
Abstract
Background
Knowledge of the frequency of rare SERPINA1 mutations could help in the management of alpha1 antitrypsin deficiency (AATD). The present study aims to assess the frequencies of rare and null alleles and their respiratory and hepatic pathogenicity.
Methods
This is a secondary analysis of a study that evaluated the viability of the Progenika diagnostic genotyping system in six different countries by analyzing 30,827 samples from cases of suspected AATD. Allele-specific genotyping was carried out with the Progenika A1AT Genotyping Test which analyses 14 mutations in buccal swabs or dried blood spots samples. SERPINA1 gene sequencing was performed for serum AAT-genotype discrepancies or by request of the clinician. Only cases with rare mutations were included in this analysis.
Results
There were 818 cases (2.6%) carrying a rare allele, excluding newly identified mutations. All were heterozygous except for 20 that were homozygous. The most frequent alleles were the M-like alleles, PI*Mmalton and PI*Mheerlen. Of the 14 mutations included in the Progenika panel, there were no cases detected of PI*Siiyama, PI*Q0granite falls and PI*Q0west. Other alleles not included in the 14-mutation panel and identified by gene sequencing included PI*Mwürzburg, PI*Zbristol, and PI*Zwrexham, and the null alleles PI*Q0porto, PI*Q0madrid, PI*Q0brescia, and PI*Q0kayseri.
Conclusions
The Progenika diagnostic network has allowed the identification of several rare alleles, some unexpected and not included in the initial diagnostic panel. This establishes a new perspective on the distribution of these alleles in different countries. These findings may help prioritize allele selection for routine testing and highlights the need for further research into their pathogenetic role.
Similar content being viewed by others

Introduction
In recent decades, the number of described mutations of the SERPINA1 gene associated with alpha1 antitrypsin deficiency (AATD) has increased considerably. Beyond the two most frequent mutations, the S mutation (c.863A > T) and the Z mutation (c.1096G > A) [1], the number of described variants has risen to more than 500 [2]. Usually, the identification of these rare mutations is initiated by a discrepancy between the serum AAT level and the mutation found by direct genotyping [3]. For this reason, rare mutations are usually considered to be pathogenetic. However, most have been described in individual cases without thorough examination of their pathogenetic capacity [4]. Moreover, the frequency of these rare mutations in a large population of AATD patients has not been consistently described. It is clear that the mutation should be identified in AATD cases with significant pulmonary or hepatic involvement [5]. Knowing the frequency of these rare mutations could help in the management of the disease and in prioritizing allele identification in routine practice. This could also highlight the gaps in our understanding of the pathophysiologic behavior of these mutations.
Recently, our group published the results of a new system for AATD diagnosis based on buccal swabs and dried blood spots samples. After analyzing more than 30,000 samples from six countries, the study showed this diagnostic procedure was feasible and suitable for the genetic diagnosis of AATD [1, 3]. The implementation of this AATD diagnostic network has revealed that there are 14 mutations that can explain the majority of the pathological cases of this disease. Using the data from this study, the present analysis describes the frequencies of rare alleles and relate them to the available data on their respiratory and hepatic pathogenicity. These results will help understand the epidemiological importance of the mutations in each geographic area and will highlight the research needed for a more complete understanding of the pathogenetic potential of these mutations.
Methods
This is a secondary analysis of the data from a study evaluating the Progenika diagnostic system (Progenika Biopharma, Derio, Vizcaya, Spain) in 30,827 samples from patients with suspected AATD from six different countries. This diagnostic network found mutations in 9,528 (30.9%) of the samples. The methodology has been previously described [1]. Briefly, this was an observational, cross-sectional analysis analyzing the anonymized data included on the Progenika web platform (https://grifolsalpha1test.com/) from March 12, 2018, to January 10, 2022. The collection kits for sampling with the dried blood spots or buccal swabs were provided to participating centers free of charge by Grifols (Barcelona, Spain) upon request from the treating physicians. For the current analysis, samples from Argentina, Brazil, Chile, Colombia, Spain, and Turkey were analyzed. The samples were registered on the web platform through a unique code associated with each sample collection kit and sent by post to the reference laboratory at the Progenika headquarters.
When registering the sample on the website, clinicians were asked to include some clinical data about the patient including age, smoking status (smoker, former smoker or never smoker), serum AAT level, and forced expiratory volume in one second (FEV1, expressed as a percentage of its predicted value), and the reasons for requesting the test. Although inclusion of these data was not mandatory, the AAT level was considered for concordance with the genotype and, if not concordant, the SERPINA1 gene was sequenced. Per the Spanish guidelines [6], AAT levels ≤ 50 mg/dl were considered a severe deficiency.
