
Abstract
Background
Fine-scale genetic structure of ethnolinguistically diverse Chinese populations can fill the gap in the missing diversity and evolutionary landscape of East Asians, particularly for anthropologically informed Chinese minorities. Hmong–Mien (HM) people were one of the most significant indigenous populations in South China and Southeast Asia, which were suggested to be the descendants of the ancient Yangtze rice farmers based on linguistic and archeological evidence. However, their deep population history and biological adaptative features remained to be fully characterized.
Objectives
To explore the evolutionary and adaptive characteristics of the Miao people, we genotyped genome-wide SNP data in Guizhou HM-speaking populations and merged it with modern and ancient reference populations via a comprehensive population genetic analysis and evolutionary admixture modeling.
Results
The overall genetic admixture landscape of Guizhou Miao showed genetic differentiation between them and other linguistically diverse Guizhou populations. Admixture models further confirmed that Miao people derived their primary ancestry from geographically close Guangxi Gaohuahua people. The estimated identity by descent and effective population size confirmed a plausible population bottleneck, contributing to their unique genetic diversity and population structure patterns. We finally identified several natural selection candidate genes associated with several biological pathways.
Conclusions
Guizhou Miao possessed a specific genetic structure and harbored a close genetic relationship with geographically close southern Chinese indigenous populations and Guangxi historical people. Miao people derived their major ancestry from geographically close Guangxi Gaohuahua people and experienced a plausible population bottleneck which contributed to the unique pattern of their genetic diversity and structure. Future ancient DNA from Shijiahe and Qujialing will provide new insights into the origin of the Miao people.
Similar content being viewed by others


Introduction
The complex human admixture history in Eastern Eurasia has resulted in the formation of Mongolic-, Turkic-, and Tungusic-speaking populations in present-day northern East Asia, Sino-Tibetan (ST)-speaking populations across East Asia, Hmong–Mien (HM), Tai–Kadai (TK), Austronesian (AN), and Austroasiatic (AA)-speaking populations in southern East Asia and Southeast Asia. Several comprehensive studies have been conducted to characterize East Asia’s linguistic and genetic history [1,2,3,4,5,6,7]. These studies found that geographic isolation, population expansion, and cultural interaction have shaped the genetic and linguistic landscape of modern and ancient Chinese populations [5, 8, 9]. Besides, agriculture innovation from the Yellow River Basin and Yangtze River Basin significantly influenced the population formation of ST and southern Chinese indigenous TK, HM, AA, and AN people [10]. The ancestor of modern HM people and their cultures (beliefs, languages, and technologies) are suggested to be associated with the origin of rice farming from the middle Yangtze River Basin. Modern HM people are predominantly living in mountainous areas (Guizhou, Hunan, Hubei, Sichuan, Yunnan, Guangxi, and Hainan) in southern East Asia [11,12,13] and northern Mainland Southeast Asia [4]. Recent archeological, genetic, and historical evidence has further revealed that HM groups might originate from the Neolithic Daxi groups (~ 5000 to 3300 BCE) in the middle reaches of the Yangtze River [14,15,16,17] and migrated westward and southward into the Yungui Plateau due to climate changes and warfare. Historic documents suggested that the migrations and admixture of ancient source populations facilitated the formation of the present-day distributions of HM groups in Southeast Asia (mainly in Laos and Vietnam), Europe, the USA, and Australia during the Ming and Qing dynasties [18,19,20,21].
Southwest China possessed rich archaeological cultures, including the Shijiahe, Qujialing, Daxi cultures and others. Much evidence has linked the origin of HM people and the ancient ancestors of Yangtze Rice farmers. Li et al. conducted a Y-chromosome-based ancient DNA study to explore the genetic relationship between HM-speaking (Miao, She, and Yao) and geographically close modern and ancient East Asians [14]. Their inferred population evolutionary model based on the Y-chromosome haplogroup O2a2a1a2-M7 suggested that the 5000-year-old Daxi people may have shared a common ancestor with the contemporary Miao people [14], which provided a critical piece of genetic evidence of common origin between ancient Daxi people and modern Miao people. HM-speaking populations currently have their specific language (Hmong and Mien) and culture. Still, few written records of the Hmong language were conserved, which was spread mainly relying on myths and legends and oral transmission. Here, we aimed to present a deeper and more comprehensive evolutionary history of the HM-speaking populations. A fine-scale genetic study focused on genetic origins is beneficial to explore the cultural roots of the HM-speaking people and their admixed history and to gain a comprehensive understanding of the migration events and admixture times in East and Southeast Asian history.
