
Abstract
Background
We analyzed the genetic causes of sensorineural hearing loss in racial and ethnic minorities of South Florida by reviewing demographic, phenotypic, and genetic data on 136 patients presenting to the Hereditary Hearing Loss Clinic at the University of Miami. In our retrospective chart review, of these patients, half self-identified as Hispanic, and the self-identified racial distribution was 115 (86%) White, 15 (11%) Black, and 6 (4%) Asian. Our analysis helps to reduce the gap in understanding the prevalence, impact, and genetic factors related to hearing loss among diverse populations.
Results
The causative gene variant or variants were identified in 54 (40%) patients, with no significant difference in the molecular diagnostic rate between Hispanics and Non-Hispanics. However, the total solve rate based on race was 40%, 47%, and 17% in Whites, Blacks, and Asians, respectively. In Non-Hispanic Whites, 16 different variants were identified in 13 genes, with GJB2 (32%), MYO7A (11%), and SLC26A4 (11%) being the most frequently implicated genes. In White Hispanics, 34 variants were identified in 20 genes, with GJB2 (22%), MYO7A (7%), and STRC-CATSPER2 (7%) being the most common. In the Non-Hispanic Black cohort, the gene distribution was evenly dispersed, with 11 variants occurring in 7 genes, and no variant was identified in 3 Hispanic Black probands. For the Asian cohort, only one gene variant was found out of 6 patients.
Conclusion
This study demonstrates that the diagnostic rate of genetic studies in hearing loss varies according to race in South Florida, with more heterogeneity in racial and ethnic minorities. Further studies to delineate deafness gene variants in underrepresented populations, such as African Americans/Blacks from Hispanic groups, are much needed to reduce racial and ethnic disparities in genetic diagnoses.
Similar content being viewed by others

Background
Hearing loss (HL) is a global health concern affecting approximately 1.5 billion people with a projected increase to 2.5 billion by 2050 [1, 2]. It affects ~ 1 newborn in every 500 births in developed countries [3, 4]. In at least 50% of these individuals, it is assessed to be of genetic etiology [5]. Genetic HL can be syndromic (30% of inherited HL) or non-syndromic (NS; 70% of inherited HL). Autosomal recessive (AR) inheritance is the most common form of inheritance, accounting for up to 80% of individuals [4, 6, 7]. Autosomal dominant (AD) inheritance is seen in approximately 20%, with the remaining 5% belonging to X-linked (XL) and mitochondrial inheritance forms [8].
Previous studies have shown that the etiological diagnostic rate after genetic testing of genes and variants varies broadly between ethnic and racial groups [9, 10]. For instance, GJB2 variants have been reported as the most common cause in people of European and Asian ancestry [11, 12]. On the other hand, GJB2 variants are rare in Black populations [13,14,15]. Florida is the third most populous state of the U.S. and two-thirds of the South Floridian population is Hispanic/Latino and half of whom are foreign-born [16]. Despite the 14.9% prevalence of hearing impairment in the state of Florida, detailed studies of deafness genes in a South Florida population have not been reported [17,18,19]. Caribbean Hispanics (Cuban, Puerto Rican, and Dominican) make up the majority of this population and the remaining is largely from Central and South America other than Mexico [20]. The Black population of South Florida is also quite diverse with one-third of Blacks being foreign-born (including Haiti and English-speaking Caribbean countries such as the Bahamas, Jamaica, and Trinidad). Other immigrant groups include people from the Middle East, and Central, South, and East Asia. In this study, we present demographic and phenotypic data in relation to causal gene variants in a diverse population of South Florida.
