacteria [54]. In the neonatal period, the composition of the gut microbiome is very dynamic, starting at low levels of distinguishable taxa, dominated by Proteobacteria and Actinobacteria and successively changing toward a more diverse population with Firmicutes and Bacteroidetes emerging and dominating the local environment in older individuals [36, 55]. These life changes are reflected in a noticeable difference in the Firmicutes/Bacteroidetes ratio between infants and adults (ratio 0.4 and 10.9, respectively). Interestingly, ratios in the elderly tend toward values more similar to those in infants [56,57,58].
However, the relationship between the gut microbiome and health and disease is complex. Our knowledge of variation in the intestinal microbiome composition across the human lifetime remains, at best, superficial.
Whereas large cohort studies provided in-depth data to generate a microbiome profile in the adult population, much less is known about the microbiome of children and adolescents [59]. A recent study involving 16S rRNA sequencing of microbiome fecal samples from 2111 children in the age range of 9 to 12 years revealed a significantly lower Shannon diversity than the adult control group suggesting that the immature microbiome fundamentally differs from that of adults. Metagenome analysis provided additional information showing that these compositional differences also reflect in functional metabolic disparities including the overrepresentation of vitamin B biosynthesis pathways (riboflavin: B2, pyridoxine: B6, and folate: B9) and a predominance of catabolic amino acid metabolism (Valin, Leucin, and Isoleucin) in children [59, 60]. Once established, the human gut microbiome remains relatively stable over time. However, a plethora of factors can impact the gut microbiome: medication, which is not limited to antibiotics [50, 61,62,63], but also includes proton pump inhibitors [64, 65] and laxatives; diet (fiber, artificial sweeteners, and sodium chloride intake) [66,67,68]; chronic autoimmune, inflammatory, metabolic, and neurodegenerative diseases [69,70,71,72]; genetics [73]; stress [74]; exercise [75]; surgeries [76]; geography [77]; and aging itself [78, 79]. In summary, it is commonly accepted that many influences shape the gut microbiome over time.
The benefits of symbiosis in the gut microbiome of healthy individuals
The host–microorganism relationship can generally be defined as symbiotic as the respective partners not only coexist without detriment but benefit from each other. The resident bacteria metabolize dietary components, which are otherwise inaccessible for humans, such as fiber. The microbiota is not only beneficial in the digestion of complex polysaccharides but also a key player in amino acid homeostasis (lysine and threonine), absorption of vitamins (vitamin K and B groups), metabolism of bile acids, and integrity of the intestine barrier and protecting against pathogens [31, 80,81,82,83,84]. In addition, the gut microbiome is crucial for host immune system maturation. This has been clearly demonstrated in animal studies, whereby mice devoid of a gut microbiota have an immature immune system phenotype with low lymphocyte counts and diminished cytokine production, which could be reversed within 3 weeks by restoring normal flora [85, 86].
Recent technological advances enabling functional readouts provide an in-depth understanding of the physiological state of an organism via genomics, proteomics, and metabolomics approaches by detecting small molecule substrates and intermediates of metabolism. In addition, the use of germ-free animal models complements the multi-omics approaches and provides insights into fundamental mechanisms of host–microbiome interactions [87]. Despite the advances in understanding microbial taxonomic composition, we are just beginning to assemble the necessary experimental and computational tools to understand the functional metabolic capacities of the gut microbiome. The bacterial genome of the gut vastly exceeds the complexity of the human genome, with many levels of potential further diversity resulting from branching and combinations of compounds like lipids and oligosaccharides. Thus, thanks to rapid advances in technology, we are at the leading edge of understanding the interplay of gut microbiota, metabolic changes, and the immune system [39, 88].
Saccharolytic and proteolytic fermentation processes
Were it not for bacterial fermentation by the gut microbiota humans would not be able to extract nutrients and healthy beneficial compounds from dietary fiber [89].
There are two main pathways of bacterial fermentation: saccharolytic and proteolytic fermentation. Saccharolytic fermentation takes place primarily in the proximal colon [90] and involves the extraction of energy from complex oligosaccharides (α-glucans and non-α-glucan oligosaccharides) and polysaccharides (resistant starch, inulin, pectin, and cellulose), all of which are characterized by beta-glycosidic links that cannot be processed by human enzymes.
The saccharolytic fermentation of complex carbohydrates produces SCFAs. The most abundant with 60% being acetate—consisting of two carbon molecules (C2), followed by propionate (C3) at 25%, and butyrate (C4) at 15% [91,92,93,94]. SCFAs are naturally produced in small quantities in the liver; however, the proximal colon is their primary site of production [95]. A growing body of evidence suggests numerous mechanisms whereby SCFAs facilitate effects on gut health, including nutrient supply [96], gut motility [97], barrier function of colonocytes [31, 98], and “competitive exclusion” (paraphrasing the limited access of pathogenic bacteria to the gut epithelium due to expanding commensal bacteria [99]). Over and above this, numerous studies suggest that SCFAs modulate an individual’s inflammation state by affecting recruitment, trans-migration, and cytokine production of immune cells [31, 100,101,102,103,104]. However, the functional effects of these gut-derived metabolites on endothelial and immune cells within the intestine remain elusive [104, 105].
Whereas the major proportion acetate and propionate are rapidly absorbed and utilized by colonocytes or metabolized by the liver [96, 99, 105, 106], a comparatively low concentration of butyrate is found in the systemic circulation. Commonly, this is explained by the fact that the four-carbon molecule is a major energy substrate of colonocytes, thus being primarily utilized locally [96]. The dependence of colonocytes on bacterial-derived SCFAs was elegantly demonstrated in a study by Donohoe et al. who showed an impaired metabolic state in the colon of germ-free mice as measured by ATP depletion and autophagy in gut epithelial cells, which could be reversed by butyrate supplementation [107].
Proteolytic fermentation, as its name implies, is the breakdown of protein in the absence of oxygen, mainly within the distal part of the colon. It has been shown that several metabolites resulting from peptides that escape digestion in the small intestine are precursors of harmful toxins including ammonia as well as thiols, phenols, and indoles [31, 99]. Consequently, it was postulated that a saccharolytic fermentation pattern is more favorable and that an imbalance toward more proteolytic bacterial activity could indeed be a pathophysiologic contributor in various diseases such as CKD.
CKD-associated alterations in the gut microbial composition: dysbiosis
It was proposed that the accumulation of uremic toxins profoundly changes the biochemical environment in the gut and exerts a selection pressure that may favor microbes capable of using these substrates [108,109,110]. Vaziri et al. were the first to describe quantitative and qualitative alterations in the composition of gut microbiota in patients with CKD [69]. Since then, significant differences in the abundance of several bacterial species have been demonstrated in patients with CKD and their adverse consequences gained increasing attention. There is a general consensus that expansion of proteolytic bacteria with urease, p-cresol-producing enzymes, and indole-forming enzymes contributes to the production of nitrogen-containing compounds and, consequently, the accumulation of uremic toxins in CKD. Relevant proteolytic bacterial families include Ruminococcacae (phylum Firmicutes), Enterobacteriaceae (phylum Proteobacteria), and Pseudomonadaceae (phylum Proteobacteria). In addition, saccharolytic bacteria with enzymes essential for SCFA production, i.e., Roseburia (phylum Firmicutes), Bifidobacterium (phylum Actinobacteria), Prevotellaceae (phylum Bacteroidetes), and Lactobacillaceae (phylum Firmicutes), are depleted in patients with ESKD in comparison to healthy controls [26, 69, 108, 111,112,113,114]. In conclusion, these findings indicate a shift toward a proteolytic fermentation pattern that further contributes to the accumulation of uremic toxins fueling inflammation and eventually resulting in CVD and disease progression.
While adaptions of the intestinal flora to CKD-characteristic micro-environmental changes have been extensively reviewed in adult populations, to the best of our knowledge, only two small cohort studies exist that focus on alterations of the intestinal flora in children with CKD.
In 2015, the Midwest Pediatric Nephrology Consortium study highlighted a significant decrease in bacterial diversity in peritoneal dialysis (PD) patients when compared to healthy controls. Additionally, the composition of bacterial communities showed marked separations between pediatric CKD patients and healthy controls. Whereas at a family level the relative abundance of Proteobacteria and Enterobacteriaceae was increased, Bifidobacteria decreased in PD and transplant patients. Both findings are generally in line with results in adult microbiome studies in CKD [115, 116]. Pediatric PD patients exhibit an increase in the relative abundance of proteolytic Proteobacteria and, at a family level, Enterobacteriaceae. Furthermore, evidence for depleted amounts of beneficial SCFA-producing bacteria, including Actinobacteria and Bifidobacteriacae, has been presented at least at the RNA level [115, 116].
Factors contributing to dysbiosis
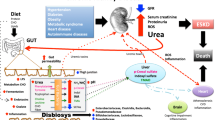
There are numerous factors contributing to intestinal dysbiosis, which are also summarized in Fig. 1 [80, 117]. Diet is perhaps the most significant environmental factor affecting the gut microbiota in both diseased and healthy individuals. In CKD, dietary restrictions are invariably imposed to prevent fluid overload, hyperkaliemia, and oxalate overload. Most significant with regard to gut health and the microbiome is the low consumption of fruit and vegetables and dietary fiber among CKD patients. This restricted intake of complex carbohydrates reduces the abundance of bacterial phyla capable of utilizing these substrates. Consequently, the normal symbiotic relationship is disturbed, markedly affecting the composition, function, and metabolism of gut microbiota and conferring the potential to significantly alter the biochemical milieu in the CKD population [31, 69, 108].