Research on the mystery of HM people and their language family has increased with advances in computational biology and molecular biology techniques. Previous studies have primarily focused on forensic-related autosomal short tandem repeats (STRs), insertion/deletions (Indels), Y-chromosomal, and mitochondrial single nucleotide polymorphisms (SNPs) markers [13, 22,23,24,25,26]. These studies have been dwarfed in studying the fine-scale genetic structure and exploring the history of the genetic mixing process. Recently, the reported 500-year-old Gaohuahua population was found to be genetically related to the HM ancestry [8]. Still, a recent study of genome-wide SNPs in the present-day HM populations showed that the Libo Yao is more representative of the ancestral lineage of HM groups than the 500-year-old Gaohuahua [27]. There is a growing number of genome-wide studies of HM-speaking populations, and they all revealed a north–south mixing pattern of HM-speaking people and the existence of genetic substructures among HM-speaking populations from different regions [27,28,29]. Gao et al. extrapolated the divergence time of HM-speaking people and found that the separation of Yao from other HM groups was earlier than that between Miao and She. These findings also supported that the Yao was closer related to populations belonging to the TK language family than the Miao and She [30]. Liu et al. collected genome-wide SNP data from three representative Miao groups in Sichuan Province and merged them with data from 144 Miaos from 13 regions to comprehensively characterize the biological adaptations of the Miao people during the migration [31]. The findings revealed high genetic similarity among HM populations from different regions in southwestern China and Southeast Asia, but subtle genetic differences between each other.
Although there are many studies on the genetic origins, admixture history and biological adaptations of Miao populations, limited genetic studies focused on the fine-scale genetic structure and admixture processes of geographically diverse Guizhou Miao people have been performed, especially complex admixture modeling based on the haplotype information. Guizhou, located on the Yungui Plateau with a high topography in the west and a low topography in the east, is known to be one of the provinces rich in ethnic diversity. Its complex topography has laid the foundation for its ethnic diversity. For example, the Miao people in Congjiang/Nayong/Longli/Duyun County from Guizhou Province are excellent sites for studying and supplementing genetic data of HM-speaking populations. Many villages still preserve the traditional Hmong language and culture, which have not been reported in detail. We collected genetic data from the Guizhou Miao people and conducted deep genomic research, which allowed Miao and other ethnic minorities to benefit from participating in precision medicine and achieve clinical precision treatment [32]. Generally, we collected 67 Guizhou Miao samples and merged the newly generated data with data from HM populations from the HGDP to analyze the genetic structure and admixture history of the HM-speaking populations and explore biological adaptations. We also estimated the identity by descent (IBD) and calculated the effective population size to explore the genetic differences between the Miao and other ethnic groups.
Results
The general structure of HM populations in the context of East and Southeast Asian populations
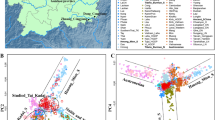
We genotyped approximately 669,896 genome-wide SNPs in 67 Miao individuals from four populations [Miao_Congjiang (CJM), Miao_Nayong (NYM), Miao_Duyun (DYM), and Miao_Longli (LLM)] in Guizhou Province (Fig. 1a). We then merged our data with published modern and ancient worldwide populations from the Whole-Genome Sequencing (WGS), Human Origins (HO), and 1240K datasets. These datasets included African, North American, South Asian, Southeast Asian, European, Central Asian, and Oceanian populations, as well as ancient populations from Northwest China, Northeast China, Middle Yellow River, Southeast China, and the ancient Gaohuahua, BaBanQinCen, and Longlin people in Guangxi; modern Altaic, ST, and HM-speaking populations. We performed a PCA analysis to explore the general relationship between the HM-speaking populations and East and Southeast Asian populations. We observed that the population clustering patterns were consistent with the geographic and linguistic distribution. In the East and Southeast Asian PCA (Fig. 1b), we observed that the four Miao populations clustered together to form a unique cluster and were closely related to the Miao and She from the HGDP project, followed by the Han, Tu, Dai, and Lahu. And compared with NYM, the other three HM-speaking populations (DYM, LLM, and CJM) gathered more closely, and they were closely related to Dai, indicating that DYM, LLM, CJM, and Dai have more genetic exchanges in the process of population evolution and cultural exchange.