Results
Demographic data
Among the patients, 68 were female and 68 were male, with both groups ranging in age from 3 to 77 years old at the time of their last visit. The study population was ethnically diverse having an even divide, 68 (50%) Hispanic and 68 (50%) Non-Hispanic. The racial diversity of the population included 115 (86%) White, 15 (11%) Black, and 6 (4%) Asian. The self-reported ancestry of patients was highly diverse (Table 1). The Non-Hispanic Black population had ancestral origins from the U.S. and the Caribbean islands including Jamaica, Haiti, Bahamas, and Dominica. The Hispanic White cohort had many countries of origin ranging from Latin America, Europe, and the Caribbean. The ancestral origins of Non-Hispanic White cohort were from different regions of the U.S. with distant backgrounds from Europe. The Hispanic Black patients self-reported that their origins were from Cuba and Venezuela.
Diagnostic yield and gene distribution by ethnicity and race
There was no significant difference in the solve rate (SR) based on ethnicity, with both Non-Hispanic and Hispanic groups having a SR of 40% (Figs. 1 and 2). Based on race, total SR was 40%, 47%, and 17% in Whites, African American/Blacks, and Asians, respectively (Figs. 1 and 2).
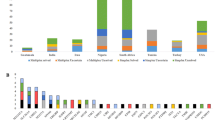
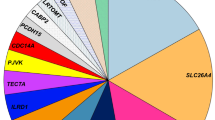
We solved (including those with possibly solved) 54 cases (40%) with 66 variants in 33 genes. Table 1, Additional file 1: Table S1 and Fig. 1 show the distribution of the genes and variants identified in different ethnic and racial groups. The overall gene distribution of the studied population shows that approximately half of the solved cases have variants in the top three genes: GJB2 (22%, n = 12), MYO7A (7%, n = 4), and SLC26A4 (4%, n = 2). A total of 16 gene variants causing sensorineural HL (SNHL) in the White Non-Hispanic cohort were GJB2 (32%), MYO7A (11%), SLC26A4 (11%) and single families (5%) in 10 other genes (Fig. 1A). In comparison, the White Hispanic cohort had a total of 34 gene variants identified as the cause of SNHL. The gene distribution showed that the most common genes are GJB2 (22%), STRC-CATSPER2 (7%), and MYO7A (7%) (Fig. 1B). The remaining 17 genes were detected in single families. The Non-Hispanic Black cohort had a total of 7 genes and 11 variants all identified as the cause of SNHL (Fig. 1C). The gene distribution in the Non-Hispanic Black Cohort was evenly dispersed, all occurring at one gene per family for variants in 7 genes (14% each). For the Asian cohort one gene variant in SIX1 in one SNHL proband out of 6 patients was diagnostic.

Causal Gene Distribution According to Racial and Ethnic Groups. A Exhibits the gene distribution in the solved White Non-Hispanic population. B Exhibits the gene distribution in the solved White Hispanic population. C Exhibits the gene distribution in the solved Non-Hispanic Black/African American population
Diagnostic yield of phenotypic subgroups
The diagnostic rate varied with phenotypic groupings such as the age of onset, severity, laterality, and family history (Fig. 2). The SR differed between the age of onset groups. The late-onset group had an SR of 24% (n = 8) compared to the SR of 45% (n = 46) in those having early onset HL. The severity of HL had a slight difference in SR. Mild to Moderate HL had a higher SR of 43% compared to Severe to Profound HL which had an SR at 36%. SNHL laterality drastically affected the diagnostic yield. Bilateral SNHL had a SR of 47% (n = 54) compared to a 0% (n = 0) SR if unilateral SNHL was present (Fig. 2).