Allele-specific genotyping was carried out with the Progenika A1AT Genotyping Test. The test uses polymerase chain reaction amplification to obtain large amounts of target sequences from the SERPINA1 gene. The Luminex® 200 system to detect previously labeled amplified fragments, as previously described [3]. The test and OCR100 buccal swabs used to collect the samples are CE marked (European Conformity) and United States Food and Drug Administration approved. The test is intended for use with genomic DNA extracted from human whole blood samples collected as dried blood spots or from human buccal swab samples using ORAcollect Dx OCD-100.
The test can identify the 14 most frequent deficiency variants of the SERPINA1 gene, namely PI*S, PI*Z, PI*I, PI*Mprocida, PI*Mmalton, PI*Siiyama, PI*Q0granite falls, PI*Q0west, PI*Q0bellingham, PI*F, PI*Plowell, PI*Q0mattawa, PI*Q0clayton, and PI*Mheerlen. When none of the 14 alleles was found, the result was noted as negative and interpreted as an M allele. The absence of any of these 14 alleles suggests with over 99% probability that the genotype corresponds to PI*M, unless there was a discrepancy with AAT levels. In those cases, gene sequencing was conducted.
For the current analysis, only cases with rare mutations identified by the Progenika diagnostic system were included. Accordingly, those cases with genotypes exclusively resulting from a combination of S or Z alleles (MS, MZ, SS, SZ and ZZ) were excluded from this analysis. Newly identified mutations not previously described were also excluded. After the identification of all rare alleles, we performed a non-systematic review of the literature looking for information on these rare alleles by searching for the name of the allele in PubMed.
Results
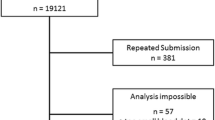
The number of patients with rare variants was 818 (2.7% out of 30,827 samples; 8.6% out of 9,528 carrying any mutation). The flowchart of the distribution of the samples is available from a previous analysis [1]. The number of patients carrying rare alleles is listed by country in Table 1. Severe AAT deficiency was seen in 572 patients (9.8% of those with serum AAT values). All cases were heterozygous except for the following (n = 20): 1 homozygous PI*Mprocida (n = 1), homozygous PI*Mmalton (n = 13), homozygous PI*Mheerlen (n = 1), homozygous PI*Plowell (n = 4), and homozygous PI*Q0mattawa (n = 1). Of the 14 mutations included in the Progenika panel, no cases of PI*Siiyama, PI*Q0granite falls and PI*Q0west were found. Other alleles not included in the initial 14-mutation panel were identified by gene sequencing. They included PI*Mwürzburg, PI*Zbristol, and PI*Zwrexham, and the null alleles PI*Q0porto, PI*Q0madrid, PI*Q0brescia, and PI*Q0kayseri.The frequency of rare and null alleles in the different countries are summarized in the Table 2.
The frequency of the different M-like rare alleles is shown in Fig. 1. The most frequent M-like allele was PI*Mmalton followed by PI*Mheerlen. Although these alleles were identified predominantly in the samples from Spain, some combinations (PI*Mmalton, PI*Mheerlen or PI*Mprocida) were found in samples from other countries. After Spain, Brazil had the most of these rare mutations.

M-like alleles distribution
The frequency of other rare alleles is shown in Fig. 2. PI*I was the most common and was predominantly found in samples from Spain. The allele PI*F was also frequently identified. Other alleles were less frequent, but some were identified in Turkey, e.g., combinations with Plowell.

Other rare alleles distribution
The null alleles are summarized in Fig. 3. The most frequent null allele was Q0mattawa. These alleles were less frequent, and homozygous combinations were extremely rare. The Q0kayseri mutation is native to Turkey, but the only homozygous case for Q0brescia was also found in a sample from that country. Information on these mutations from a non-systematic literature review is summarized in Table 3.

Null alleles distribution
Discussion
The present study assessed the frequency of rare mutations in a large sample of cases with suspected AATD in six countries. Our results show the low frequency of these alleles and their distribution in different countries and help identify which variants are more frequent in different geographical areas. Our data indicate that these so-called rare variants may not be as rare when a thorough diagnostic system is used.
AATD is an inherited disorder that increases the risk of lung and liver disease. Numerous point mutations of the SERPINA1 gene have been identified so far, although many of them are not associated with an increased risk for developing respiratory or liver disorders [2]. Consequently, the identification of less frequent, but consequential mutations and their characterization are relevant objectives for the management of AATD. Greater understanding of the underlying biologic pathways leading to cell damage in AATD will also be of benefit for the treatment of AATD [7]. This is of special importance in the current pandemic situation with potential associations between AATD and COVID19 [8, 9]. The Progenika diagnostic network is formed by those countries using the Progenika system as the diagnostic standard for AATD. Other countries have started to use a similar system including Italy [10] and Germany [11].
