
Abstract
Background
Rickettsia and related diseases have been identified as significant global public health threats. This study involved comprehensive field and systematic investigations of various rickettsial organisms in Yunnan Province.
Methods
Between May 18, 2011 and November 23, 2020, field investigations were conducted across 42 counties in Yunnan Province, China, encompassing small mammals, livestock, and ticks. Preliminary screenings for Rickettsiales involved amplifying the 16S rRNA genes, along with additional genus- or species-specific genes, which were subsequently confirmed through sequencing results. Sequence comparisons were carried out using the Basic Local Alignment Search Tool (BLAST). Phylogenetic relationships were analyzed using the default parameters in the Molecular Evolutionary Genetics Analysis (MEGA) program. The chi-squared test was used to assess the diversities and component ratios of rickettsial agents across various parameters.
Results
A total of 7964 samples were collected from small mammals, livestock, and ticks through Yunnan Province and submitted for screening for rickettsial organisms. Sixteen rickettsial species from the genera Rickettsia, Anaplasma, Ehrlichia, Neoehrlichia, and Wolbachia were detected, with an overall prevalence of 14.72%. Among these, 11 species were identified as pathogens or potential pathogens to humans and livestock. Specifically, 10 rickettsial organisms were widely found in 42.11% (24 out of 57) of small mammal species. High prevalence was observed in Dremomys samples at 5.60%, in samples from regions with latitudes above 4000 m or alpine meadows, and in those obtained from Yuanmou County. Anaplasma phagocytophilum and Candidatus Neoehrlichia mikurensis were broadly infecting multiple genera of animal hosts. In contrast, the small mammal genera Neodon, Dremomys, Ochotona, Anourosorex, and Mus were carrying individually specific rickettsial agents, indicating host tropism. There were 13 rickettsial species detected in 57.14% (8 out of 14) of tick species, with the highest prevalence (37.07%) observed in the genus Rhipicephalus. Eight rickettsial species were identified in 2375 livestock samples. Notably, six new Rickettsiales variants/strains were discovered, and Candidatus Rickettsia longicornii was unambiguously identified.
Conclusions
This large-scale survey provided further insight into the high genetic diversity and overall prevalence of emerging Rickettsiales within endemic hotspots in Yunnan Province. The potential threats posed by these emerging tick-borne Rickettsiales to public health warrant attention, underscoring the need for effective strategies to guide the prevention and control of emerging zoonotic diseases in China.
Graphical Abstract

Similar content being viewed by others

Background
The order Rickettsiales comprises a major group of obligate intracellular Gram-negative bacteria responsible for various human diseases [1]. This order primarily includes Rickettsia, Anaplasma, Ehrlichia, Neoehrlichia, Wolbachia, and Orientia genera, each of which preferentially infects different vectorial invertebrate arthropods like ticks, fleas, mosquitoes, and mites [2]. Rickettsiales are highly prevalent and globally distributed across various arthropod vectors, animals, and human [3, 4]. Tick-associated rickettsioses are recognized as significant emerging zoonoses worldwide. Pathogens from the genera Anaplasma, Ehrlichia, and Rickettsia have been documented in parasitic ticks collected from humans [5].
Currently, there are at least 25 known tick species capable of transmitting pathogenic Rickettsia species [6]. Eight formally described species within the genus Anaplasma [7, 8] and 4 species within the genus Ehrlichia that are extensively distributed among terrestrial wild animal hosts [4, 9, 10]. In China, at least 11 species of tick-associated Rickettsiales known to cause human disease have been confirmed [11,12,13,14]. Clinical manifestations of tick-associated rickettsial diseases range from mild symptoms like fever, malaise, headache, myalgia, lymphadenopathy, rash, scabs, and gastrointestinal symptoms to severe complications such as renal or respiratory failure, respiratory acidosis, hyponatremia, pericarditis, septic shock, pneumonia, pleural effusion, hemorrhage, neurological complications, and potentially death [11,12,13,14].
Over the past three decades, advances in molecular diagnostic techniques have led to the rapid and precise identification of emerging pathogens among tick-associated rickettsial agents, as well as some Candidatus species of rickettsial agents worldwide [14,15,16,17,18,19]. In particular, novel rickettsioses and newly recognized natural foci often emerge unexpectedly. The rising popularity of ecotourism, extensive global travel, and the frequent migration of wild animal populations along with parasitic ticks have significantly contributed to the increasing incidence of tick-associated rickettsioses. Therefore, addressing this issue and reducing the burden of human rickettsioses presents significant challenges for public health authorities, physicians, veterinarians, and scientists.
In China, numerous reports have documented the distribution and co-circulation of Rickettsiales bacteria among ticks, wild animals, livestock, and febrile patients [20,21,22]. Yunnan Province, located in Southwest China and bordering the Indo-Chinese peninsula, is characterized by diverse eco-climatic zones and is recognized as a biodiversity hotspot. This region is of unique medical significance for assessing potential spillover vector-borne zoonoses and the emergence of new pathogens. Previous surveys have highlighted the high diversity of Rickettsiales in limited tick and small mammals samples collected from Yunnan [20, 23,24,25,26]. It is speculated that the host and vector species of tick-associated Rickettsiales in Yunnan Province are abundant and widely dispersed. In this study, extensive field and systematic investigations of multiple rickettsial species were conducted using molecular detection methods to reveal the true nature of Rickettsiales infections and their potential threats to public health.
Methods
Study sites and sample collection
Between May 18, 2011 and November 23, 2020, field investigations covering small wildlife mammals, domestic animal hosts, and tick vectors were conducted across 42 counties of Yunnan Province, encompassing various geographical terrains such as alpine meadows, cultivated lands, bushes, woodlands, coniferous forests, broadleaved forests, mixed forests, bamboo forests, residential areas, and dry-hot valleys, spanning an altitude gradient ranging from 383 to 4000 m above sea level.