Schematic overview of CKD-specific conditions that influence the local biochemical milieu in the colon (A) and contribute to alterations in the composition and metabolism of the gut microbiome (B) leading to disruption of the epithelial barrier function (leaky gut), inflammation, and cardiovascular end-organ damage (C). A In healthy individuals, the biochemical balance in the colon is maintained through a diet rich in fiber providing an energy source for colonocytes and stabilizing local pH as well as downstream metabolism of intestinal microbiota. In contrast, in CKD, several circumstances contribute to a disturbance of biochemical hemostasis. Dietary restrictions (low-fiber diet), oral intake of medications (e.g., antibiotics and phosphate binders), and muscle wasting promote an accumulation of peptides in the gut. Additionally, urea is converted by bacterial-derived urease to ammonium hydroxide which increases the gut pH. Lastly, constipation promotes a prolonged transit time of these metabolites exerting selection pressure on intestinal bacteria. B Under normal circumstances, humans benefit from the symbiotic relationship with their gut microbiota by saccharolytic fermentation of complex carbohydrates resulting in the production of SCFA namely acetate, propionate, and butyrate. Whereas butyrate is mainly used by epithelial cells as a nutrient source, acetate and propionate enter the systemic circulation. Generally, SCFAs promote gut epithelial integrity and balance systemic regulation of adaptive immunity and modulate inflammation response. In CKD, dysbiotic changes are characterized by a shift in the fermentation pattern of bacteria from a health-promoting saccharolytic to a proteolytic fermentation. This alteration not only leads to a decrease in local and systemic SCFA concentration but also promotes fermentation of amino acid tryptophan and tyrosine to uremic acid precursors indoles and cresols respectively. C Intestinal barrier dysfunction (leaky gut), enforced by dysbiosis and local metabolite imbalance, promotes paracellular migration of bacteria and uremic toxins. Renal excretion decline inevitably induces accumulation of these bacterial-derived metabolites. Moreover, lymphocytes infiltrate the colonocytes’ lamina propria and enter systemic circulation resulting in a state of chronic low-grade inflammation. All these factors finally promote endothelial calcification and cardiovascular disease. CKD chronic kidney disease, Tyr tyrosine, Trp tryptophane, Phe phenylalanine, SCFA short-chain fatty acid, VSMC vascular smooth muscle cell
Moreover, constipation is highly prevalent in CKD. It has been shown that in states where dietary fiber intake is low, as it holds true in CKD, the expansion of proteolytic bacteria may lead to a degradation of the goblet cell-derived mucin 2 (MUC-2) layer, which under normal circumstances facilitates the nutritional transit by lubricating the gut’s surface. A slower transit time affects uremic toxin generation by increasing the availability of amino acids to be fermented by proteolytic bacteria [118, 119]. The accumulation of these toxins in turn exacerbates intestinal dysmotility via intestinal inflammation, thereby forming a vicious cycle of constipation coupled with enhanced uremic toxin production, which again increases transit time [97, 120, 121].
In addition, muscle wasting is common in CKD, further exacerbating the advancing issue of uremia, which ultimately leads to increases in colonic pH and dysbiotic alterations. Generally, protein catabolism creates problems in terms of nitrogen elimination and requires conversion to urea within the ornithine cycle in the liver in order to reduce toxicity. The intestinal microbiota can further utilize urea to produce the sulfate-containing amino acids lysine and threonine and thus play a key role in nitrogen recycling [89]. However, in CKD, the rise in retention solute concentrations in the body fluids leads to its massive influx into the gut lumen. Within the intestinal tract, urea is hydrolyzed by microbial urease leading to the formation of ammonia [CO(NH2)2 + H2O → CO2 + 2NH3]. Ammonia is, in turn, converted to ammonium hydroxide [NH3+ H2O → NH4OH] which elevates the gut’s luminal pH and causes mucosal damage and enterocolitis [109, 122]. Accordingly, it has been proposed that the accumulation of uremic toxins profoundly changes the biochemical environment in the gut and drives a selection pressure that favors microbes capable of using these uremic toxins as substrates by proteolytic instead of saccharolytic fermentation [108,109,110]. Indeed, data in adults revealed a relative increase in microbes with urease, uricase, and indole- and p-cresyl-forming enzymes [108, 113, 114]. Interestingly, it has been demonstrated that once returned to the typical pH of 5.5 from abnormal high pH levels (6.8), the production of gut-derived uremic acids could be reduced by approximately 33%. Remarkably, this effect was doubled (60% reduction) in the presence of fermentable carbohydrates [123,124,125].
In addition, pharmaceuticals are known to have an important impact on dysbiosis in CKD. Antibiotics are frequently prescribed in CKD to treat inter alia vascular access infection and account for a considerable loss in key bacteria taxa [61, 62, 126].
Finally, iron-containing phosphate binders to prevent secondary hyperparathyroidism are associated with increased production of uremic toxins and cIMT, which may be due to an iron-dependent expansion of the phylum Proteobacteria and a general decrease in the abundance of beneficial saccharolytic species such as Lactobacillus and Bifidobacterium [108, 126,127,128].
Crosstalk between the gut microbiome and target organs in CKD via microbially derived metabolites
Microbially derived uremic toxins as drivers of cardiovascular disease
In CKD, the uremic syndrome is attributed to the progressive loss of excretory function, which inevitably results in the retention of a variety of substances. These retention solutes are found to exert toxicity that affects numerous biological functions, hence referred to as uremic toxins [129]. According to the European Uremic Toxin (EUTox) Work Group, they can be classified into three physicochemical categories based on the molecular weight and kinetic behavior during dialysis. Besides small water-soluble molecules (molecular weight (MW) ≤ 500 Da—i.e., urea and phosphorus) and middle-sized molecules (MW ≥ 500 Da—i.e., parathyroid hormone and β2-microglobulin), the third group consists of plasma-bound compounds like indoxyl sulfate (IS) and p-cresol sulfate (PCS). The latter have long been neglected with respect to their pathophysiological importance despite their tendency to accumulate during dialysis due to being mostly albumin bound and therefore not filtered out [27, 130, 131].
Uremic toxins mainly result from proteolytic fermentation of amino acids in the intestine. Therefore, the generation of these toxins depends mainly on nutritional intake and on microbial dysbiosis in CKD, favoring proteolytic fermentation as described above. The role of the microbiota in the generation of uremic toxins has been emphasized in a study of hemodialysis patients who received a colectomy. The authors demonstrated the absence of more than 30 uremic toxins in this study group highlighting the colon microbes as a prime contributor to the uremic milieu present in CKD patients [132].
Among the metabolic pathways associated with uremic toxicity, tryptophan (TRP) metabolism has been the focus of numerous studies (Fig. 1). Although tryptophan is an essential amino acid, less than 1% is used for protein synthesis. The vast majority serves as a biosynthetic precursor for distinct microbial metabolization pathways generating serotonin, melatonin, kynurenine, and indoles [133]. Approximately 95% of TRP is converted to kynurenine (KYN) by the enzymes tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO). In adult CKD, metabolites of the KYN pathway such as kynurenic acid and xanthurenic acid have been linked to chronic inflammation [134, 135], mineral bone disease [136], thrombosis [137, 138], and cognitive impairment [135]. A variable portion of dietary TRP is catabolized to indole metabolites by intestinal bacteria, which express enzymes such as tryptophanase [139]. Indoles are further metabolized by gut microbiota and the liver to indoxyl-sulfate (IS), the prototype of microbially derived uremic toxins [140].
The accumulation of uremic toxins in adult CKD patients has been repeatedly described and is associated with the progression of kidney disease and its comorbidities, first and foremost CVD [141,142,143]. Subsequently, pediatric nephrologists started to pay attention to these gut-derived uremic toxins and their impact on CKD-associated comorbidities. Initial studies showed that hippuric acid (HA), indole acetic acid (IAA), IS, and PCS were indeed elevated in pediatric CKD patients [131, 144]. Recently, further investigations provided insights into the clinical implications of elevated serum levels of uremic toxins in pediatric CKD. Our work has demonstrated that IS concentration not only inversely correlates with the estimated glomerular filtration rate (eGFR) but also associates with disease progression and cardiovascular morbidity, independent of other known risk factors like proteinuria or blood pressure [127, 145].