Solve Rate Analysis of Hearing Loss in a Multicultural Population. Solve Rate Analysis of ethnicity, race, phenotype, and family history of the Total, Non-Hispanic and Hispanic cohort. (The percentages are representative of the solve rate. i.e. Asian cohort has a 17% SR, of these if late onset HL the SR increases to 50%, when mild to moderate severity HL also present the SR remains at 50%, when bilateral HL also present the SR also remains at 50%, when the case is also syndromic the SR increases to 100% and remains at a 100% SR if no family history)
Probands who had syndromic SNHL had an SR of 100% (n = 22) compared to the non-syndromic SNHL phenotype which had an SR of 28% (n = 32). Syndromic SNHL included Stickler (18%, n = 4), Usher (18%, n = 4), Alport (9%, n = 2), Baraitser-Winter (4.5%, n = 1), AIFM1-related (4.5%, n = 1), Branchiootorenal (BOR 1) (9%, n = 2), Hypoparathyroidism, Sensorineural deafness, and Renal dysplasia (HDR) (4.5%, n = 1), Perrault (4.5%, n = 1), Donnai Barrow (4.5%, n = 1), Kearns-Sayre (4.5%, n = 1), RTN4IP1-related (4.5%, n = 1), Branchiootic (BOR 3) (4.5%, n = 1), Pendred (4.5%, n = 1), and multiple sulfatase deficiency (4.5%, n = 1) syndromes (Additional file 1: Table S1).
The SNHL simplex and multiplex cases exhibited a moderate difference in SR. Simplex cases had a diagnostic yield of 35% (n = 31) in comparison to 48% (n = 23) in the multiplex cases. In cases with a family history of AR HL the SR was highest at (70%, n = 38) compared to other inheritance patterns, AD (22%, n = 12), mitochondrial (4%, n = 2), and XL (4%, n = 2) (Additional file 1: Table S1).
Discussion
In this report, we present the genetic causes of SNHL in a diverse population from South Florida. Overall, our diagnostic yield is 40%, which is similar to that previously reported in the U.S. with mixed racial and ethnic backgrounds [9]. While there appears to be a difference based on race, with African Americans/Blacks having a slightly higher SR compared to Whites, we do not see a difference in the SR between Hispanic and Non-Hispanic groups.
As with previous studies, we found variants in GJB2 as the most common cause of SNHL, explaining 10% of our probands. While this percentage is smaller compared to that of previous reports from the U.S., it should be noted that our cohort contained patients with syndromic, unilateral, and mild-moderate HL, which are not commonly caused by GJB2 variants [21]. When we included only probands with non-syndromic, early onset, severe to profound, and bilateral SNHL, GJB2 variants are present in 30% (12/40). In accordance with earlier studies, none of the 3 African American/ Black patients in this group had GJB2 variants [13].
In our cohort, GJB2 variants were seen in 6 of 65 Hispanic Whites (9%). Earlier studies reported a range varying from 4 to 22% GJB2 variants in Hispanic Whites. For instance, 22% biallelic GJB2 variants were reported from a U.S. deaf population of 121 Hispanics by Pandya et al. [9, 21], while Sloan et al. reported a prevalence of 14% in their Hispanic cohort. However, Shan et al. [22] reported a much lower prevalence of GJB2 mutations (less than 4%) in their study of Hispanic populations in the Bronx. These discrepancies in the prevalence of GJB2 mutations among different studies could be due to differences in the study populations, sample sizes, and criteria used to define SNHL.
Causal variants in MYO7A were detected in 4 probands followed by SLC26A4 and STRC/CATSPER2 variants in 2 probands each. These three genes have been reported as relatively common causes of SNHL in previous studies from the U.S. and elsewhere [23,24,25]. As we observed in our study, MYO7A and SLC26A4 are typically detected in patients with severe to profound SNHL, while STRC/CATSPER2 variants cause mild to moderate HL. Additionally, as we observed, STRC/CATSPER2 variants are predominantly copy number variants [26]. Interestingly, none of the African American/Black probands was found to have variants in these genes. Each of the other genes identified in this group involves only one proband, confirming the extreme heterogeneity of hereditary deafness. This was more prominent in Non-Hispanic Black/African Americans and Hispanic Whites, where the variants were rare and diverse. The heterogeneity in deafness gene variants were seen highest in the Hispanic Whites, which was also seen in a similar tertiary center [27]. It should be noted that the 3 Hispanic Blacks were negative for a causal gene variant. Some previous studies showed relatively more prevalent genes in certain populations. For instance, MYO15A variants were reported as one of the most frequently reported genetic causes of HL in North and Central Africa [28]. Similarly, MYO15A variants were found to be relatively common in the Middle East, Pakistan, Puerto Rico and a district of Brazil [29,30,31]. The scarcity of research on genetic HL in Africans impedes our comprehension of the distribution of causal HL genes. However, studies have shown that approximately 5% of African Americans carry variants in recurring genes, such as OTOGL, COL11A2, and OTOF [9, 32]. Our African American/Black cohort reported diverse geographical origins, including the Caribbean Islands, which may have contributed to the assortment of gene variants identified.