Small mammals were sampled using the baited snap night-trap method. Morphological identification of captured animals was performed by experienced biologists and subsequently confirmed through sequence analysis of partial cytochrome oxidase I (COI) gene of suspected rodent species, followed by aseptic dissection to obtain liver and spleen tissue samples [27]. Livestock were sampled for whole blood collection via jugular vein puncture using EDTA anticoagulant tubes. Domestic mammal-associated ticks were collected by manual scratching, while host-seeking ticks were obtained through flag-sweeping on vegetation at the same sampling sites. Morphological identification of ticks was conducted by trained entomologists and subsequently validated using molecular evidence from COI genes [28]. All collected samples, including blood, tissues, and ticks, were transported to the laboratory under cold-chain conditions and stored at -80 °C until further processed.
DNA extraction, PCR and sequencing
DNA extraction from host blood samples and small mammal tissue samples was conducted based on the instructions provided by the manufacturer with the TIANamp Genomic DNA Kit (TIANGEN, Cat. No. 4992199). The identified ticks underwent surface sterilization with 70% alcohol, followed by drying and resin coating in PBS before being crushed. Buffer ATL (a tissue lysis buffer) and protease K were added to the homogenized liquid to facilitate tissue lysis. Subsequently, after centrifugation in a water bath, buffer ATL and anhydrous ethanol were added for DNA extraction. The DNA from individual ticks was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Cat. No. 69504), and the extracted DNA was stored in a freezer set at -80 °C for future use.
For screening the presence of bacterial DNA, including Rickettsia spp., Anaplasma spp., Ehrlichia spp., Neoehrlichia spp., and Wolbachia spp., primers targeting 16S rRNA of Rickettsiaceae and Anaplasmataceae agents were used. The primers used were Eh-out1 (5’-TTGAGAGTTTGATCCTGGCTCAGAACG-3’), 3-17pan (5’-TAAGGTGGTAATCCAGC-3’), and Eh-out2 (5’-CACCTCTACACTAGGAATTCCGCTATC-3’) [29]. Positive controls comprised of DNA templates from several previously confirmed positive samples. All positive PCR samples underwent purification using the Agarose Gel DNA Purification Kit (TaKaRa, Dalian, China) and were directly forwarded to Kunming Sangon Biological Engineering Technology and Services Co., Ltd. (Kunming, China) for sequencing.
The sequences were successfully assembled using the CLC Genomics Workbench (version 3.6.11, www.clcbio.com). Based on preliminary sequencing results, positive samples carrying human pathogenicity-associated pathogens or potential novel variants underwent further confirmation using nested PCR assays targeting related functional protein genes with previously reported primers (Table S1) [30,31,32,33,34,35,36].
Sequence comparisons and phylogenetic analyses
Sequence comparisons were conducted using the Basic Local Alignment Search Tool (BLAST) available on the NCBI website (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to analyze species and gene types. All sequences were aligned using Clustal W with default parameters in the Molecular Evolutionary Genetics Analysis (MEGA) program (version 6.0, The Pennsylvania State University, PA, USA). Phylogenetic relationships were inferred using the neighbor-joining method based on 1000 bootstrapped datasets. The statistical support for individual nodes was indicated by posterior probability values.
Statistical analyses
The chi-squared test or Fisher’s exact method was used to analyze the diversities and component ratios of rickettsial agents across various parameters, their distribution among different host species, habitats, and other potential influencing factors. Variables with a P value of < 0.05 were deemed statistically significant. All analyses were performed using SPSS (version 17.0, SPSS Inc., Chicago, IL).
Results
Diverse small mammals, livestock and ticks involved in Rickettsiales natural cycles
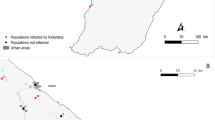
Between May 18, 2011 and November 23, 2020, a total of 7964 organisms were collected from 42 investigation counties (Table 1). Among them, 4330 small mammals were identified, representing 57 species across 21 genera, 10 families, and 4 orders. Rattus tanezumi emerged as the dominant species with the highest constituent ratio (720/4330, 16.63%) and exhibited the broadest distribution across 28 counties (Fig. 1, Table S2). Livestock samples comprised of 2375 individuals, representing 4 species, with goat (Capra aegagrus) as the predominant species (446/2375, 18.78%). Of the 1259 ticks collected, 829 were parasitized ticks and 430 were host-seeking ones, classified into 14 species under 5 genera of Ixodidae. Rhipicephalus microplus ranked as the dominant species (457/1259, 36.30%), followed by Ixodes ovatus (302/1259, 23.99%) (Fig. 1, Table S2).

Distribution of different species of samples and positive samples in different counties in Yunnan Province. Blue, red and green circles represent samples of livestock, small mammals and ticks, “ + ” represent sites, hosts and vectors with positive results for rickettsiae. The circles size of symbols represents the number of samples
Ten species under five genera emerging Rickettsiales in small mammals
In total, 2.59% (112/4330) of sampled individuals and 24 out of 57 (42.11%) species of small mammals were infected with rickettsial agents, primarily concentrated in western counties of Yunnan Province. The highest prevalence of rickettsial agents was observed in Yuanmou County (11.06%), followed by Weixi County (10.00%), with significant differences (P < 0.001) (Table 1). Among the 21 identified genera, the genus Rattus exhibited the highest frequencies of rickettsial agent infections (n = 51), with R. tanezumi having a prevalence of 3.06%. The genus Dremomys had the highest prevalence (5.60%, 1/18). Significant differences in prevalence were observed among genera of small mammals (P = 0.025) (Fig. 2A). Furthermore, the highest prevalence of rickettsial agents was observed in areas with altitudes above 4000 m (P = 0.004) (Fig. 2B). Additionally, the prevalence of rickettsiae was higher in males than in females (P = 0.004). Small animals collected from alpine meadows exhibited the highest prevalence, followed by those in mixed forests (P = 0.046) (Fig. 2C).