While not yet shown in children, detailed mechanisms of the accumulation of uremic toxins leading to CVD in adult CKD patients have been demonstrated [27, 141, 146,147,148,149]. In brief, the uremic milieu provokes endothelial production of reactive oxygen species (ROS) leading to a pro-inflammatory state in which the NOD-like receptor proteins (NLRP3) of the inflammasome are activated and downstream cytokine production promoted (e.g., IL-6). The downstream transcription of several adhesion molecules ultimately results in endothelial dysfunction [150]. IS is perhaps the best characteristic of the gut-derived uremic toxins and recent work focusing on this metabolite suggests its interfering role regarding oxidative stress levels as it was shown to stimulate the generation of free radicals by activation of NADPH oxidase [151]. Moreover, IS induces vascular smooth muscle cell (VSMC) proliferation, reduces endothelial repair, and promotes vascular calcification via its binding to the nuclear aryl hydrocarbon receptor (AhR) and activation of the NF-κB signaling pathway [152,153,154]. In the clinical context, studies point toward an association of serum IS levels with surrogate markers of CVD, namely cIMT and PWV [127].
Decrease of SCFA induces a pro-inflammatory and proatherogenic milieu in CKD
The reduced abundance of SCFA-producing bacteria in the gut of CKD patients is coupled with a marked decrease in SCFA in the blood (Fig. 1) [31]. SCFAs are key regulators of inflammation via a number of interactions. They can modify the transcription of genes responsible for leukocyte rolling and subsequent adhesion and migration of immune cells. Additionally, they play an essential role in controlling the production of pro-inflammatory cytokines. The binding of SCFA to different intracellular and extracellular receptors is discussed below, and potential mechanistic insights into the link between gut-derived metabolites and their potential to control immune function are highlighted [105].
SCFAs, particularly butyrate and propionate, are shown to be effective non-competitive inhibitors of the histone deacetylase (HDAC) enzyme in a millimolar range, which lays within the physiologic concentration of SCFA in the lumen of the colon (from 20–70 mmol/l proximally to 70–140 mmol/l distally) [94, 155, 156]. Interestingly, evidence shows that these two SCFAs can modulate endothelial and immune cell inflammation by inhibition of HDAC [99, 105, 157, 158]. Recent studies revealed the potential of SCFAs to lower levels of LPS-induced TNFα production in mononuclear cells via inhibition of NF-κB [159]. In detail, deacetylation of p65, a subunit of NF-κB, enhances its binding to the inhibiting molecule IκBα in the nucleus and may result in the export of NF-κB complexes back to the cytoplasm, where it cannot deploy its transcriptional pro-inflammatory activity [160]. Importantly, the order of potency for NF-κB suppression butyrate > propionate > acetate coincides with the order of HDAC suppression activity [161, 162].
It has been observed that HDAC inhibitors induce IL-10 gene expression in regulatory T cells (Tregs) and elevate suppression capacity of Tregs thereby providing further mechanistic insights explaining SCFA’s anti-inflammatory capacity, as colonic Tregs limit proliferation of effector CD4+ T cells (Teff) and thus control inflammation [163]. More specifically, Clostridia species known as high butyrate producers are the most potent inducers of de novo generation of inducible colonic Tregs (iTregs) in the large intestine [164,165,166]. In germ-free mice, SCFAs, in particular propionate (C3), increase both the absolute count and the proportion of Tregs and augment their immune suppressive function [104]. Current data reveal the potential of SCFA to induce the production of anti-inflammatory cytokines such as IL-10 and the repression of pro-inflammatory molecules IL-12, TNFα, IL-1β, and NO by inhibiting NF-κB activity [101, 103, 155, 159]. Moreover, the most potent inhibitor of HDACs butyrate (80% inhibitory efficiency) protects against vascular inflammation and atherosclerosis, thereby modulating oxidative stress and endothelial function [167, 168]. Still, the broad spectrum of HDACs with pleiotropic and even divergent effects on transcriptional responses should be kept in mind, bearing future topics of research, e.g., if and by which pathways different types of HDAC mediate the effects of SCFAs on colonocytes and immune cells.
SCFA may also act through binding to specific membrane-bound receptors. In this context, two free fatty acid receptors (FFA-R) are discussed in more detail. In both cases, the carbon molecule chain length dictates their respective activation potential. SCFAs acetate and propionate bind to FFA2-R (G protein-coupled receptor, GPR43) that is primarily expressed on neutrophils, eosinophils, dendritic cells, and monocytes suggesting their role in inflammatory responses [169]. Activation of FFA2-R results in an attenuated NF-κB response with downregulated release of pro-inflammatory cytokines, including IL-6 and IL-1beta. This effect may be mediated by β-arrestin—a regulator of G protein-coupled receptors, as demonstrated by restored cytokine production upon β-arrestin-2 knockout [170]. Conversely, FFA3 receptors (GPR41) are most efficiently activated by C3-C4 SCFAs; thus, propionate and butyrate present the main activators [171, 172]. FFA3 receptors are infrequently expressed on immune cells but rather on the surface of the pancreas, spleen, and adipose tissue [172]. Their implications in obesity and metabolic disorder are widely recognized [172]. Despite the lower abundance of FFA3 receptors on immune cells, studies still suggest an anti-inflammatory potential of butyrate and propionate. In short, activation of the FFA3 receptor decreased LPS-induced TNFα expression as well as MCP-1, IL-6, and inducible nitric oxide synthase (iNOS) levels naturally released by monocytes and neutrophils [105, 155, 173].
At the same time, data from recent studies emerged pointing toward a pro-inflammatory effect of FFA2-R and FFA3-R activation accompanied by increased production of IL-6, CXCL1, and CXCL2 [99].
Data on SCFAs and their influence on immune cell migration are sometimes contradictory. While some research shows induction of neutrophil recruitment by SCFAs followed by migration to the cite of inflammation via binding to FFA2-R following MAPK activation, other studies demonstrate the capacity of butyrate to suppress the expression of adhesion molecules such as VCAM-1, ICAM-1, lymphocyte function-associated antigen-3 (LFA-3), and L-selectin [100, 102, 155, 174, 175] via inhibition of histone acetylation and NF-κB-suppression. As this may lead to a lower incidence of adhesion and migration of macrophages to vascular lesion areas, it is tempting to speculate the potential of SCFAs as an influential mediator in the prevention of arteriosclerosis [176].
Concerning vascular inflammation, another GPR109A also known as hydroxyl-carboxylic acid 2 (HCA2) has been reported to decrease cytokine secretion of TNFα, IL-6, and MCP-1 and, as a result, alleviate the effect on the progression of arteriosclerosis. Of the main SCFAs, butyrate shows the highest potential for GPR109A activation and is shown to inhibit ROS production [105, 177, 178].
To conclude, inflammatory regulation properties of SCFA on endothelial and inflammatory cells within the human gut are complex. A growing body of evidence suggest their potential to modulate inflammation, cytokine production, and immune cell migration likely mediated through binding of extracellular G protein-coupled receptors and inhibition of histone deacetylases which regulate gene transcription, which is summarized in Fig. 2. However, due to the pleiotropic effects of SCFA, which are summarized in Table 1, the exact mechanisms by which they promote or inhibit inflammation and consequently CVD remain elusive.

Overview on the effect of SCFAs, LPS, and TNFα on gene transcription of inflammatory cytokines and adhesion factors involved in immune cell activation and promotion of endothelial dysfunction. LPS and TNFα bind to their receptor and activate downstream MAPK and NF-κB signaling through phosphorylation and subsequent ubiquitination of I-κB (inhibitory subunit of NF-κB) resulting in its degradation thus inducing gene transcription of pro-inflammatory molecules. SCFA pleotropic effects on inflammation are mediated through binding of extracellular G protein-coupled receptors as well as its potential to inhibit histone deacetylation. Acetate and propionate primarily bind to FFA2-R/GPR43 which are expressed on immune cells and may inhibit proteasomal degradation of I-κB via β-arrestin attenuating NF-κB response. Additionally, contrary effects of FFA2-R/GPR43 activation are shown as SCFA binding initiates downstream MAPK signaling. SCFA butyrate dictates activation of G protein-coupled receptor FFA3-R/GPR41 and GPR109 which decreases LPS-induced expression of TNFα, MCP-1, IL-6, and inducible nitric oxide synthase (iNOS). Confliction to its anti-inflammatory effect butyrate is also shown to enhance MAPK signaling via its binding to extracellular G protein-coupled receptor FFA3-R/GPR41. Despite receptor signaling, SCFAs are potential non-competitive inhibitors of histone deacetylases (HDACs). These enzymes facilitate a decrease in the interaction of histones to the DNA, hence promoting gene transcription. Furthermore, HDAC enhances acetylation status and thus inhibitory interaction potential of MKP-1 to MAPK and in this way attenuates downstream pro-inflammatory gene transcription. Lastly, SCFAs may decrease proteasome activity through HDAC and inhibit TNFα-induced NF-κB activation normally mediated by the degradation of I-κB. GPR G protein-coupled receptor, LPS lipopolysaccharides, TNFa tumor necrosis factor a, cAMP cyclic adenosine monophosphate, PKA protein kinase A, PI3K phosphoinostide 3-kinase, MAPK mitogen-activated protein kinase, MEK mitogen-activated protein kinase kinase, aMKP acetylated mitogen-activated protein kinase phosphatase, HDAC histone deacetylase, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, I-κB inhibitor of nuclear factor kappa B, MCP-1 monocyte chemoattractant protein-1, ICAM intercellular adhesion molecule, VCAM vascular adhesion molecule, iNOS inducible nitric oxide synthase, CXCL chemokine ligand
Vicious cycle of uremia, loss of gut symbiosis, and disrupted barrier functions (leaky gut)
The intestinal epithelium is at the center of interactions between the immune system and luminal content of the gut including microbes, microbial products, and dietary compounds. Hence, its primary role is to maintain balance whereby invasion of pathogens is prevented and simultaneously provide selective permeability allowing immune stimulation and nutritional uptake. Disruption of epithelial integrity leads to local and systemic inflammation and is intensively discussed to modulate the initiation and progression of several diseases [179].