The genetic affinity between HM populations and ancient and modern East Asians. a The geographical positions of our studied HM populations and reference populations; b principal component analysis (PCA) of 22 populations at the East and Southeast Asian scale; c PCA analysis of 51 ancient and modern populations at the East Asian scale; and d PCA analysis of 17 modern populations at the East Asian scale; e ADMIXTURE results showed genetic similarity between ancient and modern populations at the East Asian scale with our studied HM people in the K value = 2 and 3
To identify the genetic affinity of our studied Miao people with other geographically and linguistically distributed worldwide populations, we conducted an ADMIXTURE analysis based on the combined dataset of 121 ancient and modern worldwide populations. When K = 13, five ancestral components (dark blue, orange, light gray, light green, and red) were found in HM populations (Fig. 2). The dark blue component reached the largest proportion in the NYM and Gaohuahua in southern East Asia. The orange component reached the largest proportion in the Taiwan_Hanben_IA in southern China and the Agta_Philippines in Southeast Asia. The light gray component reached the largest proportion in the Mongolia_North_N and Mongolia_East_N, the light green component reached its maximum proportion in Nepal’s Samdzong and Mebrak populations, and the red component reached its maximum proportion in the Bellona_Solomon and Rennell_Solomon populations of Oceania. We also found that the HM populations (She and Miao) from the HGDP contained a higher proportion of ancestral components related to northern East Asian compared with the four Miao populations we studied, indicating that they were significantly influenced by incoming northern East Asian gene flow. Interestingly, we also found genetic differences among our studied Miao populations. The dark blue ancestral component in NYM, LLM, CJM, and DYM were in descending order, indicating that NYM is more representative of the ancestors of the HM-speaking populations than the other three Miao groups and even more representative of the ancestors of the HM-speaking people than Gaohuahua, as previously commonly believed.

The general structure of East and Southeast Asian populations. We merged our data with published modern and ancient global populations from the WGS and 1240K datasets. We conducted an ADMIXTURE analysis based on the combined dataset of 121 ancient and modern worldwide populations (K = 13)
The genetic affinity between HM populations and ancient East Asians
To visualize the genetic relationship between HM populations and ancient East Asians, we further performed PCA analysis on 51 populations from ancient and modern East Asia based on the merged 1240K dataset (Fig. 1c). We observed that our studied Miaos formed a unique genetic branch that clustered closely with HM populations from the HGDP and Dai and ST-speaking populations compared to ancient and modern people from northern China and ancient coastal and TK populations from southeastern China. The HM populations were more related to Gaohuahua from Guangxi 500 years ago and BaBanQinCen from Guangxi 1500 years ago than all other ancient East Asians. And we found that NYM was clustered closely with the Gaohuahua compared with DYM, LLM, and CJM.
We conducted a model-based ADMIXTURE clustering analysis to profile the ancestral components and genetic resemblance of our studied Miao with geographically close ancient and present-day populations. At K = 2 (Fig. 1e), we observed the minimum CV value. We observed two ancestral components, with the blue component reaching a maximum in Neolithic ancients from the middle reaches of the Yellow River and northwestern ancients, and the orange component reaching its maximum in populations from the southern China, especially in the ancient southeastern coastal people and Hanben_Taiwan populations. We found that the proportion of these two ancestral components in the HM populations from the HGDP was almost equal to that in Han, Tujia, and Lahu, which were genetically related to the coastal people and Han populations in southern China, indicating that the HM populations from the HGDP were heavily Sinicized. We also found that all Miaos in our study showed fewer blue ancestral components and more orange ancestral components, especially NYM, which had almost all orange ancestral components, indicating that NYM was less influenced by northern gene flow.