The main strengths of our study are the large number of samples analyzed, the simultaneous determination of several genotypes and the sequencing of samples from different countries, allowing the assessment of the geographic distribution of these mutations. However, there are some limitations that must be taken into account when interpreting our results. This is not a population-based study, but a highly selected population of patients with suspected AATD. Accordingly, the prevalence figures may overestimate the prevalence of AATD in the general population. Another note of caution should be considered in the cases with hepatopathy of unknown cause. The clinicians participating in this circuit were mostly pulmonologists or general practitioners. Therefore, cases with hepatopathy of unknown cause may be under-represented. The addition of liver disease specialists to the evaluation of these patients might contribute to the detection of cases of AATD in this clinical context. Additionally, not all samples were sequenced, only those with a discrepancy between the serum level of AAT and the mutation found. There was a considerable number of cases with no AAT level available. Therefore, there may be an underestimation of some alleles. Finally, serum AAT and FEV1 reported in Table 1 are influenced by the other accompanying allele in heterozygosis. Consequently, these data may lead to a false picture of the impact of these alleles on AAT levels or the resulting functional impairment. Interestingly, the majority of cases with AAT values available presented as non-severe AATD, suggesting that these alleles cannot be ruled out by the level of serum AAT alone.
Despite these limitations, this is the largest study to date that includes analysis of the frequency of rare variants in a sample of patients with suspected AATD. The frequency of these rare alleles has been previously reported in several individual countries including Germany [12], Italy [13, 14], Tunisia [15], Switzerland [16], Spain [17], Poland [18], Turkey [19] and the USA [20]. In these studies, the frequency of rare alleles ranged from 0.5% of all screened patients in Germany [12] to 4.1% in Tunisia [15] corresponding to 1.7% of cases with any mutation in Germany [12] and 20% in Tunisia [15]. In Turkey, our data showed a higher frequency of rare alleles, in line with recently published data from this country [19] within the Progenika network.
The information obtained from our literature review should be interpreted with caution since some mutations have low case numbers, and their effects may be influenced by an accompanying mutation. Additionally, some mutations have been assigned more than one name. There were two major allele complexes that are worth noting. The Mmalton complex includes the Mmalton (c.227_229delTCT on M2 variant), Mpalermo (same mutation on M1V variant) and Mnichinan (same mutation with an additional mutation c.514G > A that does not seem to have a deleterious effect on its own). The Plowell complex includes Plowell (c.839A > T on M3 variant) and Pduarte, (same mutation on M1 variant; also known as Q0cardiff). The Plowell mutation is also seen in Ybarcelona which results from the combination of Plowell and Yorzinuovi in the same gene [21].
PI*I and PI*F were first alleles described in 1967 [22]. PI*I allele has been associated with moderate AATD with serum concentrations similar to those observed with the S allele [23]. PI*II homozygotes usually have AAT levels around 50 mgr/dL [24, 25]. Liver involvement is not usually seen with PI*I unless it is accompanied by an allele associated with liver involvement [26]. The serum concentration and function associated with the PI*F allele are at least 80% of that of the M allele [27, 28]. However, the PI*F allele shows a decreased ability to bind and less time-dependent inhibition of human neutrophil elastase compared to the M phenotype and similar inhibition to that of the Z phenotype [29]. The PI*F allele has a reduced functional ability to inhibit neutrophil elastase but not proteinase 3 [30], suggesting that inheritance of the F variant may increase a person's susceptibility to elastase-induced lung damage, but not necessarily to emphysema. Due to normal hepatic secretion, it does not produce intrahepatic accumulation and therefore, does not increase the risk of liver injury.
According to our results, M-like alleles are the most frequent in patients with suspected AATD. PI*Mmalton complex (PI*Mmalton, PI*Mpalermo and PI*Mnichinan) have a similar behavior. PI*Mmalton was first described in 1975 in a 2-year child with a minor infection [31]. PI*Mnichinan was first described in 1990 in a Japanese individual with severe AATD (18 mg/dl), associated with aggregated AAT molecules in the hepatocytes [32]. Finally, PI*Mpalermo was first described in 1994 [33]. Their presence is associated with serum AAT levels below 15%. These mutations are characterized by conformational abnormalities that result in polymerized/aggregated insoluble forms of AAT that accumulate in the endoplasmic reticulum of hepatocytes. Therefore, all three variants meet the requirements for endoplasmic reticulum storage diseases and conformational diseases [34, 35]. Interestingly, the c.514G > A additional mutation of the PI*Mnichinan does not contribute to AATD [32].