As paracellular transport is generally more permeable than a transcellular pathway, structural components, particularly the tight junction proteins occludin and claudin, are principal determinants of gut integrity and prevent unregulated passing of nutritional antigens and microbes [31, 180, 181]. In uremic CKD patients, barrier dysfunction is common and there is mounting evidence for its pivotal role in the pathogenesis of systemic low-grade inflammation as well as its contribution to end-organ damage. Leaky gut facilitates endotoxemia with no clinical signs of infection and massive infiltration of mononuclear leukocytes in the lamina propria accompanied by a marked thickening of the colonic wall [182]. Further evidence for the leaky gut phenotype in CKD includes the detection of bacteria DNA fragments in the intestinal wall [26, 183, 184], reduced transepithelial electrical resistance in vitro following incubation with a pre-dialysis plasma of ESKD patients which can be rescued with post-dialysis plasma [185], and depletion of transcellular and intracellular protein constituents of the tight junction [31].
Several mechanisms are likely to mediate the progressive breakdown of the apical junctional complex. Firstly, as SCFA production declines, the colonic epithelia are effectively starved of the essential nutrition that jeopardizes the health of the mucosal cells. Secondly, alterations in the biochemical milieu are associated with an accumulation of urea-derived NH3 and ammonium hydroxide. In fact, twelve of the 19 microbial families (63%) with the greater abundances in ESKD patients were among the urease-possessing families. Recent studies demonstrated their potential to be a caustic compound capable of dissolving proteins, which subsequently weakens their ability to seal the intercellular space. Hence, it allows a now unrestricted transport of large solutes like dietary antigens and bacterial lipopolysaccharides thus aggravating systemic inflammation [185,186,187].
Furthermore, fluid overload, generalized edema, and congestive heart failure are common complications in CKD all of which can aggravate endotoxemia and contribute to a disturbance in mucosal barrier function [188, 189]. In addition, aggressive ultrafiltration or generous use of diuretics can lead to hypotension and bowel ischemia impacting the intestinal epithelia [190]. As the microbial flora is highly sensitive to any changes in the iron concentration, gastrointestinal micro-bleeding followed by uremic platelet dysfunction and high incidence of angiodysplasia and systemic anticoagulation disrupt the barrier function [191].
The molecular events that cause increased permeability are poorly defined. Recent work suggests that myosin light chain kinase (MLCK) plays a central role in epithelial barrier hemostasis, which is disrupted by chronic inflammation both in vivo and in vitro [192].
Of note, inflammation and disruption of the epithelial tight junction in CKD is associated with an impaired anti-oxidative system demonstrated by a decrease in the key antioxidant enzymes catalase and Cu-Zn superoxide dismutase on the one hand, and increased plasma level of nitric oxide synthase, monocyte chemotactic protein 1, and COX-2 on the other. These oxidative stress mediators induce the depletion of epithelial tight junction proteins, i.e., ZO-1, occludin, and claudin-1 [193].
In summary, leaky gut is an important hallmark in CKD pathophysiology, causally linked to dysbiosis and a loss of local metabolite hemostasis, being itself aggravating the detrimental interaction between the gut microbiota and the host by means of a vicious cycle (Fig. 1).
Conclusions and outlook: targeting the gut microbiome to attenuate CVD and progression of CKD in children
A novel additional approach in the treatment strategy of CKD could target gut health rather than the primary diseased organ [194]. In light of all the mechanistic evidence described here, a logical next phase in improving health outcomes for CKD patients would be to treat the gut microbiome dysbiosis, reduce bacterial production of uremic toxins, and boost SCFA production. Potential exists for dietary strategies to mitigate the downstream consequences of microbiome dysbiosis that drive disease progression in CKD, such as the gut barrier dysfunction, systemic low-grade inflammation, and accumulating uremic toxins [77, 195, 196].
A large-scale epidemiological study with CKD patients has recently identified a diet low in fruits and vegetables as a significant risk factor for ESKD [197]. Fiber shortens intestinal transit time and promotes the growth of saccharolytic bacteria. The particular benefit of this for CKD patients is by reducing the time for absorption of the metabolites of proteolytic fermentation and most significantly by increasing SCFAs to support a healthy intact epithelial barrier. Fiber deficiency is common in CKD patients, who tend to avoid fruits and vegetable in a misguided attempt to prevent diet-induced hyperkalemia that can have severe consequences for patients with impaired renal function [198].
Therefore, in the simplest terms, more fruits, vegetables, and dietary fiber should be included in the typical CKD diet, but caution should be implemented due to the risk of hyperkalemia. Individualized dietary plans are required with restriction to low potassium vegetables and fruits when appropriate.
Furthermore, pre- and probiotic supplementation could potentially help to improve gut health in CKD as well as other chronic diseases. Prebiotics have been defined as “substrates that are selectively utilized by host microorganisms conferring a health benefit” [199]. Indeed, a small trial in hemodialytic adults showed a significant reduction in circulating indoxyl sulfate and p-cresyl sulfate levels following oligofructose-inulin or resistant starch supplementation [200, 201]. However, attempts to restore the desired saccharolytic bacteria by introducing favorable microorganisms (probiotics) failed to reduce plasma concentrations of uremic solutes. The lack of benefit with probiotics may be partially explained by a persistence of uremia-induced biochemical milieu; thus, attempts with probiotic formulations without simultaneously improving the biochemical environment in the gut will be of no avail [31, 202]. The use of pre- and probiotics in patients with CKD has been reviewed recently confirming these contradictory findings [203].
Another promising approach is the use of oral adsorbents. AST-120, a highly potent charcoal that is widely known as a decontaminant, has been shown to markedly reduce the plasma concentration of indoxyl sulfate and p-cresol sulfate [204]. In animals, the adsorbent partially restored expression of tight junction proteins in the colon, reduced monocyte activation, and lowered inflammatory markers such as endotoxin, IL-6, and TNF-α [187]. However, AST-120 had no significant effect in terms of slowing CKD progression [205, 206].
To the best of our knowledge, no large-scale interventional studies have investigated the use of pre- or probiotic supplementation in children with CKD. Nevertheless, a small observational study in children with CKD revealed an inverse association between fiber consumption and serum concentrations of several protein-bound uremic toxins such as indoxyl sulfate, p-cresyl sulfate, indole acetic acid, and p-cresyl glucuronide [202]. In the absence of other contributing diseases which are commonly seen in adults, pre- or probiotic supplementation and improved, individualized nutrition seem to be a very promising treatment to slow CKD progression and prevent CKD-associated comorbidities.
Availability of data and materials
Not applicable.