The diagnostic yield of our small Asian patient cohort was similar to that of a study by Sloan et al. [9] which evaluated 40 self-identified Asians and resulted in a diagnostic yield of 4%. This suggests that the low diagnostic yield may be due to the lack of high-quality studies with large numbers of Asian Americans. It is noteworthy that the only variant we detected in our Asian cohort, SIX1 c.533G > C (p.Arg178Thr), was previously reported to cause HL in another Asian family [33].
Our study found that the phenotype related to the lowest diagnostic yield is unilateral HL. This is consistent with other studies showing that individuals with unilateral HL were less likely to receive a genetic diagnosis [34, 35]. Based on the evidence available, it can be assumed that genetic testing may not be the most appropriate first-line option in the diagnostic workup for individuals with unilateral HL. Instead, it may be more beneficial to focus on assessing potential environmental factors that may have contributed to HL. Therefore, before considering genetic testing, it is essential to evaluate the patient's medical history, environmental exposures, and any underlying medical conditions that may contribute to their HL.
We show that the SR for early-onset HL is higher compared to late-onset HL. This is consistent with previous research, that has shown that underlying genetic cause was more likely to be found in patients with congenital/prelingual HL, compared to those with post-lingual HL [35]. Furthermore, our study found no significant difference in SR between mild to moderate and severe to profound HL. This suggests that the severity of HL does not necessarily affect the likelihood of obtaining a diagnosis. Interestingly, our study found a difference in the diagnostic rate between syndromic and non-syndromic HL. The diagnostic rate for syndromic cases was 100%, while the diagnostic rate for non-syndromic cases was only 30%. This is likely due to the fact that environmental factors, such as CMV, can be more difficult to etiologically diagnose in non-syndromic HL [36]. Our study also found that the SR for a genetic HL diagnosis in cases with a positive family history was higher compared to cases with a negative family history. This is not surprising, as there is a greater chance of a genetic etiology for HL in multiplex families. Our study provides valuable insights into the factors that can affect the SR for HL etiology diagnosis. Figure 2 can be used as a guide to assess the diagnostic possibilities through genetic testing based on race and ethnicity as well as phenotypic variables of HL.
Some limitations of our study is that we obtained genetic test results from different laboratories instead of utilizing the same panel in all patients. This may have introduced a variance in diagnostic rates in different groups (Additional file 1: Table S2). Additionally, while our study provides valuable insights into the genetic causes of SNHL in racial and ethnic minorities of South Florida, it is essential to acknowledge the limitations associated with the small cohort size. The analysis was based on data from 136 patients presenting to the Hereditary Hearing Loss Clinic at the University of Miami, with a distribution of 115 (86%) White, 15 (11%) Black, and 6 (4%) Asian individuals, half of whom self-identified as Hispanic. The relatively small sample size, particularly in the Asian cohort, may not fully represent the diverse genetic landscape of the populations studied. Consequently, caution should be exercised in generalizing our findings to broader demographic groups. Larger and more comprehensive studies involving a more extensive and diverse patient population are warranted to enhance the reliability of our conclusions, particularly in addressing the genetic factors contributing to hearing loss in underrepresented populations.
Conclusion
In conclusion, our study reveals that genetic testing for hearing loss in South Florida's Minority Population uncovers diverse DNA variants. We observe a greater variety of causative variants in racial and ethnic minorities compared to Non-Hispanic Whites. The identified variants in known hearing loss genes are less commonly found in racial/ethnic minorities, highlighting genetic heterogeneity. Our findings also indicate that the diagnostic rate of genetic studies varies by race in South Florida, with a 40% diagnostic rate for the genetic basis of hearing loss in this highly diverse population. Importantly, we note a persistently low diagnostic yield in some racial minorities, emphasizing the need to bridge the discovery gap in these groups. [22, 37].