Comparison the positivity rate of rickettsiae in different altitudes, hosts, and habitats in Yunnan Province. A Host genus (P = 0.025) B Altitude (P = 0.004) C Habitat types (P = 0.046)
To better understand the potential transmission of rickettsial agents in small mammals, we constructed host-rickettsial correlation networks. Among the small mammals studied, 21 genera were found in 10 families. Of these, 14 genera in 6 families of small animals were found to be harboring rickettsial agents. Some rickettsial agents were specifically found to infect only one genus, such as Wolbachia sp. strain YL020 in the genus Apodemus, Anaplasma bovis in the genus Niniventer, and Ehrlichia sp. YN04 in the genus Rattus. Conversely, the genera Neodon, Dremomys, Ochatona, and Anourosorex were found to carry A. phagocytophilum, and the genus Mus was detected to host only Wolbachia sp. strain YM011, indicating host tropism, a preference towards certain rickettsia. However, both A. phagocytophilum and Candidatus Neoehrlichia mikurensis (CNM) were detected in more than 7 genera of small animals (Fig. 3), indicating a broad host spectrum.

Host-rickettsial correlation network topology. Rectangles represent the genera of the small mammals; the size of the circle represents the level of rickettsial agents’ positivity and the thickness of the straight line represents the level of host infection with rickettsiae
As revealed in Fig. 4, 10 different species of rickettsial agents were detected. Among these, A. phagocytophilum, A. ovis, CNM, and Rickettsia typhi were confirmed as human pathogens. CNM exhibited the highest positivity rate (1.04%, 45/4330), followed by A. phagocytophilum (0.97%, 42/4330) in small mammals. Notably, the sequences of samples DQ098 and DQ292 were identical and revealed the closest similarity (97.65%) to an uncultured Anaplasma sp. clone T7 (GenBank No. KU189193). This was provisionally termed Anaplasma sp. strain DQ098. Additionally, YL-020 and the other eight identical sequences represented by YM-011 had the highest similarities of 99.68% with OX366385 and 98.72% with CP116767, respectively, representing two distinct Wolbachia spp. (Table S3).

Phylogenetic tree based on partial sequences of 16S rRNA (660 bp) gene of the rickettsiae species derived from small mammals. Phylogenetic analysis was performed using the neighbor-joining method and trees were tested by bootstrapping (1000 replicates)
The constructed phylogenetic tree revealed that the rickettsial species detected in the small mammals clustered into five major groups: Neoehrlichia, Ehrlichia, Anaplasma, Wolbachia, and Rickettsia (Fig. 4). Within the Wolbachia cluster, the sequences identified were significantly different from known species and were categorized into two clades, termed Wolbachia sp. strain YL020 and Wolbachia sp. strain YM011.
High prevalence of eight species emerging Rickettsiales in livestocks
The positivity rate in livestock blood samples collected from 16 counties was 33.68% (800/2375). The highest prevalence was observed in Yingjiang County (81.65%, 89/109), followed by Huaping County (66.02%, 68/103), with significant differences (P < 0.001). Among the four types of livestock sampled, Bovine taurus exhibited the highest number of infected individuals and the highest prevalence of rickettsial agent infections (45.03%, 317/704, P < 0.001). The phylogenetic tree indicated that the rickettsial strains detected in livestock clustered into three distinct groups: Ehrlichia, Anaplasma, and Wolbachia. The predominant species identified were A. ovis, A. marginale, and A. phagocytophilum. Notably, A. phagocytophilum displayed considerable diversity, with at least 3 variants differing by 1 to 30 base pairs, though they were located within the same clade in the phylogenetic tree (Fig. 5).

Phylogenetic tree based on partial sequences of 16S rRNA (660 bp) gene of the rickettsiae species derived from livestock. Phylogenetic analysis was performed using the neighbor-joining method and trees were tested by bootstrapping (1000 replicates)
High prevalence and wide distribution of Rickettsiales in ticks
The overall positivity rate among 1259 ticks, encompassing 14 species in 5 genera, was 20.65% (260/1259). Rickettsiales agents were observed in all 14 sampling counties, with infestations of 8 out of 14 (57.14%) tick species. The genus Rhipicephalus had the highest prevalence (37.07%, 175/472, P < 0.001). Specifically, the prevalence of Rickettsiales in the dominant tick species R. microplus and Haemaphysalis montgomeryi was 36.54% (167/457) and 28.86% (58/201), respectively. H. yeni exhibited the highest prevalence (85.71%, 12/14, P < 0.001). A total of 13 species of rickettsiae were detected in the tick samples, of which two were identified as pathogenic to humans (Fig. 5). The main tick-associated rickettsial agents were Candidatus R. longicornii (n = 147) and A. ovis (n = 67). Among the 14 sampling sites, Jianchuan County had the highest number of positive ticks (n = 126), while Heqing County had the highest prevalence (85.71%, 12/14, P < 0.001), reflecting their different population sizes. Additionally, ticks collected at altitudes of 2000 to 2499 m had the highest prevalence of rickettsiae (27.08%, 88/325, P = 0.004). The prevalence of parasitic ticks (28.47%, 236/829) was significantly higher than that of host-seeking ticks (5.58%, 24/430, P < 0.001). Notably, ticks parasitizing Bos taurus exhibited the highest prevalence (53.84%, 49/91, P < 0.001).
BLAST analysis revealed that the 16S rRNA of HQ-T-1 had 98.49% identity with uncultured Anaplasma sp. clone D9_6 (GenBank No. MK814441), JC-T-222 had complete identity with uncultured Anaplasma sp. clone Dedessa (GenBank No. KY924886), and TC-T-400 had 99% identity with OL690561, forming a distinct branch in the phylogenetic tree. Combined with phylogenetic analyses, HQ-T-1, JC-T-222, and TC-T-400 were confirmed as monophyletic groups, named Anaplasma sp. strain HQT1, Anaplasma sp. strain JCT222, and Anaplasma sp. strain TCT400, respectively (Fig. 6).