References
Ardissino G, Dacco V, Testa S, Bonaudo R, Claris-Appiani A, Taioli E et al (2003) Epidemiology of chronic renal failure in children: data from the ItalKid project. Pediatrics. 111(4 Pt 1):e382–e387
Hamed RM (2002) The spectrum of chronic renal failure among Jordanian children. J Nephrol 15(2):130–135
Ahn SY, Moxey-Mims M (2018) CKD in children: the importance of a national epidemiologic study. Am J Kidney Dis 72(5):628–630
Mitsnefes MM (2012) Cardiovascular disease in children with chronic kidney disease. J Am Soc Nephrol 23(4):578–585
Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, Jafar TH, Heerspink HJ, Mann JF et al (2013) Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet. 382(9889):339–352
McDonald SP, Craig JC, Australian, New Zealand Paediatric Nephrology A (2004) Long-term survival of children with end-stage renal disease. N Engl J Med 350(26):2654–2662
Mitsnefes MM, Laskin BL, Dahhou M, Zhang X, Foster BJ (2013) Mortality risk among children initially treated with dialysis for end-stage kidney disease, 1990-2010. JAMA. 309(18):1921–1929
Oh J, Wunsch R, Turzer M, Bahner M, Raggi P, Querfeld U et al (2002) Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation. 106(1):100–105
Lilien MR, Groothoff JW (2009) Cardiovascular disease in children with CKD or ESRD. Nat Rev Nephrol 5(4):229–235
Kavey RE, Allada V, Daniels SR, Hayman LL, McCrindle BW, Newburger JW et al (2006) Cardiovascular risk reduction in high-risk pediatric patients: a scientific statement from the American Heart Association Expert Panel on Population and Prevention Science; the Councils on Cardiovascular Disease in the Young, Epidemiology and Prevention, Nutrition, Physical Activity and Metabolism, High Blood Pressure Research, Cardiovascular Nursing, and the Kidney in Heart Disease; and the Interdisciplinary Working Group on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation 114(24):2710–2738
Weaver DJ, Mitsnefes M (2018) Cardiovascular disease in children and adolescents with chronic kidney disease. Semin Nephrol 38(6):559–569
Civilibal M, Caliskan S, Adaletli I, Oflaz H, Sever L, Candan C et al (2006) Coronary artery calcifications in children with end-stage renal disease. Pediatr Nephrol 21(10):1426–1433
Lilien MR, Koomans HA, Schroder CH (2005) Hemodialysis acutely impairs endothelial function in children. Pediatr Nephrol 20(2):200–204
Kari JA, Donald AE, Vallance DT, Bruckdorfer KR, Leone A, Mullen MJ et al (1997) Physiology and biochemistry of endothelial function in children with chronic renal failure. Kidney Int 52(2):468–472
London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H (2003) Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 18(9):1731–1740
Kaddourah A, Uthup S, Madueme P, O'Rourke M, Hooper DK, Taylor MD et al (2015) Prevalence and predictors of aortic dilation as a novel cardiovascular complication in children with end-stage renal disease. Clin Nephrol 83(5):262–271
Rubin MF, Rosas SE, Chirinos JA, Townsend RR (2011) Surrogate markers of cardiovascular disease in CKD: what’s under the hood? Am J Kidney Dis 57(3):488–497
Groothoff JW, Gruppen MP, Offringa M, de Groot E, Stok W, Bos WJ et al (2002) Increased arterial stiffness in young adults with end-stage renal disease since childhood. J Am Soc Nephrol 13(12):2953–2961
Wong CJ, Moxey-Mims M, Jerry-Fluker J, Warady BA, Furth SL (2012) CKiD (CKD in children) prospective cohort study: a review of current findings. Am J Kidney Dis 60(6):1002–1011
Ross R (1999) Atherosclerosis--an inflammatory disease. N Engl J Med 340(2):115–126
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C et al (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377(12):1119–1131
Kato S, Chmielewski M, Honda H, Pecoits-Filho R, Matsuo S, Yuzawa Y et al (2008) Aspects of immune dysfunction in end-stage renal disease. Clin J Am Soc Nephrol 3(5):1526–1533
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B et al (2011) Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 472(7341):57–63
Stock J (2013) Gut microbiota: an environmental risk factor for cardiovascular disease. Atherosclerosis. 229(2):440–442
Rak K, Rader DJ (2011) Cardiovascular disease: the diet-microbe morbid union. Nature. 472(7341):40–41
Wang F, Jiang H, Shi K, Ren Y, Zhang P, Cheng S (2012) Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrology (Carlton) 17(8):733–738
Velasquez MT, Centron P, Barrows I, Dwivedi R, Raj DS (2018) Gut microbiota and cardiovascular uremic toxicities. Toxins 10(7):287. https://doi.org/10.3390/toxins10070287
Lin CJ, Wu V, Wu PC, Wu CJ (2015) Meta-analysis of the associations of p-cresyl sulfate (PCS) and indoxyl sulfate (IS) with cardiovascular events and all-cause mortality in patients with chronic renal failure. PLoS One 10(7):e0132589
Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G et al (2017) Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 5(1):14
Wu PH, Lin YT, Chiu YW, Baldanzi G, Huang JC, Liang SS et al (2021) The relationship of indoxyl sulfate and p-cresyl sulfate with target cardiovascular proteins in hemodialysis patients. Sci Rep 11(1):3786
Vaziri ND, Zhao YY, Pahl MV (2016) Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: the nature, mechanisms, consequences and potential treatment. Nephrol Dial Transplant 31(5):737–746
Berg G, Rybakova D, Fischer D, Cernava T, Verges MC, Charles T et al (2020) Microbiome definition re-visited: old concepts and new challenges. Microbiome. 8(1):103
Group NHW, Peterson J, Garges S, Giovanni M, McInnes P, Wang L et al (2009) The NIH Human Microbiome Project. Genome Res 19(12):2317–2323
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI (2007) The human microbiome project. Nature. 449(7164):804–810
Human Microbiome Project C (2012) Structure, function and diversity of the healthy human microbiome. Nature. 486(7402):207–214
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M et al (2005) Diversity of the human intestinal microbial flora. Science. 308(5728):1635–1638
Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL et al (2013) The long-term stability of the human gut microbiota. Science. 341(6141):1237439
Martin FP, Dumas ME, Wang Y, Legido-Quigley C, Yap IK, Tang H et al (2007) A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol Syst Biol 3:112
Dorrestein PC, Mazmanian SK, Knight R (2014) Finding the missing links among metabolites, microbes, and the host. Immunity. 40(6):824–832
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 464(7285):59–65
Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB et al (2017) Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 550(7674):61–66
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M et al (2012) Human gut microbiome viewed across age and geography. Nature. 486(7402):222–227
Tiihonen K, Ouwehand AC, Rautonen N (2010) Human intestinal microbiota and healthy ageing. Ageing Res Rev 9(2):107–116
Kennedy KM, Gerlach MJ, Adam T, Heimesaat MM, Rossi L, Surette MG et al (2021) Fetal meconium does not have a detectable microbiota before birth. Nat Microbiol 6(7):865–873. https://doi.org/10.1038/s41564-021-00904-0. Epub 2021 May 10
Martin R, Makino H, Cetinyurek Yavuz A, Ben-Amor K, Roelofs M, Ishikawa E et al (2016) Early-life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PLoS One 11(6):e0158498
Jakobsson HE, Abrahamsson TR, Jenmalm MC, Harris K, Quince C, Jernberg C et al (2014) Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut. 63(4):559–566
Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R (2011) Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 140(6):1713–1719
Penders J, Thijs C, van den Brandt PA, Kummeling I, Snijders B, Stelma F et al (2007) Gut microbiota composition and development of atopic manifestations in infancy: the KOALA Birth Cohort Study. Gut. 56(5):661–667
Vu K, Lou W, Tun HM, Konya TB, Morales-Lizcano N, Chari RS et al (2021) From birth to overweight and atopic disease: multiple and common pathways of the infant gut microbiome. Gastroenterology. 160(1):128–144 e10
Hviid A, Svanstrom H, Frisch M (2011) Antibiotic use and inflammatory bowel diseases in childhood. Gut. 60(1):49–54
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature. 444(7122):1022–1023
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 444(7122):1027–1031
Jost T, Lacroix C, Braegger CP, Rochat F, Chassard C (2014) Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ Microbiol 16(9):2891–2904
Cheng L, Akkerman R, Kong C, Walvoort MTC, de Vos P (2021) More than sugar in the milk: human milk oligosaccharides as essential bioactive molecules in breast milk and current insight in beneficial effects. Crit Rev Food Sci Nutr 61(7):1184–1200
Tuikhar N, Keisam S, Labala RK, Imrat RP, Arunkumar MC et al (2019) Comparative analysis of the gut microbiota in centenarians and young adults shows a common signature across genotypically non-related populations. Mech Ageing Dev 179:23–35
Silveira-Nunes G, Durso DF, Olieveira L Jr, Cunha EHM, Maioli TU, Vieira AT et al (2020) Hypertension is associated with intestinal microbiota dysbiosis and inflammation in a Brazilian population. Front Pharmacol 11:258
Mariat D, Firmesse O, Levenez F, Guimaraes V, Sokol H, Dore J et al (2009) The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol 9:123
Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM et al (2015) Gut dysbiosis is linked to hypertension. Hypertension. 65(6):1331–1340
Radjabzadeh D, Boer CG, Beth SA, van der Wal P, Kiefte-De Jong JC, Jansen MAE et al (2020) Diversity, compositional and functional differences between gut microbiota of children and adults. Sci Rep 10(1):1040