Methods
Study population
A retrospective chart review was completed for 136 patients presenting for an etiological evaluation to the Hereditary HL clinic with a diagnosis of sensorineural hearing loss (SNHL) at the University of Miami Health System from 2017 to 2022. Patients were seen by an otologist, audiologist, and clinical geneticist during clinic encounters. Clinical evaluations included past medical and family histories and an otology exam as well as a thorough physical exam and eye exams. Investigations included a CT scan or MRI of the temporal bone, an EKG, and a kidney ultrasound. Patients with SNHL with or without additional findings were included. Patients with a clear environmental cause of HL were excluded. Only one affected person (proband) per family was included in the review.
Self-reported data on sex, family history, family origin, and phenotype were obtained from the patients` electronic medical records. Self-reported ethnicity included options of Hispanic and Non-Hispanic groups and race options were Asian, Black, White, American Indian/Alaska Native or Native Hawaiian/Pacific Islander.
Phenotypic data
Clinical data collected from electronic medical records included audiological evaluations, pedigrees, HL age of onset, severity, and laterality. The onset of HL was categorized into two groups as early if the patient presented with HL before the age of 10, and late if the patient presented with HL at the ages above 11 years old. The most recent audiogram was used to group patients for unilateral vs bilateral HL categories. The severity was calculated from the pure tone average (PTA) hearing threshold between 500 and 4000 Hz (PTA500-400) in the most recent audiograms based on the better ear for bilateral HL. HL was defined based on PTA500-400 as follows: mild-moderate (20–70 dB) and severe-profound (> 71 dB) [38].
Genetic data
Genetic testing was performed following pretest counseling by a certified genetic counselor or clinical geneticist. The utilized gene panels were from four different CLIA-certified laboratories and included 264 different genes. The range of genes included in each panel was 92–239 (Additional file 1: Table S2). Sequencing of coding regions and splice junctions and copy-number variant detection were performed. Confirmation of variants identified via next-generation sequencing was performed by the CLIA laboratories following their standard operational procedures and included Sanger sequencing, MLPA, and microarrays. Each variant reported by the laboratory was interpreted again by the study authors according to ACMG 2015 Guidelines and ClinGen HL Expert Panel (HL-EP) Specifications [39, 40]. A definitive genetic diagnosis was made based on the presence of pathogenic (P) or likely pathogenic (LP) variant(s) in a proband in accordance with the expected inheritance pattern. In AD or XL inheritance, if the proband is heterozygous or hemizygous for an LP or P variant, we considered this family solved. In AR inheritance, if the proband is homozygous or confirmed compound heterozygous by testing family members for LP/P variants, we considered this family solved. In mitochondrial inheritance, if the variant was LP/P in the proband, we considered this family solved. In AR inheritance, if the proband is heterozygous for two LP/P variants in the same gene but parental testing is not available, we considered this family possibly solved. If the proband has unique findings for a rare syndrome consistent with the gene-related phenotype, even if the variant is a variant of uncertain significance (VUS), we considered this family possibly solved.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
GBD 2019 Hearing Loss Collaborators. Hearing loss prevalence and years lived with disability, 1990–2019: findings from the Global Burden of Disease Study 2019. Lancet. 2021;397(10278):996–1009. https://doi.org/10.1016/S0140-6736(21)00516-X.
World Health Organization. World report on hearing. 2021; Accessed from [https://apps.who.int/iris/handle/10665/339913. License: CC BY-NC-SA 3.0 IGO]
Fortnum HM, Summerfield AQ, Marshall DH, Davis AC, Bamford JM. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ. 2001;323(7312):536–40. https://doi.org/10.1136/bmj.323.7312.536.