Phylogenetic tree based on partial sequences of 16S rRNA (660 bp) gene of the rickettsiae species derived from ticks. Phylogenetic analysis was performed using the neighbor-joining method and trees were tested by bootstrapping (1000 replicates)
Characterization of seven pathogenic rickettsial and Candidatus Rickettsia longicornii with multigene analysis
Seven tick samples (WX-T-85 to WX-T-91) positive for Candidatus R. longicornii from Weixi County were selected for sequencing of the entire ompA, gltA, ompB, 17 kDa, 16S rRNA (nearly full length), sca4, and sca1 genes (Fig. 7A-G). The ompA, ompB, 16S rRNA, and sca1 genes displayed 100% homology with the sequences of Candidatus R. longicornii or Rickettsia endosymbiont of Haemaphysalis longicornis. The gltA and 17 kDa genes from WX-T-90 and WX-T-91 had 100% homology with the sequences of Candidatus R. jingxinensis (GenBank Nos. MW114883 and MW114879). The sca4 gene sequence revealed 99.51% homology with Candidatus R. longicornii (GenBank Nos. MK620855, MG906677). Despite some conflicting sequences, the rickettsial organism is likely Candidatus R. longicornii.

Phylogenetic trees constructed by the MEGA v.6.0 software based on the neighbor-joining method of multigene detected in ticks infected by Candidatus Rickettsia longicornii. A ompA gene (384 bp); B gltA gene (381 bp); C ompB gene (400 bp); D 17 kDa gene (395 bp); E 16S rRNA gene (nearly full length); F sca4 gene (843 bp); G sca1 gene (429 bp)
Both the full-length 16S rRNA gene (Fig. S1A) and the groEL gene of CNM (Fig. S1B) detected in this study were remarkably distinct from other sequences detected in China (Heilongjiang), Germany, and Switzerland. Among the three clusters of CNM based on the groEL gene the phylogenetic tree indicated that the present sequences belong to cluster I (southwest) and cluster III (southeast) [37] (Fig. S1B). Although the sequences from Lushui and Weixi were close to the southwest branch (I), they clustered into small branches, indicating they differ from previous ones in Yunnan.
Five Anaplasma-associated pathogens (A. capra, A. ovis, A. bovis, A. marginale, and A. phagocytophilum) were confirmed in samples. The groEL (Fig. S2B) and gltA (Fig. S2C) genes of A. capra revealed 100% homology between Capra aegagrus in Tengchong and patients in Heilongjiang [8]. The msp4 gene (Fig. S2D) of A. capra had 99.8% homology, indicating pronounced similarity with the pathogen infecting patients. Variations of A. capra were also detected in Tengchong, Deqin, and Shangri-La (Fig. S2). The phylogenetic tree based on the msp4 and entire 16S rRNA gene (Fig. S3A) of A. ovis formed a cohesive branch closely aligned with reference sequences. Sequences detected in Yunlong consistently clustered within a separate branch, demonstrating a close genetic relationship across different hosts within this geographic region. The gltA gene (Fig. S3C) of A. ovis distributed into two small branches. The groEL gene of A. bovis, A. marginale, and A. phagocytophilum also indicated diversity and distinctive distribution among their host ticks and animals (Fig. S4). The groEL and gltA genes of E. canis clustered in a distinct branch along with other sequences identified in ticks in China, with sister branches from Thailand, the Philippines, Spain, and France. The Dsb and TRP36 genes of E. canis formed unique, singular clades (Fig. S5).
Discussion
We identified the co-circulation of multiple species of Rickettsiales bacteria, including previously uncharacterized species, among small mammals, livestock, and ticks in Yunnan Province. This underscores the enduring importance of Rickettsiales infections in southwestern China and the need for ongoing surveillance of local arthropods, mammals, and humans.
The extensive diversity of tick-associated Rickettsia species globally is well documented, with instances such as R. tillamookensis in the United States and various species found in wild mammals in Morocco and Mauritania [38, 39]. Advances in phylogenetics and molecular detection techniques have greatly facilitated the identification of numerous novel Rickettsiales species associated with mammals and arthropods [5]. In China, new and uncharacterized rickettsial species continue to be discovered such as Rickettsia sp. sw. in Yunnan Province and Ehrlichia sp. in Inner Mongolia [23, 40]. Recent reports indicate a high diversity of Rickettsia spp. and Ehrlichia spp. in five species of tick from Yunnan Province [26]. We further identified six novel strains and one new record in Yunnan along with known Rickettsiales agents, emphasizing the remarkable diversity of Rickettsiales bacteria in ticks, as well as in small mammals and livestock.
There are variations in the reported prevalence of rickettsial agent infections among wild small mammals in different regions such as Southwest China and the Indo-China peninsula (2.02%), Slovakia (19.10%), and Kazakhstan (2.72%) [41,42,43]. In our study, the genus Dremomys exhibited a significantly high rate of Rickettsiales positivity among small mammals, while the highest frequency was observed in the genus Rattus, which is widely distributed and commonly found near local residences. Therefore, these two genera should be focused on when monitoring rickettsial infections. Additionally, our observations indicated changes in rickettsial prevalence based on sex and eco-habitats. The higher infection rates in males may be attributed to their more competitive and aggressive behaviors, leading to increased contact and susceptibility to infection. Furthermore, samples collected from alpine meadows revealed higher prevalence rates of Rickettsiales agents, possibly due to the presence of dominant host animals in these habitats. These observations warrant further detailed investigation. In summary, the rickettsial agents detected in these small mammals exhibited distinctive genetic diversity characteristics both interspecifically and intraspecifically.
Moreover, diverse rickettsial species, such as Rickettsia spp., Anaplasma spp., Ehrlichia spp., Neoehrlichia spp., and Wolbachia spp., were identified in 33.68% (800/2375) of livestock and were distributed across almost all surveyed counties. Given the close contact between humans and livestock, this highlights a potentially high risk of human exposure to these rickettsial agents. Among the rickettsial agents detected in livestock, A. phagocytophilum, A. capra, A. platys, and A. bovis variant were confirmed to possess human pathogenicity in earlier studies [7, 8, 44, 45]. Additionally, A. bovis and A. bovis-like infections have recently been identified in patients from China and the United States [16, 46]. Conversely, A. marginale and E. canis are recognized as pathogenic to animals rather than humans [47, 48].