Tremaroli V, Backhed F (2012) Functional interactions between the gut microbiota and host metabolism. Nature. 489(7415):242–249
Vangay P, Ward T, Gerber JS, Knights D (2015) Antibiotics, pediatric dysbiosis, and disease. Cell Host Microbe 17(5):553–564
Risnes KR, Belanger K, Murk W, Bracken MB (2011) Antibiotic exposure by 6 months and asthma and allergy at 6 years: findings in a cohort of 1,401 US children. Am J Epidemiol 173(3):310–318
Schwartz DJ, Langdon AE, Dantas G (2020) Understanding the impact of antibiotic perturbation on the human microbiome. Genome Med 12(1):82
Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE et al (2018) Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 555(7698):623–628
Vich Vila A, Collij V, Sanna S, Sinha T, Imhann F, Bourgonje AR et al (2020) Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat Commun 11(1):362
Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H et al (2017) Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 551(7682):585–589
Mafra D, Borges NA, Lindholm B, Shiels PG, Evenepoel P, Stenvinkel P (2021) Food as medicine: targeting the uraemic phenotype in chronic kidney disease. Nat Rev Nephrol 17(3):153–171
Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O et al (2014) Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature. 514(7521):181–186
Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan J, DeSantis TZ et al (2013) Chronic kidney disease alters intestinal microbial flora. Kidney Int 83(2):308–315
Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E (2017) Dysbiosis and the immune system. Nat Rev Immunol 17(4):219–232
Rutsch A, Kantsjo JB, Ronchi F (2020) The gut-brain axis: how microbiota and host inflammasome influence brain physiology and pathology. Front Immunol 11:604179
Li B, Selmi C, Tang R, Gershwin ME, Ma X (2018) The microbiome and autoimmunity: a paradigm from the gut-liver axis. Cell Mol Immunol 15(6):595–609
Russell JT, Roesch LFW, Ordberg M, Ilonen J, Atkinson MA, Schatz DA et al (2019) Genetic risk for autoimmunity is associated with distinct changes in the human gut microbiome. Nat Commun 10(1):3621
Foster JA, Rinaman L, Cryan JF (2017) Stress & the gut-brain axis: regulation by the microbiome. Neurobiol Stress 7:124–136
Quiroga R, Nistal E, Estebanez B, Porras D, Juarez-Fernandez M, Martinez-Florez S et al (2020) Exercise training modulates the gut microbiota profile and impairs inflammatory signaling pathways in obese children. Exp Mol Med 52(7):1048–1061
Zheng S, Shao S, Qiao Z, Chen X, Piao C, Yu Y et al (2017) Clinical parameters and gut microbiome changes before and after surgery in thoracic aortic dissection in patients with gastrointestinal complications. Sci Rep 7(1):15228
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S et al (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 107(33):14691–14696
Wilmanski T, Diener C, Rappaport N, Patwardhan S, Wiedrick J, Lapidus J et al (2021) Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat Metab 3(2):274–286
Kim M, Benayoun BA (2020) The microbiome: an emerging key player in aging and longevity. Transl Med Aging 4:103–116
Castillo-Rodriguez E, Fernandez-Prado R, Esteras R, Perez-Gomez MV, Gracia-Iguacel C, Fernandez-Fernandez B et al (2018) Impact of altered intestinal microbiota on chronic kidney disease progression. Toxins 10(7):300. https://doi.org/10.3390/toxins10070300
Hooper LV, Gordon JI (2001) Commensal host-bacterial relationships in the gut. Science. 292(5519):1115–1118
Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS (2014) Bile acids and the gut microbiome. Curr Opin Gastroenterol 30(3):332–338
Metges CC (2000) Contribution of microbial amino acids to amino acid homeostasis of the host. J Nutr 130(7):1857S–1864S
Ramakrishna BS (2013) Role of the gut microbiota in human nutrition and metabolism. J Gastroenterol Hepatol 28(Suppl 4):9–17
Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ et al (2010) Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol 3(2):148–158
Gordon HA (1959) Morphological and physiological characterization of germfree life. Ann N Y Acad Sci 78:208–220
Anders HJ, Andersen K, Stecher B (2013) The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int 83(6):1010–1016
Shevchenko A, Simons K (2010) Lipidomics: coming to grips with lipid diversity. Nat Rev Mol Cell Biol 11(8):593–598
Hooper LV, Midtvedt T, Gordon JI (2002) How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr 22:283–307
Ouwehand AC, Derrien M, de Vos W, Tiihonen K, Rautonen N (2005) Prebiotics and other microbial substrates for gut functionality. Curr Opin Biotechnol 16(2):212–217
Snelson M, Kellow NJ, Coughlan MT (2019) Modulation of the gut microbiota by resistant starch as a treatment of chronic kidney diseases: evidence of efficacy and mechanistic insights. Adv Nutr 10(2):303–320
Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A (2011) Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol 17(12):1519–1528
Tazoe H, Otomo Y, Kaji I, Tanaka R, Karaki SI, Kuwahara A (2008) Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J Physiol Pharmacol 59(Suppl 2):251–262
Topping DL, Clifton PM (2001) Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 81(3):1031–1064
Snelson M, Coughlan MT (2019) Dietary advanced glycation end products: digestion, metabolism and modulation of gut microbial ecology. Nutrients 11(2):215. https://doi.org/10.3390/nu11020215
Annison G, Topping DL (1994) Nutritional role of resistant starch: chemical structure vs physiological function. Annu Rev Nutr 14:297–320
Ikee R, Sasaki N, Yasuda T, Fukazawa S (2020) Chronic kidney disease, gut dysbiosis, and constipation: a burdensome triplet. Microorganisms 8(12):1862. https://doi.org/10.3390/microorganisms8121862
Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K et al (2011) Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 469(7331):543–547
Thorburn AN, Macia L, Mackay CR (2014) Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity. 40(6):833–842
Bohmig GA, Krieger PM, Saemann MD, Wenhardt C, Pohanka E, Zlabinger GJ (1997) n-Butyrate downregulates the stimulatory function of peripheral blood-derived antigen-presenting cells: a potential mechanism for modulating T-cell responses by short-chain fatty acids. Immunology. 92(2):234–243
Chang PV, Hao L, Offermanns S, Medzhitov R (2014) The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A 111(6):2247–2252
Miller SJ, Zaloga GP, Hoggatt AM, Labarrere C, Faulk WP (2005) Short-chain fatty acids modulate gene expression for vascular endothelial cell adhesion molecules. Nutrition. 21(6):740–748
Saemann MD, Bohmig GA, Osterreicher CH, Burtscher H, Parolini O, Diakos C et al (2000) Anti-inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB J 14(15):2380–2382
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM et al (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 341(6145):569–573
Li M, van Esch B, Wagenaar GTM, Garssen J, Folkerts G, Henricks PAJ (2018) Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur J Pharmacol 831:52–59
Morrison DJ, Preston T (2016) Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 7(3):189–200
Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK et al (2011) The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13(5):517–526
Wong J, Piceno YM, DeSantis TZ, Pahl M, Andersen GL, Vaziri ND (2014) Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am J Nephrol 39(3):230–237
Kang JY (1993) The gastrointestinal tract in uremia. Dig Dis Sci 38(2):257–268
Vaziri ND, Freel RW, Hatch M (1995) Effect of chronic experimental renal insufficiency on urate metabolism. J Am Soc Nephrol 6(4):1313–1317
Jiang S, Xie S, Lv D, Zhang Y, Deng J, Zeng L et al (2016) A reduction in the butyrate producing species Roseburia spp. and Faecalibacterium prausnitzii is associated with chronic kidney disease progression. Antonie Van Leeuwenhoek 109(10):1389–1396
Barrios C, Beaumont M, Pallister T, Villar J, Goodrich JK, Clark A et al (2015) Gut-microbiota-metabolite axis in early renal function decline. PLoS One 10(8):e0134311
Dawson LF, Donahue EH, Cartman ST, Barton RH, Bundy J, McNerney R et al (2011) The analysis of para-cresol production and tolerance in Clostridium difficile 027 and 012 strains. BMC Microbiol 11:86
Ward LA, Johnson KA, Robinson IM, Yokoyama MT (1987) Isolation from swine feces of a bacterium which decarboxylates p-hydroxyphenylacetic acid to 4-methylphenol (p-cresol). Appl Environ Microbiol 53(1):189–192
Hsu CN, Lu PC, Hou CY, Tain YL (2019) Blood pressure abnormalities associated with gut microbiota-derived short chain fatty acids in children with congenital anomalies of the kidney and urinary tract. J Clin Med 8(8):1090. https://doi.org/10.3390/jcm8081090
Crespo-Salgado J, Vehaskari VM, Stewart T, Ferris M, Zhang Q, Wang G et al (2016) Intestinal microbiota in pediatric patients with end stage renal disease: a Midwest Pediatric Nephrology Consortium study. Microbiome. 4(1):50
Al Khodor S, Shatat IF (2017) Gut microbiome and kidney disease: a bidirectional relationship. Pediatr Nephrol 32(6):921–931
Sumida K, Yamagata K, Kovesdy CP (2020) Constipation in CKD. Kidney Int Rep 5(2):121–134
Evenepoel P, Poesen R, Meijers B (2017) The gut-kidney axis. Pediatr Nephrol 32(11):2005–2014
Ohama T, Hori M, Momotani E, Iwakura Y, Guo F, Kishi H et al (2007) Intestinal inflammation downregulates smooth muscle CPI-17 through induction of TNF-alpha and causes motility disorders. Am J Physiol Gastrointest Liver Physiol 292(5):G1429–G1438
De Winter BY, van den Wijngaard RM, de Jonge WJ (2012) Intestinal mast cells in gut inflammation and motility disturbances. Biochim Biophys Acta 1822(1):66–73