Morton CC, Nance WE. Newborn hearing screening–a silent revolution. N Engl J Med. 2006;354(20):2151–64. https://doi.org/10.1056/NEJMra050700.
Thorpe RK, Smith RJH. Future directions for screening and treatment in congenital hearing loss. Precis Clin Med. 2020;3(3):175–86. https://doi.org/10.1093/pcmedi/pbaa025.
Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Human Mol Genet. 1997;6(12):2173–7. https://doi.org/10.1093/hmg/6.12.2173.
Shearer AE, Smith RJ. Genetics: advances in genetic testing for deafness. Curr Opin Pediatr. 2012;24(6):679–86. https://doi.org/10.1097/MOP.0b013e3283588f5e.
Young A, Ng M. Genetic hearing loss. Treasure Island: StatPearls Publishing; 2023.
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, Ranum PT, Weaver AE, Black-Ziegelbein EA, Wang D, Azaiez H, Smith RJH. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135(4):441–50. https://doi.org/10.1007/s00439-016-1648-8.
Yan D, Tekin D, Bademci G, Foster J 2nd, Cengiz FB, Kannan-Sundhari A, Guo S, Mittal R, Zou B, Grati M, Kabahuma RI, Kameswaran M, Lasisi TJ, Adedeji WA, Lasisi AO, Menendez I, Herrera M, Carranza C, Maroofian R, Crosby AH, et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Human Genet. 2016;135(8):953–61. https://doi.org/10.1007/s00439-016-1697-z.
Chan DK, Chang KW. GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope. 2014;124(2):E34–53. https://doi.org/10.1002/lary.24332.
Zheng J, Ying Z, Cai Z, Sun D, He Z, Gao Y, Zhang T, Zhu Y, Chen Y, Guan MX. GJB2 mutation spectrum and genotype-phenotype correlation in 1067 Han Chinese subjects with non-syndromic hearing loss. PLoS ONE. 2015;10(6):e0128691. https://doi.org/10.1371/journal.pone.0128691.
Lasisi AO, Bademci G, Foster J 2nd, Blanton S, Tekin M. Common genes for non-syndromic deafness are uncommon in sub-Saharan Africa: a report from Nigeria. Int J Pediatr Otorhinolaryngol. 2014;78(11):1870–3. https://doi.org/10.1016/j.ijporl.2014.08.014.
Lebeko K, Bosch J, Noubiap JJ, Dandara C, Wonkam A. Genetics of hearing loss in Africans: use of next generation sequencing is the best way forward. Pan Afr Med J. 2015;20:383. https://doi.org/10.11604/pamj.2015.20.383.5230.
Rudman JR, Kabahuma RI, Bressler SE, Feng Y, Blanton SH, Yan D, Liu XZ. The genetic basis of deafness in populations of African descent. J Genet Genom. 2017;44(6):285–94. https://doi.org/10.1016/j.jgg.2017.03.008.
U.S. Census Bureau. QuickFacts Miami-Dade County, Florida; Broward County, Florida; Palm Beach County, Florida; Florida. 2021; Accessed from https://www.census.gov/quickfacts/fact/table/miamidadecountyflorida,browardcountyflorida,palmbeachcountyflorida,FL/RHI725221#RHI725221
Florida Health. Population with hearing difficulty (Aged 65 Years and Older). 2021; Accessed from https://www.flhealthcharts.gov/ChartsDashboards/rdPage.aspx?rdReport=NonVitalIndNoGrp.Dataviewer&cid=8686
Florida Health. Population with hearing difficulty (Aged 18–64 Years) (Census). 2021; Accessed from https://www.flhealthcharts.gov/ChartsDashboards/rdPage.aspx?rdReport=NonVitalIndNoGrp.Dataviewer&cid=8685
Florida Health. Population with hearing difficulty (Aged 0–17 Years) (Census). 2021; Accessed from https://www.flhealthcharts.gov/ChartsDashboards/rdPage.aspx?rdReport=NonVitalIndNoGrp.Dataviewer&cid=8684
U.S. Census Bureau. Census 2000 brief: the Hispanic population. 2000; Accessed from https://www2.census.gov/library/publications/decennial/2000/briefs/c2kbr01-03.pdf
Pandya A, Arnos KS, Xia XJ, Welch KO, Blanton SH, Friedman TB, Garcia Sanchez G, Liu MD, et al. Frequency and distribution of GJB2 (connexin 26) and GJB6 (connexin 30) mutations in a large North American repository of deaf probands. Genet Med Off J Am Coll Med Genet. 2003;5(4):295–303. https://doi.org/10.1097/01.GIM.0000078026.01140.68.