Numerous reports in previous literatures have highlighted the risk posed by Rickettsiales, such as A. ovis infections in Iran and Anaplasma spp. in Pakistan [49, 50]. Our large-scale study confirmed that locations in Yuanmou County, areas at latitudes over 4000 m, and alpine meadow regions exhibited significantly higher Rickettsiales positivity rates among small mammals. Additionally, A. phagocytophilum was frequently detected in small mammals, livestock, and ticks, confirming its widespread distribution in Yunnan Province, particularly in Deqin County, where there is a higher potential for forming transmission chains.
Certain rickettsial species may exhibit specific host or vector associations, indicating host tropism for these agents. While some Rickettsiales are host-specific, others switch hosts or circulate regularly among different hosts, particularly mammals and blood-sucking arthropods [5]. Since most emerging human infectious diseases originate from spillover events from vectors or animal hosts, assessing infections among hosts, vectors, and febrile patients in local representative areas is crucial for understanding tick-associated Rickettsiales [4, 6, 7]. Therefore, the characterization and identification of novel Rickettsiales are of significant importance for both human and animal health.
Multigene analysis of E. canis from representative samples revealed a clustering pattern, distinct from international strains, indicating that the E. canis detected in Yunnan Province has unique genetic characteristics. Similarly, the multigene analysis of CNM from Weixi samples revealed close genetic similarities to those from southeastern China, yet notable divergence from strains previously detected in Yunnan, Heilongjiang, and abroad. Based on the previous classification standards for CNM, the CNM from Weixi should be considered distinct variants [44]. Our results also indicated that A. ovis was the most prevalent species, with a high positivity rate, widespread distribution, and interspecific differences in the survey region. However, sequences from Yunlong consistently clustered within a separate branch, demonstrating close genetic relationships across different hosts within this specific geographic region.
A comprehensive analysis of multiple genes for our Rickettsia agent, which was closely related to Candidatus R. longicornii, Rickettsia YN03, and R. jingxinensis, revealed that the nomenclature of “Candidatus R. jingxinensis” is not rigorous [51,52,53,54]. In fact, “Candidatus R. jingxinensis” and “Candidatus R. longicornii” should be considered synonyms. Based on the rules of species nomenclature, the name “Candidatus R. longicornii” should be accepted as valid, hence it should be referred to as R. longicornii. Similarly, the name Rickettsia YN03 should also be synonymized as R. longicornii, despite the observed degree of variation and considerable diversity.
Our primary limitation was the absence of human case records and samples, which precluded the confirmation of potential infections among local residents. Most study areas are located in remote mountainous regions of Yunnan Province, characterized by a high population of ethnic minorities. These areas are economically underdeveloped, and the local population generally has low awareness of self-protection measures and limited access to medical services. Furthermore, medical services at the grassroots level are inadequate, lacking the capacity for clinical diagnosis and management of emerging tick-associated rickettsioses, leading to frequent missed or misdiagnosed cases. Additionally, the prevalence of tick-associated rickettsioses could negatively impact ecotourism and economic development in Yunnan Province. Therefore, early surveillance of these emerging Rickettsiales among hosts, vectors, and exposed populations in these hotspots is crucial for effective prevention and control.
Conclusions
This large-scale study underscores the genetic diversity and overall prevalence of emerging tick-associated Rickettsiales within biodiversity hotspots in Yunnan Province. These findings reveal the substantial threats posed by emergent Rickettsiales and serve as a crucial reference for the prevention and control of related zoonotic diseases in southwestern China. Given the impact of these emerging Rickettsiales in these hotspots, urgent surveillance and implementation of prevention and control measures for the local human populations is warranted.
Availability of data and materials
Sequencing data generated during study was submitted to China National Microbiology Data Center under the BioProject. For each biological sample, the accession number as well as the corresponding link can be found in Supplementary Table.
References
Dantas-Torres F, Chomel BB, Otranto D. Ticks and tick-borne diseases: a One Health perspective. Trends Parasitol. 2012;28(10):437–46. https://doi.org/10.1016/j.pt.2012.07.003.
Salje J. Cells within cells: Rickettsiales and the obligate intracellular bacterial lifestyle. Nat Rev Microbiol. 2021;19(6):375–90. https://doi.org/10.1038/s41579-020-00507-2.
Rifkin RF, Vikram S, Alcorta J, Ramond JB, Cowan DA, Jakobsson M, et al. Rickettsia felis DNA recovered from a child who lived in southern Africa 2000 years ago. Commun Biol. 2023;6(1):240. https://doi.org/10.1038/s42003-023-04582-y.
Kolo A. Anaplasma species in Africa-A century of discovery: A Review on molecular epidemiology, genetic diversity, and control. Pathogens. 2023;12(5):702. https://doi.org/10.3390/pathogens12050702.
Seo JY, Kim YJ, Kim SY, Lee HI. Molecular detection of Anaplasma, Ehrlichia and Rickettsia pathogens in ticks collected from humans in the Republic of Korea, 2021. Pathogens. 2023;12(6):802. https://doi.org/10.3390/pathogens12060802.
Piotrowski M, Rymaszewska A. Expansion of tick-borne rickettsioses in the world. Microorganisms. 2020;8(12):1906. https://doi.org/10.3390/microorganisms8121906.
Dumler JS, Barbet AF, Bekker CP, Dasch GA, Palmer GH, Ray SC, et al. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and “HGE agent” as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol. 2001;51(6):2145–65. https://doi.org/10.1099/00207713-51-6-2145.
Li H, Zheng YC, Ma L, Jia N, Jiang BG, Jiang RR, et al. Human infection with a novel tick-borne Anaplasma species in China: a surveillance study. Lancet Infect Dis. 2015;15(6):663–70. https://doi.org/10.1016/S1473-3099(15)70051-4.