Bourke E, Milne MD, Stokes GS (1966) Caecal pH and ammonia in experimental uraemia. Gut. 7(5):558–561
Smith EA, Macfarlane GT (1997) Formation of phenolic and indolic compounds by anaerobic bacteria in the human large intestine. Microb Ecol 33(3):180–188
Smith EA, Macfarlane GT (1996) Enumeration of human colonic bacteria producing phenolic and indolic compounds: effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J Appl Bacteriol 81(3):288–302
Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT (1987) Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 28(10):1221–1227
Kortman GAM, Reijnders D, Swinkels DW (2017) Oral iron supplementation: potential implications for the gut microbiome and metabolome in patients with CKD. Hemodial Int 21(Suppl 1):S28–S36
Holle J, Querfeld U, Kirchner M, Anninos A, Okun J, Thurn-Valsassina D et al (2019) Indoxyl sulfate associates with cardiovascular phenotype in children with chronic kidney disease. Pediatr Nephrol 34(12):2571–2582
Drueke T, Witko-Sarsat V, Massy Z, Descamps-Latscha B, Guerin AP, Marchais SJ et al (2002) Iron therapy, advanced oxidation protein products, and carotid artery intima-media thickness in end-stage renal disease. Circulation. 106(17):2212–2217
Meyer TW, Hostetter TH (2007) Uremia. N Engl J Med 357(13):1316–1325
Vanholder R, De Smet R, Glorieux G, Argiles A, Baurmeister U, Brunet P et al (2003) Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int 63(5):1934–1943
Snauwaert E, Holvoet E, Van Biesen W, Raes A, Glorieux G, Vande Walle J et al (2019) Uremic toxin concentrations are related to residual kidney function in the pediatric hemodialysis population. Toxins 11(4):235. https://doi.org/10.3390/toxins11040235
Aronov PA, Luo FJ, Plummer NS, Quan Z, Holmes S, Hostetter TH et al (2011) Colonic contribution to uremic solutes. J Am Soc Nephrol 22(9):1769–1776
Cheng Y, Li Y, Benkowitz P, Lamina C, Kottgen A, Sekula P (2020) The relationship between blood metabolites of the tryptophan pathway and kidney function: a bidirectional Mendelian randomization analysis. Sci Rep 10(1):12675
Debnath S, Velagapudi C, Redus L, Thameem F, Kasinath B, Hura CE et al (2017) Tryptophan metabolism in patients with chronic kidney disease secondary to type 2 diabetes: relationship to inflammatory markers. Int J Tryptophan Res 10:1178646917694600
Karu N, McKercher C, Nichols DS, Davies N, Shellie RA, Hilder EF et al (2016) Tryptophan metabolism, its relation to inflammation and stress markers and association with psychological and cognitive functioning: Tasmanian Chronic Kidney Disease pilot study. BMC Nephrol 17(1):171
Kalaska B, Pawlak K, Domaniewski T, Oksztulska-Kolanek E, Znorko B, Roszczenko A et al (2017) Elevated levels of peripheral kynurenine decrease bone strength in rats with chronic kidney disease. Front Physiol 8:836
Kaminski TW, Pawlak K, Karbowska M, Mysliwiec M, Grzegorzewski W, Kuna J et al (2018) Association between uremic toxin-anthranilic acid and fibrinolytic system activity in predialysis patients at different stages of chronic kidney disease. Int Urol Nephrol 50(1):127–135
Addi T, Dou L, Burtey S (2018) Tryptophan-derived uremic toxins and thrombosis in chronic kidney disease. Toxins 10(10):412. https://doi.org/10.3390/toxins10100412
Zhang LS, Davies SS (2016) Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions. Genome Med 8(1):46
Poesen R, Viaene L, Verbeke K, Claes K, Bammens B, Sprangers B et al (2013) Renal clearance and intestinal generation of p-cresyl sulfate and indoxyl sulfate in CKD. Clin J Am Soc Nephrol 8(9):1508–1514
Bryniarski MA, Hamarneh F, Yacoub R (2019) The role of chronic kidney disease-associated dysbiosis in cardiovascular disease. Exp Biol Med (Maywood) 244(6):514–525
Holmar J, de la Puente-Secades S, Floege J, Noels H, Jankowski J, Orth-Alampour S (2020) Uremic toxins affecting cardiovascular calcification: a systematic review. Cells 9(11):2428. https://doi.org/10.3390/cells9112428
Lim YJ, Sidor NA, Tonial NC, Che A, Urquhart BL (2021) Uremic toxins in the progression of chronic kidney disease and cardiovascular disease: mechanisms and therapeutic targets. Toxins 13(2):142. https://doi.org/10.3390/toxins13020142
Snauwaert E, Van Biesen W, Raes A, Glorieux G, Van Bogaert V, Van Hoeck K et al (2018) Concentrations of representative uraemic toxins in a healthy versus non-dialysis chronic kidney disease paediatric population. Nephrol Dial Transplant 33(6):978–986
Holle J, Kirchner M, Okun J, Bayazit AK, Obrycki L, Canpolat N et al (2020) Serum indoxyl sulfate concentrations associate with progression of chronic kidney disease in children. PLoS One 15(10):e0240446
Zhu K, Meng Q, Zhang Z, Yi T, He Y, Zheng J et al (2019) Aryl hydrocarbon receptor pathway: role, regulation and intervention in atherosclerosis therapy (Review). Mol Med Rep 20(6):4763–4773
Carracedo J, Alique M, Vida C, Bodega G, Ceprian N, Morales E et al (2020) Mechanisms of cardiovascular disorders in patients with chronic kidney disease: a process related to accelerated senescence. Front Cell Dev Biol 8:185
Sarnak MJ, Amann K, Bangalore S, Cavalcante JL, Charytan DM, Craig JC et al (2019) Chronic kidney disease and coronary artery disease: JACC State-of-the-Art Review. J Am Coll Cardiol 74(14):1823–1838
Jovanovich A, Isakova T, Stubbs J (2018) Microbiome and cardiovascular disease in CKD. Clin J Am Soc Nephrol 13(10):1598–1604
Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC et al (2013) Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 128(6):632–642
Niwa T (2010) Uremic toxicity of indoxyl sulfate. Nagoya J Med Sci 72(1-2):1–11
Yamamoto H, Tsuruoka S, Ioka T, Ando H, Ito C, Akimoto T et al (2006) Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int 69(10):1780–1785
He X, Jiang H, Gao F, Liang S, Wei M, Chen L (2019) Indoxyl sulfate-induced calcification of vascular smooth muscle cells via the PI3K/Akt/NF-kappaB signaling pathway. Microsc Res Tech 82(12):2000–2006
Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK et al (2008) Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A 105(43):16767–16772
Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R (2011) Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem 22(9):849–855
Licciardi PV, Ververis K, Karagiannis TC (2011) Histone deacetylase inhibition and dietary short-chain fatty acids. ISRN Allergy 2011:869647
Li M, van Esch B, Henricks PAJ, Folkerts G, Garssen J (2018) The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor alpha-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front Pharmacol 9:533
Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L (2014) The role of short-chain fatty acids in health and disease. Adv Immunol 121:91–119
Usami M, Kishimoto K, Ohata A, Miyoshi M, Aoyama M, Fueda Y et al (2008) Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr Res 28(5):321–328
Chen L, Fischle W, Verdin E, Greene WC (2001) Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 293(5535):1653–1657
Tedelind S, Westberg F, Kjerrulf M, Vidal A (2007) Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: a study with relevance to inflammatory bowel disease. World J Gastroenterol 13(20):2826–2832
Miyoshi M, Usami M, Ohata A (2008) Short-chain fatty acids and trichostatin A alter tight junction permeability in human umbilical vein endothelial cells. Nutrition. 24(11-12):1189–1198
Waldecker M, Kautenburger T, Daumann H, Veeriah S, Will F, Dietrich H et al (2008) Histone-deacetylase inhibition and butyrate formation: fecal slurry incubations with apple pectin and apple juice extracts. Nutrition. 24(4):366–374
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H et al (2013) Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 500(7461):232–236
Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y et al (2011) Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 331(6015):337–341
Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y et al (2010) Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol 184(7):3433–3441
Kasubuchi M, Hasegawa S, Hiramatsu T, Ichimura A, Kimura I (2015) Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients. 7(4):2839–2849
Davie JR (2003) Inhibition of histone deacetylase activity by butyrate. J Nutr 133(7 Suppl):2485S–2493S
Kim S, Kim JH, Park BO, Kwak YS (2014) Perspectives on the therapeutic potential of short-chain fatty acid receptors. BMB Rep 47(3):173–178
Lee SU, In HJ, Kwon MS, Park BO, Jo M, Kim MO et al (2013) beta-Arrestin 2 mediates G protein-coupled receptor 43 signals to nuclear factor-kappaB. Biol Pharm Bull 36(11):1754–1759
Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME et al (2003) Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem 278(28):25481–25489
Ulven T (2012) Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front Endocrinol (Lausanne) 3:111
Ohira H, Fujioka Y, Katagiri C, Mamoto R, Aoyama-Ishikawa M, Amako K et al (2013) Butyrate attenuates inflammation and lipolysis generated by the interaction of adipocytes and macrophages. J Atheroscler Thromb 20(5):425–442
Sun M, Wu W, Liu Z, Cong Y (2017) Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J Gastroenterol 52(1):1–8
Sina C, Gavrilova O, Forster M, Till A, Derer S, Hildebrand F et al (2009) G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J Immunol 183(11):7514–7522
Aguilar EC, Leonel AJ, Teixeira LG, Silva AR, Silva JF, Pelaez JM et al (2014) Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFkappaB activation. Nutr Metab Cardiovasc Dis 24(6):606–613
Chai JT, Digby JE, Choudhury RP (2013) GPR109A and vascular inflammation. Curr Atheroscler Rep 15(5):325
Digby JE, Martinez F, Jefferson A, Ruparelia N, Chai J, Wamil M et al (2012) Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler Thromb Vasc Biol 32(3):669–676