When K = 3, three ancestral components of blue, orange, and pink were observed in the HM populations (Fig. 1e). The blue component reached its maximum in the ancients from Neolithic Northern Inland regions, Western Liao River, and Shaanxi Province in the middle reaches of the Yellow River. And the orange component reached its maximum in the southern Chinese islanders and the Hanben_Taiwan, and the emerging pink component reached its maximum in the Gaohuahua. Compared to all reference populations, our studied Miaos had more pink ancestral components, suggesting that the Gaohuahua population was likely to be the ancestor proxy of the HM populations. Compared with HM populations from HGDP, our studied Miaos had less gene flow from the ancient genetic lineage from Northern East Asia, especially NYM. NYM is more representative of the ancestral source of HM populations than DYM, LLM, and CJM.
Genetic affinity of HM populations with southern East Asians and Southeast Asian populations
The PCA analysis in the context of East Asians revealed that HM-speaking populations formed a separate genetic branch (Fig. 1d). In general, our studied Miaos clustered closely with the HM populations (Miao and She), Dai, and Lahu from HGDP. We also found that the HM populations from the HGDP largely overlapped with Han-associated populations, indicating that Han-associated genetic materials largely influenced the HM populations from the HGDP compared to our studied Miaos. Interestingly, NYM was distant from the other three Miao populations and Han-related genetic lineages. DYM and CJM were located at the intersection of HM and Han-related genetic lineages, suggesting that Han populations influenced these two studied Miao populations significantly.
We carried out admixture-f3 in the form f3(X, Y; studied Miao) to test mixed signals using all possible ancestral source pairs. LLM produced significant negative Z-values (Z < −3) when combined with the northern sources related to ST (Han, Tujia, Naxi, and Yi), Altaic (Daur, Mongolian, Hezhen, and Xibo), and southern sources related to Guizhou aborigines (Guizhou Han, Chuanqing, etc.) and Southeast Asian populations (Additional file 1: Table S1). When DYM was used as the targeted population, NYM, LLM, and CJM as one source and combined with other East Asians could produce significant negative Z-values (Additional file 1: Table S2). When we used CJM as the targeted, NYM as one source and combined with the above mentioned source populations, which could yield significant negative Z-values. When we combined northern East Asian populations (Japanese, Han, Yi, Daur, Mongolian, Hezhen, Xibo, and Northern_Han) and southern East Asians (Dai, Lahu, and She) or some Guizhou aboriginals (Shui_Duyun and Dong_Congjiang) as two ancestral sources, tests focused on LLM could produce significant negative Z-values (Additional file 1: Table S3). Interestingly, we did not find significant negative Z-values when we used NYM as the target group (Additional file 1: Table S4). The above results suggested that NYM might be the purest representative of the ancestral component of the HM populations, which was consistent with the ADMIXTURE results. We also found consistent results in the pairwise Fst (Fig. 3a).

Genetic affinity of HM populations with East and Southeast Asian populations. a, Heatmap of the pairwise Fst genetic distances among 51 worldwide populations. NA: Not applicable due to the small sample size of the ancient population. b Shared allele counts, including worldwide representative 54 populations. + + means a strong positive result with the IBD number over 50. c Shared genetic drift inferred from out-group f3-statistics in the form f3(studied Miao, East Asian/Southeast Asian; Mbuti), including 27 populations in East Asia/Southeast Asia. d TreeMix-based maximum likelihood tree with three migration events revealed the genetic relationship between East Asians/Southeast Asians
We used pairwise IBD to measure shared haplotype counts, including worldwide representative populations (Fig. 3b), and we observed that the Miao people shared more IBD chunks with geographically close East Asians. We further performed out-group f3(studied Miao, East Asian/Southeast Asian; Mbuti) (Fig. 3c) and found that the Miaos shared more genetic drift with Atayal_Taiwan and Paiwan_Taiwan, followed by Miao from HGDP, and then the TK-speaking Dai and ST-speaking Han, Tujia, and Lahu.