André MR. Diversity of Anaplasma and Ehrlichia/Neoehrlichia agents in terrestrial wild carnivores worldwide: Implications for human and domestic animal health and wildlife conservation. Front Vet Sci. 2018;5:293. https://doi.org/10.3389/fvets.2018.00293.
Karshima SN, Ahmed MI, Mohammed KM, Pam VA, Momoh-Abdullateef H, Gwimi BP. Worldwide meta-analysis on Anaplasma phagocytophilum infections in animal reservoirs: Prevalence, distribution and reservoir diversity. Vet Parasitol Reg Stud Reports. 2023;38:100830. https://doi.org/10.1016/j.vprsr.2022.100830.
Fang LQ, Liu K, Li XL, Liang S, Yang Y, Yao HW, Sun RX, et al. Emerging tick-borne infections in mainland China: an increasing public health threat. Lancet Infect Dis. 2015;15(12):1467–79. https://doi.org/10.1016/S1473-3099(15)00177-2.
Li H, Li XM, Du J, Zhang XA, Cui N, Yang ZD, et al. Candidatus Rickettsia xinyangensis as cause of spotted fever group rickettsiosis, Xinyang, China, 2015. Emerg Infect Dis. 2020;26(5):985–8. https://doi.org/10.3201/eid2605.170294.
Lu M, Qin XC, Jiang YZ, Guo Q, Jin XJ, Teng ZQ, et al. Emergence of ehrlichiosis by a new tick-borne Ehrlichia species in China. Int J Infect Dis. 2023;131:32–9. https://doi.org/10.1016/j.ijid.2023.03.038.
Xu N, Gai W, Zhang Y, Wang W, Wang G, Dasch GA, et al. Confirmation of Rickettsia conorii subspecies indica infection by next-generation sequencing, Shandong, China. Emerg Infect Dis. 2021;27(10):2691–4. https://doi.org/10.3201/eid2710.204764.
Vieira TSWJ, Collere FCM, Ferrari LDR, Baggio RA, Lange RR, Ferrari MV, et al. Novel Anaplasmataceae agents Candidatus Ehrlichia hydrochoerus and Anaplasma spp. infecting Capybaras, Brazil. Emerg Infect Dis. 2022;28(2):480–2. https://doi.org/10.3201/eid2802.210705.
Karpathy SE, Kingry L, Pritt BS, Berry JC, Chilton NB, Dergousoff SJ, et al. Anaplasma bovis-like infections in humans, United States, 2015–2017. Emerg Infect Dis. 2023;29(9):1904–7. https://doi.org/10.3201/eid2909.230559.
Lu M, Meng C, Li Y, Zhou G, Wang L, Xu X, et al. Rickettsia sp. and Anaplasma spp. in Haemaphysalis longicornis from Shandong Province of China, with evidence of a novel species “Candidatus Anaplasma Shandongensis.” Ticks Tick Borne Dis. 2023;14(1):102082. https://doi.org/10.1016/j.ttbdis.2022.102082.
Krawczak FS, Binder LC, Gregori F, et al. 'Candidatus Rickettsia andeanae’ and probable exclusion of Rickettsia parkeri in ticks from dogs in a natural area of the pampa biome in Brazil. Pathogens. 2023;12(3):446. https://doi.org/10.3390/pathogens12030446.
Chaloemthanetphong A, Ahantarig A, Apanaskevich DA, Martins TF, Pádua GT, Sponchiado J, et al. A novel Rickettsia, Candidatus Rickettsia takensis, and the first record of Candidatus Rickettsia laoensis in dermacentor from Northwestern Thailand. Sci Rep. 2023;13(1):10044. https://doi.org/10.1038/s41598-023-37206-w.
Lu M, Tian J, Pan X, Qin X, Wang W, Chen J, et al. Identification of Rickettsia spp., Anaplasma spp., and an Ehrlichia canis-like agent in Rhipicephalus microplus from southwest and south-central China. Ticks Tick Borne Dis. 2022;13(2):101884. https://doi.org/10.1016/j.ttbdis.2021.101884.
Jin X, Liao J, Chen Q, Ding J, Chang H, Lyu Y, et al. Diversity of Rickettsiales bacteria in five species of ticks collected from Jinzhai county, Anhui Province, China in 2021–2022. Front Microbiol. 2023;14:1141217. https://doi.org/10.3389/fmicb.2023.1141217.
Xu J, Gu XL, Jiang ZZ, Cao XQ, Wang R, Peng QM, et al. Pathogenic Rickettsia, Anaplasma, and Ehrlichia in Rhipicephalus microplus ticks collected from cattle and laboratory hatched tick larvae. PLoS Negl Trop Dis. 2023;17(8):e0011546. https://doi.org/10.1371/journal.pntd.0011546.
Wang Q, Guo WB, Pan YS, Jiang BG, Du CH, Que TC, et al. Detection of novel spotted fever group Rickettsiae (Rickettsiales: Rickettsiaceae) in ticks (Acari: Ixodidae) in southwestern China. J Med Entomol. 2021;58(3):1363–9. https://doi.org/10.1093/jme/tjaa294.
Bian CL, Gong ZD, Zhang LY, Li DM, Ge JQ, Li SQ, et al. Identification of Anaplasma phagocytophilum in small mammals from Hengduan mountains of southwest China. Chin J Epidemiol. 2009;30(12):1277–80. https://doi.org/10.3760/cma.j.issn.0254-6450.2009.12.017. (InChinese).
Yuan TT, Du CH, Xia LY, Que TC, von Fricken ME, Jiang BG, et al. Molecular evidence of Candidatus Rickettsia longicornii and a novel Rickettsia strain from ticks in Southern China. Ticks Tick Borne Dis. 2021;12(3):101679. https://doi.org/10.1016/j.ttbdis.2021.101679.
Zhang J, Ni XB, Chen ZY, Zhang Y, Wei R, Gong ZD, et al. High diversity of tick-associated microbiota from five tick species in Yunnan. China Zoonoses. 2023;3(1):1–9. https://doi.org/10.15212/ZOONOSES-2023-0005.