Turner JR (2009) Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9(11):799–809
Nusrat A, Turner JR, Madara JL (2000) Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am J Physiol Gastrointest Liver Physiol 279(5):G851–G857
Van Itallie CM, Fanning AS, Bridges A, Anderson JM (2009) ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell 20(17):3930–3940
Vaziri ND, Dure-Smith B, Miller R, Mirahmadi MK (1985) Pathology of gastrointestinal tract in chronic hemodialysis patients: an autopsy study of 78 cases. Am J Gastroenterol 80(8):608–611
de Almeida Duarte JB, de Aguilar-Nascimento JE, Nascimento M, Nochi RJ Jr (2004) Bacterial translocation in experimental uremia. Urol Res 32(4):266–270
Wang F, Zhang P, Jiang H, Cheng S (2012) Gut bacterial translocation contributes to microinflammation in experimental uremia. Dig Dis Sci 57(11):2856–2862
Vaziri ND, Goshtasbi N, Yuan J, Jellbauer S, Moradi H, Raffatellu M et al (2012) Uremic plasma impairs barrier function and depletes the tight junction protein constituents of intestinal epithelium. Am J Nephrol 36(5):438–443
Vaziri ND, Yuan J, Norris K (2013) Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am J Nephrol 37(1):1–6
Vaziri ND, Yuan J, Khazaeli M, Masuda Y, Ichii H, Liu S (2013) Oral activated charcoal adsorbent (AST-120) ameliorates chronic kidney disease-induced intestinal epithelial barrier disruption. Am J Nephrol 37(6):518–525
Peschel T, Schonauer M, Thiele H, Anker SD, Schuler G, Niebauer J (2003) Invasive assessment of bacterial endotoxin and inflammatory cytokines in patients with acute heart failure. Eur J Heart Fail 5(5):609–614
Sandek A, Rauchhaus M, Anker SD, von Haehling S (2008) The emerging role of the gut in chronic heart failure. Curr Opin Clin Nutr Metab Care 11(5):632–639
Rossi UG, Petrocelli F, Seitun S, Ferro C (2012) Nonocclusive mesenteric ischemia in a dialysis patient with extensive vascular calcification. Am J Kidney Dis 60(5):843–846
Gerson SL, Talbot GH, Hurwitz S, Lusk EJ, Strom BL, Cassileth PA (1985) Discriminant scorecard for diagnosis of invasive pulmonary aspergillosis in patients with acute leukemia. Am J Med 79(1):57–64
Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR (2005) Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol 166(2):409–419
Kim HJ, Vaziri ND (2010) Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Ren Physiol 298(3):F662–F671
Lau WL, Savoj J, Nakata MB, Vaziri ND (2018) Altered microbiome in chronic kidney disease: systemic effects of gut-derived uremic toxins. Clin Sci (Lond) 132(5):509–522
Vanholder R, Glorieux G (2015) The intestine and the kidneys: a bad marriage can be hazardous. Clin Kidney J 8(2):168–179
Evenepoel P, Meijers BK, Bammens BR, Verbeke K (2009) Uremic toxins originating from colonic microbial metabolism. Kidney Int Suppl 114:S12–S19
Banerjee T, Carrero JJ, McCulloch C et al (2021) Dietary factors and prevention: risk of end-stage kidney disease by fruit and vegetable consumption. Am J Nephrol 52(5):356–367. https://doi.org/10.1159/000514754
Heindel J, Baid-Agrawal S, Rebholz CM, Nadal J, Schmid M, Schaeffner E et al (2020) Association between dietary patterns and kidney function in patients with chronic kidney disease: a cross-sectional analysis of the German Chronic Kidney Disease Study. J Ren Nutr 30(4):296–304
Gibson GR, Hutkins R, Sanders ME, Prescott SL, Reimer RA, Salminen SJ et al (2017) Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat Rev Gastroenterol Hepatol 14(8):491–502
Meijers BK, De Preter V, Verbeke K, Vanrenterghem Y, Evenepoel P (2010) p-Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol Dial Transplant 25(1):219–224
Marzocco S, Fazeli G, Di Micco L et al (2018) Supplementation of short-chain fatty acid, sodium propionate, in patients on maintenance hemodialysis: beneficial effects on inflammatory parameters and gut-derived uremic toxins, a pilot study (PLAN Study). J Clin Med 7(10). https://doi.org/10.3390/jcm7100315
El Amouri A, Snauwaert E, Foulon A, Vande Moortel C, Van Dyck M, Van Hoeck K et al (2021) Dietary fibre intake is associated with serum levels of uraemic toxins in children with chronic kidney disease. Toxins 13:225. https://doi.org/10.3390/toxins13030225
McFarlane C, Ramos CI, Johnson DW, Campbell KL (2019) Prebiotic, probiotic, and synbiotic supplementation in chronic kidney disease: a systematic review and meta-analysis. J Ren Nutr 29(3):209–220
Goto S, Yoshiya K, Kita T, Fujii H, Fukagawa M (2011) Uremic toxins and oral adsorbents. Ther Apher Dial 15(2):132–134
Schulman G, Berl T, Beck GJ, Remuzzi G, Ritz E, Shimizu M et al (2016) The effects of AST-120 on chronic kidney disease progression in the United States of America: a post hoc subgroup analysis of randomized controlled trials. BMC Nephrol 17(1):141
Liu W-C, Tomino Y, Lu K-C (2018) Impacts of indoxyl sulfate and p-cresol sulfate on chronic kidney disease and mitigating effects of AST-120. Toxins 10(9):367. https://doi.org/10.3390/toxins10090367
Vinolo MA, Rodrigues HG, Hatanaka E, Hebeda CB, Farsky SH, Curi R (2009) Short-chain fatty acids stimulate the migration of neutrophils to inflammatory sites. Clin Sci (Lond) 117(9):331–338
Wang L, de Zoeten EF, Greene MI, Hancock WW (2009) Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells. Nat Rev Drug Discov 8(12):969–981
Seljeset S, Siehler S (2012) Receptor-specific regulation of ERK1/2 activation by members of the “free fatty acid receptor” family. J Recept Signal Transduct Res 32(4):196–201
Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH (2013) Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 145(2):396–406 e1-10
Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T et al (2011) Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 117(4):1205–1217
Jeong Y, Du R, Zhu X, Yin S, Wang J, Cui H et al (2014) Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen-activated protein kinase phosphatase-1. J Leukoc Biol 95(4):651–659
Ashburner BP, Westerheide SD, Baldwin AS Jr (2001) The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol 21(20):7065–7077
Kendrick SF, O'Boyle G, Mann J, Zeybel M, Palmer J, Jones DE et al (2010) Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology. 51(6):1988–1997
Zou F, Qiu Y, Huang Y, Zou H, Cheng X, Niu Q et al (2021) Effects of short-chain fatty acids in inhibiting HDAC and activating p38 MAPK are critical for promoting B10 cell generation and function. Cell Death Dis 12(6):582
Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, de Roos P et al (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 504(7480):451–455
Lucas JL, Mirshahpanah P, Haas-Stapleton E, Asadullah K, Zollner TM, Numerof RP (2009) Induction of Foxp3+ regulatory T cells with histone deacetylase inhibitors. Cell Immunol 257(1-2):97–104
Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM et al (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 13(11):1299–1307
Akimova T, Ge G, Golovina T, Mikheeva T, Wang L, Riley JL et al (2010) Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3+ Tregs. Clin Immunol 136(3):348–363
Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R et al (2007) FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci U S A 104(11):4571–4576
Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C et al (2014) Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 20(2):159–166
Tazoe T, Sakamoto M, Nakajima T, Endoh T, Shiozawa S, Komiyama T (2009) Remote facilitation of supraspinal motor excitability depends on the level of effort. Eur J Neurosci 30(7):1297–1305
Yin L, Laevsky G, Giardina C (2001) Butyrate suppression of colonocyte NF-kappa B activation and cellular proteasome activity. J Biol Chem 276(48):44641–44646
Aoyama M, Kotani J, Usami M (2010) Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition. 26(6):653–661
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D et al (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 461(7268):1282–1286
Zapolska-Downar D, Siennicka A, Kaczmarczyk M, Kolodziej B, Naruszewicz M (2004) Butyrate inhibits cytokine-induced VCAM-1 and ICAM-1 expression in cultured endothelial cells: the role of NF-kappaB and PPARalpha. J Nutr Biochem 15(4):220–228
Vinolo MA, Hatanaka E, Lambertucci RH, Newsholme P, Curi R (2009) Effects of short chain fatty acids on effector mechanisms of neutrophils. Cell Biochem Funct 27(1):48–55
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E et al (2012) Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 61(2):364–371
Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD (2010) The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 32(3):379–391
Poralla L, Stroh T, Erben U, Sittig M, Liebig S, Siegmund B et al (2015) Histone deacetylase 5 regulates the inflammatory response of macrophages. J Cell Mol Med 19(9):2162–2171
Lukasova M, Malaval C, Gille A, Kero J, Offermanns S (2011) Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest 121(3):1163–1173
Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H et al (2014) Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 40(1):128–139
Acknowledgements
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Initiation and structure of text and figures: JS and JH; writing the first draft: JS; review and editing: all authors; figures: JS and FB. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schlender, J., Behrens, F., McParland, V. et al. Bacterial metabolites and cardiovascular risk in children with chronic kidney disease. Mol Cell Pediatr 8, 17 (2021). https://doi.org/10.1186/s40348-021-00126-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-021-00126-8