PI*Mwürzburg was first described in 1999 on a M1Val basis [36], and the same mutation was identified one year after as PI*Mvall d’hebron but on a M1Ala basis [37]. These defective alleles produce a change in the amino acid sequence at position 369 which is associated with a complete intracellular transport block in cell. Interestingly, the allele PI*Mheerlen has a different amino acid substitution in the same position which is also shown to cause complete retention of the mutant protein in the hepatocytes.
PI*Mheerlen was first described in 1981 [38]. Homozygous cases have serum AAT levels 2% of normal and very low antitrypsin activity. The tertiary structure of the Mheerlen protein is significantly altered resulting in intracellular proteolysis. Therefore, there is no accumulation of Mheerlen protein in hepatocytes [39].
PI*Mprocida was first described in 1988 [40]. This rare allele encoding AAT synthesis is associated with reduced serum AAT levels (below 10 mg/dl). The Mprocida molecule behaves normally in vivo with a half-life similar to normal M1 AAT. Neutrophil elastase inhibitory activity of Mprocida protein is slightly reduced. Evaluation of the crystallographic structure suggests that the mutation may alter alpha-helix A, suggesting that the molecule is unstable and degrades intracellularly prior to secretion. The tertiary structure of the protein is significantly altered resulting in intracellular proteolysis and, therefore, not associated with risk of liver injury. The risk of lung disease is high, but the risk of liver disease is low [40].
Although P-type mutations have been known since 1968 [41], it was not until 1990 that the PI*Plowell genotype began to be characterized [42]. In 1993, a new P-allele was identified as Pduarte which carried the same mutation as Plowell but on a M4 basis [43]. These alleles have similar behavior. Homozygous Plowell exhibits decreased AAT serum concentration—around 40% of normality [44]. However, Plowell has near normal function as an inhibitor of human neutrophil elastase [45]. Therefore, increased risk for lung involvement depends on the accompanying alleles [41]. The Plowell substitution has a profound effect on intracellular processing of the AAT molecule resulting in deficiency. This variant has been associated with increased intracellular degradation of newly synthetized protein and to serum levels 24% of normal [42]. Therefore, the risk for liver disease is low. PI*Pduarte is similar to Plowell but on M4. AAT levels in Pduarte are 41% of normal, similar to Plowell [43]. Thus, the Pduarte allele differs from the Plowell allele only by the normal allelic background in which the mutation occurs.
Ybarcelona was first described in 1998 as the combination of PI*Plowell + another mutation (c.1244C > A) [21]. In 2012, the mutation c.1244C > A was reported to have a pathogenetic effect by itself, i.e., a case with mild hyper-transaminasemia reported in Orzinuovi (Brescia, Italy). The allele was named as PI*Yorzinuovi [46]. Consequently, Ybarcelona results from a combination of PI*Plowell plus PI*Yorzinuovi. In heterozygous cases, the risk of lung disease is likely to be similar to that of MZ heterozygotes [47].
Zbristol was first reported in 1997 in a woman with an obstetric history of three perinatal deaths from fulminant liver disease and no living offspring [48]. Only a few cases have been reported in children with low levels of AAT if accompanied by a Z allele and near to normal if accompanied by an M allele, with frequent liver involvement in children [49, 50]. The Zwrexhan allele has only been described in a family with severe AATD which also carried the common mutation causing Z deficiency [51]. Individuals with such a deficiency are, therefore, compound heterozygotes. The behavior of these particular mutations in the absence of the Z mutation is not known.
Null (Q0) alleles encode a truncated protein with large conformational changes that is degraded intracellularly without having the opportunity to aggregate. These patients have undetectable serum concentrations of AAT. The protein is retained in the rough endoplasmic reticulum or pre-Golgi compartment and is degraded. This means that homozygotes are at very high risk for emphysema, but not liver disease.
In conclusion, the present report informs on the frequency of rare and null alleles updating their distribution in a large sample population from six countries. The Progenika diagnostic network has allowed the identification of several rare alleles providing a new view of the distribution of these alleles in different countries. Due to the efficacy in both the detection of AATD cases and the identification of new variants, in the future we believe that Progenika's system could continue to expand to other countries. Consequently, future studies should focus on the characterization of these and other new mutations as they emerge in the context of patients with suspected AATD. These findings may help prioritize allele selection for routine testing and highlights the need for continuing research into their pathogenetic roles.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- AATD:
-
Alpha1 antitrypsin deficiency
- FEV1 :
-
Forced expiratory volume in one second
References
Lopez-Campos JL, Osaba L, Czischke K, Jardim JR, Fernandez Acquier M, Ali A, Günen H, Rapun N, Drobnic E, Miravitlles M. Feasibility of a genotyping system for the diagnosis of alpha1 antitrypsin deficiency: a multinational cross-sectional analysis. Respir Res. 2022;23(1):152.