Robins JH, Hingston M, Matisoo-Smith L, Ross HA. Identifying Rattus species using mitochondrial DNA. Mol Ecol Notes. 2007;7(5):717–29. https://doi.org/10.1111/j.1471-8286.2007.01752.x.
Lu X, Lin XD, Wang JB, Qin XC, Tian JH, Guo WP, et al. Molecular survey of hard ticks in endemic areas of tick-borne diseases in China. Ticks Tick Borne Dis. 2013;4(4):288–96. https://doi.org/10.1016/j.ttbdis.2013.01.003.
Lu M, Tian JH, Yu B, Guo WP, Holmes EC, Zhang YZ. Extensive diversity of rickettsiales bacteria in ticks from Wuhan. China Ticks Tick Borne Dis. 2017;8(4):574–80. https://doi.org/10.1016/j.ttbdis.2017.03.006.
Arroyave E, Rodas-González JD, Zhang X, Labruna MB, González MS, Fernández-Silva JA, et al. Ehrlichia canis TRP36 diversity in naturally infected-dogs from an urban area of Colombia. Ticks Tick Borne Dis. 2020;11(3):101367. https://doi.org/10.1016/j.ttbdis.2019.101367.
de la Fuente J, Moraga-Fernández A, Alberdi P, Díaz-Sánchez S, García-Álvarez O, Fernández-Melgar R, et al. A quantum vaccinomics approach for the design and production of MSP4 chimeric antigen for the control of Anaplasma phagocytophilum infections. Vaccines (Basel). 2022;10(12):1995. https://doi.org/10.3390/vaccines10121995.
Inokuma H, Brouqui P, Drancourt M, Raoult D. Citrate synthase gene sequence: a new tool for phylogenetic analysis and identification of Ehrlichia. J Clin Microbiol. 2001;39(9):3031–9. https://doi.org/10.1128/JCM.39.9.3031-3039.2001.
Teja MMS, Mamatha GS, Lakkundi JN, Chandranaik BM, Murthy CMK, Gomes AR. Multiplex PCR for detection of Anaplasma marginale, A. bovis and A. platys in cattle. J Parasit Dis. 2023;47(3):659–63. https://doi.org/10.1007/s12639-023-01606-6.
Wang SW, Zhang LJ, Wang YY, Yu HL, Liang CW, Chen Q. Establishment and comparison of real-time PCR assays to detect Anaplasma msp-2 gene with general TaqMan probs and TaqMan-MGB probe. Dis Surv. 2011;26(1):12–4. https://doi.org/10.3784/j.issn.1003-9961.2011.01.005.
Wei F, Song M, Liu H, Wang B, Wang S, Wang Z, et al. Molecular detection and characterization of zoonotic and veterinary pathogens in ticks from Northeastern China. Front Microbiol. 2016;7:1913. https://doi.org/10.3389/fmicb.2016.01913.
Zhan L, Cao WC, de Vlas S, Xie SY, Zhang PH, Wu XM, et al. A newly discovered Anaplasma phagocytophilum variant in rodents from southeastern China. Vector Borne Zoonotic Dis. 2008;8(3):369–80. https://doi.org/10.1089/vbz.2007.0211.
Li H, Jiang J, Tang F, Sun Y, Li Z, Zhang W, et al. Wide distribution and genetic diversity of “Candidatus Neoehrlichia mikurensis” in rodents from China. Appl Environ Microbiol. 2013;79(3):1024–7. https://doi.org/10.1128/AEM.02917-12.
Gauthier DT, Karpathy SE, Grizzard SL, Batra D, Rowe LA, Paddock CD. Characterization of a novel transitional group Rickettsia species (Rickettsia tillamookensis sp. nov.) from the western black-legged tick, Ixodes pacificus. Int J Syst Evol Microbiol. 2021;71(7):004880. https://doi.org/10.1099/ijsem.0.004880.
Santos-Silva S, Santos N, Boratyński Z, Mesquita JR, Barradas PF. Diversity of Rickettsia spp. in ticks from wild mammals of Morocco and Mauritania. Ticks Tick Borne Dis. 2023;14(6):102235. https://doi.org/10.1016/j.ttbdis.2023.102235.
Li J, Zhang C, Lu M, Wang Y, Liu F, Wang W, et al. Infection by a previously uncharacterized Ehrlichia species in rodents from Inner Mongolia, Northern China. Ticks Tick Borne Dis. 2023;14(2):102116. https://doi.org/10.1016/j.ttbdis.2022.102116.
Rungrojn A, Chaisiri K, Paladsing Y, Morand S, Junjhon J, Blacksell SD, et al. Prevalence and molecular characterization of Rickettsia spp. from wild small mammals in public parks and urban areas of Bangkok Metropolitan, Thailand. Trop Med Infect Dis. 2021;6(4):199. https://doi.org/10.3390/tropicalmed6040199.
Berthová L, Slobodník V, Slobodník R, Olekšák M, Sekeyová Z, Svitálková Z, et al. The natural infection of birds and ticks feeding on birds with Rickettsia spp. and Coxiella burnetii in Slovakia. Exp Appl Acarol. 2016;68(3):299–314. https://doi.org/10.1007/s10493-015-9975-3.
Wagner E, Tukhanova N, Shin A, Turebekov N, Shapiyeva Z, Shevtsov A, et al. Incidence of tick-borne spotted fever group Rickettsia species in rodents in two regions in Kazakhstan. Sci Rep. 2022;12(1):14872. https://doi.org/10.1038/s41598-022-19145-0.
Arraga-Alvarado CM, Qurollo BA, Parra OC, Berrueta MA, Hegarty BC, Breitschwerdt EB. Case report: Molecular evidence of Anaplasma platys infection in two women from Venezuela. Am J Trop Med Hyg. 2014;91(6):1161–5. https://doi.org/10.4269/ajtmh.14-0372.
Chochlakis D, Ioannou I, Tselentis Y, Psaroulaki A. Human Anaplasmosis and Anaplasma ovis variant. Emerg Infect Dis. 2010;16(6):1031–2. https://doi.org/10.3201/eid1606.090175.