To confirm the genetic patterns in PCA and ADMIXTURE and discover potential ancestral origins for studied Miao people, we conducted f4-statistics to explore the genetic affinities between the Miaos and modern East Asian populations in the form of f4(Han_Northern/Han_Southern, studied Miao; East Asian/Southeast Asian, Mbuti). The studied Miaos showed more allele sharing with geographically close populations compared to the Han_Northern (Additional file 1: Tables S5–S6). Furthermore, the studied Miao people also shared more alleles with geographically close TK, ST, AN-speaking populations, and Southeast Asian populations compared to Han_Southern. The results indicated that our studied Miao populations shared more alleles with geographically close people than with Han Chinese and these studied populations were less influenced by north-to-south gene flow and more influenced by gene flow from TK and ST-speaking populations. We also found that the studied Miao populations were influenced by Han-associated gene flow in descending order of DYM, CJM, LLM, and NYM.
We ran TreeMix to study the population split and mixing patterns between selected East and Southeast Asian populations (Fig. 3d). We found that our studied Miaos clustered with HM populations from HGDP and located between the northern Chinese populations represented by Hezhen, Daur, Xibo, and Tu ethnic groups and southern Chinese and Southeast Asian populations represented by Dai, Lahu, Paiwan_Taiwan, Atayal_Taiwan, Cebuano_Philippines, and Agta_Philippines. Specifically, our studied Miaos first gathered together and then mixed with the Altaic-speaking populations from northern East Asia and the ST-speaking populations from southern East Asia. NYM was less influenced by the indigenous people of the surrounding area than the other three Miao populations. We observed gene flow from East Asia to Agta_Philippines, from Yi to Lahu, and from ST-speaking populations to Miao, indicating that Miao from HGDP was influenced by ST-speaking populations. At the same time, the four Miao groups were less affected by ST-speaking populations, which was consistent with the distribution patterns observed in PCA, ADMIXTURE, and f4-statistics. It was also compatible with a mixed north–south pattern of HM populations and migration of HM populations to Southeast Asia in response to historical events.
Genetic substructure among HM-speaking populations
We performed f4-statistics of the form f4(study1, study2; reference, Mbuti) to analyze whether genetic substructure existed among the four Miao populations (Additional file 1: Table S7). We found that no significant Z-values (Z > 3 or Z < −3) were observed in any pairs of f4(CJM, DYM/LLM/NYM; Eurasians, Mbuti), suggesting that the HM populations were genetically homogeneous relative to the northern East Asian, Southeast Asian, and southeast coastal Chinese populations. We observed significant negative Z-values in the f4(CJM, LLM/NYM; Chuanqing_Nayong, Mbuti), indicating that LLM/NYM shared more alleles with Chuanqing_Nayong compared to CJM. And we observed that previously reported Yi and Miao shared more alleles with LLM compared with CJM, as the observed negative values in f4(CJM, LLM; Yi/Miao, Mbuti). We also found that Dai shared more alleles with CJM compared to NYM, with significant positive Z-values for f4(CJM, NYM; Dai, Mbuti). These results suggested the existence of a genetic substructure among the four studied HM populations.
We further inferred the fine-scale genetic structure of studied HM populations using fineSTRUCTURE (Fig. 4). The PCA based on the sharing co-ancestry matrix and ADMIXTURE results based on allele frequency spectrum indicated that our studied Miao populations formed a separate genetic branch, which was closely related to the HM populations from HGDP, Dai, and southern Han Chinese. It was distinguished from the Altaic- and ST-speaking populations in northern China and ST-speaking populations in south China (Fig. 4a–d). We further confirmed these genetic distribution patterns in a clustering model based on shared IBD fragments at the population and individual levels (Fig. 4e–f). Similar to the ADMIXTURE results, NYM and LLM were genetically closely related to each other (Fig. 4g–i).