Seixas S, Marques PI. Known mutations at the cause of alpha-1 antitrypsin deficiency an updated overview of SERPINA1 variation spectrum. Appl Clin Genet. 2021;14:173–94.
Lopez-Campos JL, Casas-Maldonado F, Torres-Duran M, Medina-Gonzalvez A, Rodriguez-Fidalgo ML, Carrascosa I, Calle M, Osaba L, Rapun N, Drobnic E, et al. Results of a diagnostic procedure based on multiplex technology on dried blood spots and buccal swabs for subjects with suspected alpha1 antitrypsin deficiency. Arch Bronconeumol (Engl Ed). 2021;57(1):42–50.
Matamala N, Lara B, Gomez-Mariano G, Martinez S, Retana D, Fernandez T, Silvestre RA, Belmonte I, Rodriguez-Frias F, Vilar M, et al. Characterization of novel missense variants of SERPINA1 gene causing alpha-1 antitrypsin deficiency. Am J Respir Cell Mol Biol. 2018;58(6):706–16.
Hernández PJM, López CCV. Alpha-1 antitrypsin deficiency severe and no severe. Is it benefit or risk? Archivos de bronconeumologia. 2022;58(10):731–2.
Casas F, Blanco I, Martinez MT, Bustamante A, Miravitlles M, Cadenas S, Hernandez JM, Lazaro L, Rodriguez E, Rodriguez-Frias F, et al. Indications for active case searches and intravenous alpha-1 antitrypsin treatment for patients with alpha-1 antitrypsin deficiency chronic pulmonary obstructive disease: an update. Arch Bronconeumol. 2015;51(4):185–92.
Matamala N, Lara B, Gómez-Mariano G, Martínez S, Vázquez-Domínguez I, Otero-Sobrino Á, Muñoz-Callejas A, Sánchez E, Esquinas C, Bustamante A, et al. miR-320c regulates SERPINA1 expression and is induced in patients with pulmonary disease. Arch Bronconeumol. 2021;57(7):457–63.
Parr DG, Chorostowska-Wynimko J, Corsico A, Esquinas C, McElvaney GN, Sark AD, Sucena M, Tanash H, Turner AM, Miravitlles M. Impact of COVID-19 in patients with severe alpha-1 antitrypsin deficiency: the IMCA1 study of the EARCO clinical research collaboration. Arch Bronconeumol. 2022;58(12):840–2.
Strassmair M, Stangl M. Alpha-1 antitrypsin deficiency and COVID-19 infection. Arch Bronconeumol. 2021;57:97.
Ottaviani S, Barzon V, Buxens A, Gorrini M, Larruskain A, El Hamss R, Balderacchi AM, Corsico AG, Ferrarotti I. Molecular diagnosis of alpha1-antitrypsin deficiency: a new method based on Luminex technology. J Clin Lab Anal. 2020;34(7):e23279.
Veith M, Klemmer A, Anton I, El Hamss R, Rapun N, Janciauskiene S, Kotke V, Herr C, Bals R, Vogelmeier CF, et al. Diagnosing alpha-1-antitrypsin deficiency using a PCR/Luminescence-based technology. Int J Chron Obstruct Pulmon Dis. 2019;14:2535–42.
Bals R, Koczulla R, Kotke V, Andress J, Blackert K, Vogelmeier C. Identification of individuals with alpha-1-antitrypsin deficiency by a targeted screening program. Respir Med. 2007;101(8):1708–14.
Ferrarotti I, Baccheschi J, Zorzetto M, Tinelli C, Corda L, Balbi B, Campo I, Pozzi E, Faa G, Coni P, et al. Prevalence and phenotype of subjects carrying rare variants in the Italian registry for alpha1-antitrypsin deficiency. J Med Genet. 2005;42(3):282–7.
Ferrarotti I, Gorrini M, Scabini R, Ottaviani S, Mazzola P, Campo I, Zorzetto M, Luisetti M. Secondary outputs of alpha1-antitrypsin deficiency targeted detection programme. Respir Med. 2008;102(3):354–8.
Denden S, Zorzetto M, Amri F, Knani J, Ottaviani S, Scabini R, Gorrini M, Ferrarotti I, Campo I, Chibani JB, et al. Screening for alpha 1 antitrypsin deficiency in Tunisian subjects with obstructive lung disease: a feasibility report. Orphanet J Rare Dis. 2009;4:12.