Lu M, Chen Q, Qin X, Lyu Y, Teng Z, Li K, et al. Anaplasma bovis infection in fever and thrombocytopenia patients - Anhui Province, China, 2021. China CDC Wkly. 2022;4(12):249–53. https://doi.org/10.46234/ccdcw2022.053.
Atif FA. Anaplasma marginale and Anaplasma phagocytophilum: Rickettsiales pathogens of veterinary and public health significance. Parasitol Res. 2015;114(11):3941–57. https://doi.org/10.1007/s00436-015-4698-2.
Mylonakis ME, Harrus S, Breitschwerdt EB. An update on the treatment of canine monocytic ehrlichiosis (Ehrlichia canis). Vet J. 2019;246:45–53. https://doi.org/10.1016/j.tvjl.2019.01.015.
Noaman V, Sazmand A. Anaplasma ovis infection in sheep from Iran: molecular prevalence, associated risk factors, and spatial clustering. Trop Anim Health Prod. 2021;54(1):6. https://doi.org/10.1007/s11250-021-03007-4.
Niaz S, Ur Rahman Z, Ali I, Cossío-Bayúgar R, Amaro-Estrada I, Alanazi AD, et al. Molecular prevalence, characterization and associated risk factors of Anaplasma spp. and Theileria spp. in small ruminants in Northern Pakistan. Parasite. 2021;28:3. https://doi.org/10.1051/parasite/2020075.
Truong AT, Yun BR, Yoo MS, Lim J, Min S, Yoon SS, et al. Utility of ultra-rapid real-time PCR for detection and prevalence of Rickettsia spp. in ticks. BMC Vet Res. 2022;18(1):199. https://doi.org/10.1186/s12917-022-03311-7.
Fournier PE, Dumler JS, Greub G, Zhang J, Wu Y, Raoult D. Gene sequence-based criteria for identification of new rickettsia isolates and description of Rickettsia heilongjiangensis sp. nov. J Clin Microbiol. 2003;41(12):5456–65. https://doi.org/10.1128/JCM.41.12.5456-5465.2003.
Jiang J, An H, Lee JS, O’Guinn ML, Kim HC, Chong ST, et al. Molecular characterization of Haemaphysalis longicornis-borne rickettsiae, Republic of Korea and China. Ticks Tick Borne Dis. 2018;9(6):1606–13. https://doi.org/10.1016/j.ttbdis.2018.07.013.
Liu H, Li Q, Zhang X, Li Z, Wang Z, Song M, et al. Characterization of rickettsiae in ticks in Northeastern China. Parasit Vectors. 2016;9(1):498. https://doi.org/10.1186/s13071-016-1764-2.
Acknowledgements
Not applicable.
Funding
This study was funded the National Natural Science Foundation of China (U2002219) and National Key Research and Development Program (2019YFC1200501 and 2022YFC2305001).
Author information
Authors and Affiliations
Contributions
Conceptualization: JJF, DCH and CWC; Sample collection: DCH, BSS, YMG, SZT, PEN, LYQ, DCB; Sample processing: BSS, HZH, WF, LZ, ZJ, LM, SY; Data analysis: XR, YJH, LCF, ZY; First draft of the manuscript: XR, YX; Manuscript review and editing: JJF, XR, and SY. All authors have reviewed the manuscript and agreed to the submitted version for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The research protocol for trapping wild small animals and collecting samples was approved by the Animal Subjects Research Review Boards at the Yunnan Institute of Endemic Diseases Control and Prevention (2017–001), in accordance with the medical research regulations of China and the Regulation of the People's Republic of China for the Implementation of the Protection of Terrestrial Wildlife.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
40249_2024_1213_MOESM1_ESM.docx
Supplementary Material 1: Supplementary Table 1. Primers used for multigene testing. Supplementary Table 2. Overall prevalence of animals and vectors from different species with emerging Rickettsiales in Yunnan Province. Supplementary Table 3. The diversity of BLAST-based sequence analysis of tick associated Rickettsiales in animals and vectors. Supplementary Fig. 1. Phylogenetic trees constructed by the MEGA v.6.0 software based on the neighbor-joining method of multigene detected in small mammals infected by Candidatus Neoehrlichia mikurensis. A: 16S rRNA gene(1550 bp); B: groEL gene (891 bp). Supplementary Fig. 2. Phylogenetic trees constructed by the MEGA v.6.0 software based on the neighbor-joining method of multigene detected in livestock infected by Anaplasma capra. A: 16S rRNA gene (660 bp); B: groEL gene (1008 bp); C: gltA gene (636 bp); D: msp4 gene (613 bp). Supplementary Fig. 3. Phylogenetic trees constructed by the MEGA v.6.0 software based on the neighbor-joining method of multigene detected in different samples infected Anaplasma ovis. Triangles, circles and rectangles represent livestock, small mammals, and ticks in this study respectively. A: 16S rRNA gene (1850 bp); B: groEL gene (2066 bp); C: gltA gene (792 bp); D: msp4 gene (597 bp). Supplementary Fig. 4. Phylogenetic trees constructed by the MEGA v.6.0 software based on the neighbor-joining method of groEL gene (372 bp) detected in different samples. Triangles, circles and rectangles represent livestock, small mammals, and ticks in this study respectively. Supplementary Fig. 5. Phylogenetic trees constructed by the MEGA v.6.0 software based on the neighbor-joining method of multigene detected in different samples infected Ehrlichia canis. Circles and rectangles represent small mammals and ticks in this study respectively. A: Dsb gene (409 bp); B: gltA gene (125 bp); C: TRP36 gene (800–1000 bp); D: groEL gene (364 bp).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Du, CH., Xiang, R., Bie, SS. et al. Genetic diversity and prevalence of emerging Rickettsiales in Yunnan Province: a large-scale study. Infect Dis Poverty 13, 54 (2024). https://doi.org/10.1186/s40249-024-01213-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40249-024-01213-4