Shan J, Chobot-Rodd J, Castellanos R, Babcock M, Shanske A, Parikh SR, Morrow BE, Samanich J. GJB2 mutation spectrum in 209 hearing impaired individuals of predominantly Caribbean Hispanic and African descent. Int J Pediatr Otorhinolaryngol. 2010;74(6):611–8. https://doi.org/10.1016/j.ijporl.2010.03.004.
Del Castillo I, Morín M, Domínguez-Ruiz M, Moreno-Pelayo MA. Genetic etiology of non-syndromic hearing loss in Europe. Hum Genet. 2022;141(3–4):683–96. https://doi.org/10.1007/s00439-021-02425-6.
Kabahuma RI, Schubert WD, Labuschagne C, Yan D, Blanton SH, Pepper MS, Liu XZ. Spectrum of MYO7A mutations in an indigenous South African population further elucidates the nonsyndromic autosomal recessive phenotype of DFNB2 to include both homozygous and compound heterozygous mutations. Genes. 2021;12(2):274. https://doi.org/10.3390/genes12020274.
Sommen M, Schrauwen I, Vandeweyer G, Boeckx N, Corneveaux JJ, van den Ende J, Boudewyns A, De Leenheer E, Janssens S, Claes K, Verstreken M, Strenzke N, Predöhl F, Wuyts W, Mortier G, Bitner-Glindzicz M, Moser T, Coucke P, Huentelman MJ, Van Camp G. DNA diagnostics of hereditary hearing loss: a targeted resequencing approach combined with a mutation classification system. Hum Mutat. 2016;37(8):812–9. https://doi.org/10.1002/humu.22999.
Nishio SY, Usami SI. Frequency of the STRC-CATSPER2 deletion in STRC-associated hearing loss patients. Sci Rep. 2022;12(1):634. https://doi.org/10.1038/s41598-021-04688-5.
Angeli SI. Phenotype/genotype correlations in a DFNB1 cohort with ethnical diversity. Laryngoscope. 2008;118(11):2014–23. https://doi.org/10.1097/MLG.0b013e31817fb7ad.
Adadey SM, Wonkam-Tingang E, Aboagye ET, Quaye O, Awandare GA, Wonkam A. Hearing loss in Africa: current genetic profile. Hum Genet. 2022;141(3–4):505–17. https://doi.org/10.1007/s00439-021-02376-y.
Bademci G, Foster J 2nd, Mahdieh N, Bonyadi M, Duman D, Cengiz FB, Menendez I, Diaz-Horta O, Shirkavand A, Zeinali S, Subasioglu A, Tokgoz-Yilmaz S, Huesca-Hernandez F, de la Luz Arenas-Sordo M, Dominguez-Aburto J, Hernandez-Zamora E, Montenegro P, Paredes R, Moreta G, Vinueza R, et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet Med Off J Am Coll Med Genet. 2016;18(4):364–71. https://doi.org/10.1038/gim.2015.89.
Manzoli GN, Bademci G, Acosta AX, Félix TM, Cengiz FB, Foster J 2nd, Da Silva DS, Menendez I, Sanchez-Pena I, Tekin D, Blanton SH, Abe-Sandes K, Liu XZ, Tekin M. Targeted resequencing of deafness genes reveals a founder MYO15A variant in Northeastern Brazil. Ann Hum Genet. 2016;80(6):327–31. https://doi.org/10.1111/ahg.12177.