Zorzetto M, Russi E, Senn O, Imboden M, Ferrarotti I, Tinelli C, Campo I, Ottaviani S, Scabini R, von Eckardstein A, et al. SERPINA1 gene variants in individuals from the general population with reduced alpha1-antitrypsin concentrations. Clin Chem. 2008;54(8):1331–8.
Rodriguez-Frias F, Miravitlles M, Vidal R, Camos S, Jardi R. Rare alpha-1-antitrypsin variants: are they really so rare? Ther Adv Respir Dis. 2012;6(2):79–85.
Duk K, Zdral A, Szumna B, Roży A, Chorostowska-Wynimko J. Frequency of rare alpha-1 antitrypsin variants in polish patients with chronic respiratory disorders. Adv Exp Med Biol. 2016;910:47–53.
Çörtük M, Demirkol B, Arslan MA, İlhan U, Kalkan YE, Turan D, Gül Ş, Çinarka H, Baydili KN, Çetinkaya E. Frequency of alpha-1 antitrypsin deficiency and unexpected results in COPD patients in Turkey; rare variants are common. Turkish J Med Sci. 2022;52(5):1478–85.
Wiesemann GS, Oshins RA, Flagg TO, Brantly ML. Novel SERPINA1 alleles identified through a large alpha-1 antitrypsin deficiency screening program and review of known variants. Chronic Obstruct Pulm Dis. 2022;5:689.
Jardi R, Rodriguez F, Miravitlles M, Vidal R, Cotrina M, Quer J, Pascual C, Weidinger S. Identification and molecular characterization of the new alpha-1-antitrypsin deficient allele PI Y barcelona (Asp256-->Val and Pro391-->His). Mutations in brief no. 174. Hum Mutat. 1998;12(3):213.
Fagerhol MK. Serum Pi types in Norwegians. Acta Pathol Microbiol Scand. 1967;70(3):421–8.
Mahadeva R, Chang WS, Dafforn TR, Oakley DJ, Foreman RC, Calvin J, Wight DG, Lomas DA. Heteropolymerization of S, I, and Z alpha1-antitrypsin and liver cirrhosis. J Clin Investig. 1999;103(7):999–1006.
Bornhorst JA, Greene DN, Ashwood ER, Grenache DG. α1-Antitrypsin phenotypes and associated serum protein concentrations in a large clinical population. Chest. 2013;143(4):1000–8.
Arnaud P, Chapuis-Cellier C, Vittoz P, Fudenberg HH. Genetic polymorphism of serum alpha-1-protease inhibitor (alpha-1-antitrypsin): Pi i, a deficient allele of the Pi system. J Lab Clin Med. 1978;92(2):177–84.
Seri M, Magi B, Cellesi C, Olia PM, Renieri A, De Marchi M. Molecular characterization of the P and I variants of alpha 1-antitrypsin. Int J Clin Lab Res. 1992;22(2):119–21.
Okayama H, Brantly M, Holmes M, Crystal RG. Characterization of the molecular basis of the alpha 1-antitrypsin F allele. Am J Hum Genet. 1991;48(6):1154–8.
Donato LJ, Jenkins SM, Smith C, Katzmann JA, Snyder MR. Reference and interpretive ranges for α(1)-antitrypsin quantitation by phenotype in adult and pediatric populations. Am J Clin Pathol. 2012;138(3):398–405.
Cook L, Burdon JG, Brenton S, Knight KR, Janus ED. Kinetic characterisation of alpha-1-antitrypsin F as an inhibitor of human neutrophil elastase. Pathology. 1996;28(3):242–7.
Sinden NJ, Koura F, Stockley RA. The significance of the F variant of alpha-1-antitrypsin and unique case report of a PiFF homozygote. BMC Pulm Med. 2014;14:132.
Cox DW. A new deficieny allele of alpha1-antitrypsin: Pi Mmalton. In: Protides of the biological fluids Proceedings of the twenty-third colloquium. Brugge: Pergamon Press, Ltd.; 1975. p. 375–378.
Matsunaga E, Shiokawa S, Nakamura H, Maruyama T, Tsuda K, Fukumaki Y. Molecular analysis of the gene of the alpha 1-antitrypsin deficiency variant, Mnichinan. Am J Hum Genet. 1990;46(3):602–12.
Faber JP, Poller W, Weidinger S, Kirchgesser M, Schwaab R, Bidlingmaier F, Olek K. Identification and DNA sequence analysis of 15 new alpha 1-antitrypsin variants, including two PI*Q0 alleles and one deficient PI*M allele. Am J Hum Genet. 1994;55(6):1113–21.