Fine-scale population genetic structure in East Asian populations. a–c PCA results based on the co-ancestry matrix revealed the genetic relationship between our studied Miao and other modern East Asians. The color showed the re-classification of the homogenous population label. d ADMIXTURE analysis revealed a genetic relationship between our studied Miao and other modern East Asians. e and f Clustering patterns of East Asians based on population dendrogram and the pairwise coincidence matrixes. g Shared genetic drift based on the pairwise Fst showed the genetic relationship between our studied HM people. h and i Number and length of shared alleles based on IBD between our studied HM people
qpAdm-based admixture models and admixture times
The intense genetic relationship between the historic populations from southern China and the studied HM people has been demonstrated in the descriptive analyses and the quantitative f-statistics. We further performed two-way qpAdm models to explore the ancestral composition of our studied Miao people, with southern East Asians as the southern surrogates and ancient northern East Asians as the northern surrogates. We tested two-way admixture models and found that ancestry proportion from the ancient southern populations ranged from 0.864 ± 0.077 in the AR14K-Yiyang model to 0.372 ± 0.036 in the China_YR_LN-Cebuano_Philippines model (Additional file 1: Table S8). To further discuss whether distinct genetic contributions derived from the island and inland southern East Asians and Southeast Asians contributed to the studied populations, we then performed three-way admixture models with one source from southern East Asia (inland and island), one from northern East Asia, and another from Southeast Asia. The inferred northern sources included Neolithic middle and upper Yellow River farmers of the China_YR_MN, China_YR_LN, China_YR_LBIA, Upper_YR_LN, Shimao_LN, AR_EN, and AR19K, AR14K, AR13_10K; the southern East Asian inland sources were made up of Lahu, Dushan, Baojianshan, Longlin, Shenxian, Layi, Yiyang, BaBanQinCen and island sources of Paiwan_Taiwan, Atayal_Taiwan, Taiwan_Hanben_IA; Southeast Asian ancestral sources comprised of Thailand_IA, Laos_BA, Vietnam_N, Laos_LN_BA, Vietnam_BA. The fitted admixture models revealed that studied populations derived their primary ancestry from southern island sources and modern southern East Asian, a second from the northern sources, and the final one from the ancient southern and Southeast Asian sources (Additional file 1: Table S9). The admixture times with different ancestral populations based on MALDER suggested that north–south population interaction and admixture occurred at least in the Neolithic period and large-scale population admixture occurred between 1100 and 1600 years ago. The admixture of southern populations began at least 3500 years ago, with large-scale population admixture occurring between 1100 and 2800 years ago (Additional file 1: Table S10).
Complex population events contributed to the patterns of genetic diversity of HM-speaking populations
Population admixture models and descriptive analysis have shown that the Miao people were an admixture population with both northern and southern East Asian sources. Like other East Asians, admixture served as the major force to drive the formation of the gene pool of HM people. We further explored whether other additional forces contributed to the observed patterns of genetic diversity. We calculated the Runs of Homozygosity (ROH) and found that Miao people had the most significant ROH values compared with other populations. The estimated IBD fragments in different length ranges showed that Miao people had high genetic communication with each other in the past two thousand years as the more shared IBD in different categories (Fig. 5a–e). The estimated effective population size showed a plausible population bottleneck in the ancient Miao people, consistent with the observed patterns of ROH (Fig. 5f). Finally, we conducted an IBD-based admixture time estimation via fastGLOBETROTTER. We only found one population that showed the identified admixture signatures with gene flow from Han Chinese, and the other three populations showed unclear signal. The identified signatures showed that both complex admixture and population bottleneck contributed to the formation of the genetic diversity of modern HM people.