Rehman AU, Bird JE, Faridi R, Shahzad M, Shah S, Lee K, Khan SN, Imtiaz A, Ahmed ZM, Riazuddin S, Santos-Cortez RL, Ahmad W, Leal SM, Riazuddin S, Friedman TB. Mutational spectrum of MYO15A and the molecular mechanisms of DFNB3 human deafness. Hum Mutat. 2016;37(10):991–1003. https://doi.org/10.1002/humu.23042.
Florentine MM, Rouse SL, Stephans J, Conrad D, Czechowicz J, Matthews IR, Meyer AK, Nadaraja GS, Parikh R, Virbalas J, Weinstein JE, Chan DK. Racial and ethnic disparities in diagnostic efficacy of comprehensive genetic testing for sensorineural hearing loss. Hum Genet. 2022;141(3–4):495–504. https://doi.org/10.1007/s00439-021-02338-4.
Li A, Liu S, Zhang P, Hu X, Li G, Gu W, Jiang Y. A novel heterozygous SIX1 missense mutation resulted in non-syndromic unilateral hearing loss. Front Genet. 2022;13:1047230. https://doi.org/10.3389/fgene.2022.1047230.
Johansson M, Karltorp E, Asp F, Berninger E. A prospective study of genetic variants in infants with congenital unilateral sensorineural hearing loss. J Clin Med. 2023;12(2):495. https://doi.org/10.3390/jcm12020495.
Tropitzsch A, Schade-Mann T, Gamerdinger P, Dofek S, Schulte B, Schulze M, Battke F, Fehr S, Biskup S, Heyd A, Müller M, Löwenheim H, Vona B, Holderried M. Diagnostic yield of targeted hearing loss gene panel sequencing in a large german cohort with a balanced age distribution from a single diagnostic center: an eight-year study. Ear Hear. 2022;43(3):1049–66. https://doi.org/10.1097/AUD.0000000000001159.
Al-Kowari M, Espino-Guarch M. Genetics and acquired hearing loss. Geriat Med Geront. 2019. https://doi.org/10.5772/intechopen.86664.
Gasmelseed NMA, Schmidt M, Magzoub MMA, Macharia M, Elmustafa OM, Ototo B, Winkler E, Ruge G, Horstmann RD, Meyer CG. Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum Mutat. 2004;23(2):206–7. https://doi.org/10.1002/humu.9216.
Mazzoli M, Van Camp G, Newton V, Giarbini N, Declau F, Parving A. Recommendations for the description of genetic and audiological data for families with nonsyndromic hereditary hearing impairment. Audiol Med. 2003;1(2):148–50. https://doi.org/10.1080/16513860301713.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Tavtigian SV, Harrison SM, Boucher KM, Biesecker LG. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat. 2020;41(10):1734–7. https://doi.org/10.1002/humu.24088.
Acknowledgements
We are grateful to the participating families included in this study. We thank all the participating physicians, genetic counselors, and audiologists at the University of Miami Miller School of Medicine for providing all the relevant information.
Funding
This work was supported by the National Institutes of Health, Grant number [R01DC009645] Author M.T. has received research support from the National Institute on Deafness and other Communication Disorders. Dr. Liu was supported by NIH Grants R01DC005575, R01DC012115, and NIDCD R01DC019404.
Author information
Authors and Affiliations
Contributions
MT, XZL, AE, MH, SA and FT were involved in patient referral. LP wrote the original draft and MT oversaw the reviews and editing. LP, DMS, JG, DD, TS, GB, and MT were involved in data analysis and interpretation. All authors read, commented, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board at the University of Miami.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
List of variants identified in families considered to be solved. Table S2. Hearing Loss Panels.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Peart, L., Gonzalez, J., Morel Swols, D. et al. Dispersed DNA variants underlie hearing loss in South Florida’s minority population. Hum Genomics 17, 103 (2023). https://doi.org/10.1186/s40246-023-00556-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-023-00556-7