Silva D, Oliveira MJ, Guimarães M, Lima R, Gomes S, Seixas S. Alpha-1-antitrypsin (SERPINA1) mutation spectrum: three novel variants and haplotype characterization of rare deficiency alleles identified in Portugal. Respir Med. 2016;116:8–18.
Figueira GJM, Martínez BF, Díaz PD, Martín MMD, García-Talavera I, Pitti PR. Clinical manifestations of the Mmalton alpha-1 antitrypsin deficiency variant. Pulmonology. 2017;5:708.
Poller W, Merklein F, Schneider-Rasp S, Haack A, Fechner H, Wang H, Anagnostopoulos I, Weidinger S. Molecular characterisation of the defective alpha 1-antitrypsin alleles PI Mwurzburg (Pro369Ser), Mheerlen (Pro369Leu), and Q0lisbon (Thr68Ile). Eur J Hum Genet. 1999;7(3):321–31.
Jardi R, Rodriguez-Frias F, Lopez-Talavera JC, Miravitlles M, Cotrina M, Costa X, Pascual C, Vidal R. Characterization of the new alpha-1-antitrypsin-deficient PI M-type allele, PI M(vall d’hebron) (Pro(369)–>Ser). Hum Hered. 2000;50(5):320–1.
Kramps JA, Brouwers JW, Maesen F, Dijkman JH. PiMheerlen, alpha PiM allele resulting in very low alpha 1-antitrypsin serum levels. Hum Genet. 1981;59(2):104–7.
Hofker MH, Nukiwa T, van Paassen HM, Nelen M, Kramps JA, Klasen EC, Frants RR, Crystal RG. A Pro––Leu substitution in codon 369 of the alpha-1-antitrypsin deficiency variant PI MHeerlen. Hum Genet. 1989;81(3):264–8.
Takahashi H, Nukiwa T, Satoh K, Ogushi F, Brantly M, Fells G, Stier L, Courtney M, Crystal RG. Characterization of the gene and protein of the alpha 1-antitrypsin “deficiency” allele Mprocida. J Biol Chem. 1988;263(30):15528–34.
Fagerhol MK, Hauge HE. The Pi phenotype MP. Discovery of a ninth allele belonging to the system of inherited variants of serum alpha 1 antitrypsin. Vox Sang. 1968;15(5):396–400.
Holmes MD, Brantly ML, Crystal RG. Molecular analysis of the heterogeneity among the P-family of alpha-1-antitrypsin alleles. Am Rev Respir Dis. 1990;142(5):1185–92.
Hildesheim J, Kinsley G, Bissell M, Pierce J, Brantly M. Genetic diversity from a limited repertoire of mutations on different common allelic backgrounds: alpha 1-antitrypsin deficiency variant Pduarte. Hum Mutat. 1993;2(3):221–8.
Bornhorst JA, Calderon FR, Procter M, Tang W, Ashwood ER, Mao R. Genotypes and serum concentrations of human alpha-1-antitrypsin “P” protein variants in a clinical population. J Clin Pathol. 2007;60(10):1124–8.
Cook L, Burdon J, Brenton S, Janus ED, Knight K. Alpha-1-antitrypsin PLowell: a normally functioning variant present in low concentration. Aust N Z J Med. 1995;25(6):695–7.
Fra AM, Gooptu B, Ferrarotti I, Miranda E, Scabini R, Ronzoni R, Benini F, Corda L, Medicina D, Luisetti M, et al. Three new alpha1-antitrypsin deficiency variants help to define a C-terminal region regulating conformational change and polymerization. PLoS ONE. 2012;7(6):e38405.
Miravitlles M, Vilà S, Jardí R, de la Roza C, Rodríguez-Frías F, Vidal R. Emphysema due to alpha-antitrypsin deficiency: familial study of the YBARCELONA variant. Chest. 2003;124(1):404–6.
Lovegrove JU, Jeremiah S, Gillett GT, Temple IK, Povey S, Whitehouse DB. A new alpha 1-antitrypsin mutation, Thr-Met 85, (PI Zbristol) associated with novel electrophoretic properties. Ann Hum Genet. 1997;61(Pt 5):385–91.
Bates KJ, Puxley M, Hill M, Kalsheker N, Barlow A, Clark BE, Sherwood RA. A patient with the rare alpha-1-antitrypsin variant (Z)bristol in compound heterozygosity with the Z mutation. Ann Clin Biochem. 2013;50(Pt 6):618–21.
Hoàng TH, Phạm TN, Nguyễn GK, Lê QH. A rare variant of α 1 antitrypsin mutations detected in Vietnamese children with liver disease. Ann Clin Biochem. 2013;50(Pt 4):339–44.
Graham A, Kalsheker NA, Bamforth FJ, Newton CR, Markham AF. Molecular characterisation of two alpha-1-antitrypsin deficiency variants: proteinase inhibitor (Pi) Null(Newport) (Gly115-Ser) and (Pi) Z Wrexham (Ser-19-Leu). Hum Genet. 1990;85(5):537–40.
Garver RI Jr, Mornex JF, Nukiwa T, Brantly M, Courtney M, LeCocq JP, Crystal RG. Alpha 1-antitrypsin deficiency and emphysema caused by homozygous inheritance of non-expressing alpha 1-antitrypsin genes. N Engl J Med. 1986;314(12):762–6.
Nukiwa T, Takahashi H, Brantly M, Courtney M, Crystal RG. Alpha 1-antitrypsin nullGranite Falls, a nonexpressing alpha 1-antitrypsin gene associated with a frameshift to stop mutation in a coding exon. J Biol Chem. 1987;262(25):11999–2004.
Lara B, Martínez MT, Blanco I, Hernández-Moro C, Velasco EA, Ferrarotti I, Rodriguez-Frias F, Perez L, Vazquez I, Alonso J, et al. Severe alpha-1 antitrypsin deficiency in composite heterozygotes inheriting a new splicing mutation QOMadrid. Respir Res. 2014;15(1):125.
Cox DW, Billingsley GD. Rare deficiency types of alpha 1-antitrypsin: electrophoretic variation and DNA haplotypes. Am J Hum Genet. 1989;44(6):844–54.
Seixas S, Mendonça C, Costa F, Rocha J. Alpha1-antitrypsin null alleles: evidence for the recurrence of the L353fsX376 mutation and a novel G–>A transition in position + 1 of intron IC affecting normal mRNA splicing. Clin Genet. 2002;62(2):175–80.
Laubach VE, Ryan WJ, Brantly M. Characterization of a human alpha 1-antitrypsin null allele involving aberrant mRNA splicing. Hum Mol Genet. 1993;2(7):1001–5.
Brantly M, Lee JH, Hildesheim J, Uhm CS, Prakash UB, Staats BA, Crystal RG, Hildeshiem J. Alpha1-antitrypsin gene mutation hot spot associated with the formation of a retained and degraded null variant [corrected; erratum to be published]. Am J Respir Cell Mol Biol. 1997;16(3):225–31.
Seyama K, Nukiwa T, Takabe K, Takahashi H, Miyake K, Kira S. Siiyama (serine 53 (TCC) to phenylalanine 53 (TTC)). A new alpha 1-antitrypsin-deficient variant with mutation on a predicted conserved residue of the serpin backbone. J Biol Chem. 1991;266(19):12627–32.
Acknowledgements
The authors want to show their appreciation to all the clinicians responsible for the collection of samples and their determined effort to improve the diagnosis of patients with AATD.
Funding
This project is entirely funded by Grifols.
Author information
Authors and Affiliations
Contributions
JLLC performed the statistical analysis, included cases and wrote the manuscript; KC, JRJ, MAF, AA, HG, MM included cases, revised the draft and contributed with the writing; LO and NR performed the laboratory analysis, revised the draft and contributed with the writing, ED performed the statistical analysis, revised the draft and contributed with the writing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study reports the results of a diagnostic procedure that is approved for use in all the participating countries and, therefore, constitutes standard clinical practice. Consequently, since it is not an experimental procedure, approval by an ethics committee was not considered necessary. All patients included signed a written informed consent form.
Consent for publication
Not applicable.
Competing interests
JLLC has received honoraria during the last 3 years for lecturing, scientific advice, participation in clinical studies or writing for publications for (alphabetical order): AstraZeneca, Bial, Boehringer, Chiesi, CSL Behring, Faes, Ferrer, Gebro, Grifols, GSK, Megalabs, Menarini, Novartis. MM has received speaker fees from AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, Menarini, Kamada, Takeda, Zambon, CSL Behring, Specialty Therapeutics, Janssen, Grifols and Novartis, consulting fees from AstraZeneca, Atriva Therapeutics, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, CSL Behring, Inhibrx, Ferrer, Menarini, Mereo Biopharma, Spin Therapeutics, ONO Pharma, Palobiofarma SL, Takeda, Novartis, Sanofi and Grifols and research grants from Grifols. LO works as geneticist at Progenika Biopharma, a Grifols Company. ED works in Scientific and Medical Affairs at Grifols.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lopez-Campos, J.L., Rapun, N., Czischke, K. et al. Distribution of alpha1 antitrypsin rare alleles in six countries: Results from the Progenika diagnostic network. Hum Genomics 17, 48 (2023). https://doi.org/10.1186/s40246-023-00497-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-023-00497-1