Complex population events contributed to the patterns of genetic diversity of HM groups. a–e Shared IBD fragments in different length ranges of Miaos and other populations in East Asia. f The effective population size of Miaos from 150 generations before the present, the Miao_HGDP has increased exponentially at about 20 generations, and our studied Miao has increased exponentially at about 40 generations
Natural selection signatures and biological adaptation
Genetic evidence has identified many biologically adaptive genes or pathways between ethnolinguistically diverse populations. With the increase in initial allele frequencies, adaptive mutations could confer higher immunity in target populations relative to other unexposed populations. More pathogen exposure will inevitably result in more frequent adaptive gene mutations. We screened candidate genes subjected to positive selection using integrated haplotype score (iHS) and cross-population extended haplotype homozygosity (XPEHH). We first computed XPEHH values for the Miaos with the Northern Han Chinese as a reference population and identified selection signatures on chromosomes 1, 2, 4, 6, 8, 10, 11, 13, and 15 (Fig. 6a). We observed that chromosome 1 exhibited a positive selection signal for the CDK18 gene, which maintains genomic stability [33]. And we found positive selection on chromosome 2 for the SMYD1 gene associated with hypertrophic cardiomyopathy stability [34] and the potassium channel arrhythmia-associated gene KCNJ3. We also screened for its overexpression on chromosome 4 related to the cancer gene PALLD [35]. We further observed strong selection signals on chromosome 6, including MUC22, PSORS1C1, PSORS1C3, PSORS1C1|PSORS1C2, CDSN|PSORS1C1, CCHCR1, TCF19, KHDRBS2. Among them, PSORS1C1, PSORS1C3, PSORS1C1|PSORS1C2, CDSN|PSORS1C1, and CCHCR1 are genes involved in autoimmune diseases and are expressed in psoriasis, rheumatoid arthritis ankylosing spondylitis [36,37,38] and PSORS1C3 is also linked to major depressive disorder suicide [38]. MUC22 is upregulated or downregulated in lung adenocarcinoma and squamous cell carcinoma [39] and is also seen in childhood asthma Expression [40]. TCF19 may act as a susceptibility gene for non-small cell carcinoma in the Chinese populations, interacting with tumor suppressor protein (P53) and playing a role in various metabolic pathways, including cancer and diabetes [41]. KHDRBS2 has a regulatory role in microenvironmental permeability, hereditary retinopathy, and cancer [42, 43]. We also identified a positive selection signal for a tumor suppressor-associated gene (DLC1) on chromosome 8 [44]. We further identified positive selection signals for zinc finger protein family members (ZNF25, ZNF248, ZNF33BP|ZNF248, ZNF33A) on chromosome 10 [45]. And we identified a positive selection signal for the ATP-binding cassette transporter subfamily C member 8 (ABCC8) gene on chromosome 11, a variant that causes hereditary diabetes and hyperinsulinemia [46]. We also observed SLC15A1, a peptide transporter protein associated with digestion and absorption, on chromosome 13 [47] and a positive selection signal for the NIPA1 gene, which causes neurodegenerative diseases in chromosome 15 [48].

Manhattan plots showed the natural selection signatures and the results of enrichment analysis. a XPEHH value in the Miao populations with Han_Northern as the reference population. b Overlapped pattern among three selected gene lists, where purple lines link the same gene that is shared by multiple gene lists, and blue lines link genes that belong to under the same ontology term. On the inside, each arc represents a gene list, where each gene member of that list is assigned a spot on the arc. The dark orange color represents the genes shared by multiple lists, and the light orange color represents genes unique to that gene list. c Heatmap and dendrogram of the three gene lists for the top 20 enriched terms, colored by P value. d Enriched terms network colored by cluster-ID. e The enriched terms for the top 20 clusters and their representation
We further counted XPEHH values with the southern Han Chinese as the reference group and iHS values of four Miao populations to exploit all potential candidate genes and adaptive pathways (Fig. 6b–e). We screened the top 1% of XPEHH values and normalized iHS values and enriched these genes using Metascape online software. More overlapping motifs were observed from the XPEHH screen and less from the iHS screen in the three candidate gene lists, and we could observe more functional overlap in the three candidate gene lists. Heatmap based on enrichment pathway p values showed genes associated with regulation of transmembrane transport (GO:0034762), actin filament-based process (GO:0030029), regulation of membrane potential (GO:0042391), regulation of cell morphogenesis (GO:0022604), regulation of cell adhesion (GO:0030155), Neuronal System (R-HSA-112316), metal ion transport (GO:0030001), cell–cell adhesion (GO:0098609), circulatory system process (GO:0003013), cell morphogenesis (GO:0000902), heart development (GO:0007507), positive regulation of locomotion (GO:0040017), NABA CORE MATRISOME (M5884), transmembrane receptor protein tyrosine kinase signaling pathway (GO:0007169), antigen processing and presentation of peptide antigen (GO:0048002), and iHS shows a strong positive selection signal for extracellular matrix organization (R-HSA-1474244), developmental cell growth (GO:0048588), modulation of chemical synaptic transmission (GO:0050804), head development (GO:0060322), regulation of axonogenesis (GO:0050770).
Discussion
It is well known that East Asian populations account for one-fifth of the worldwide populations and have high genetic diversity. Although studies on the genetic structure and population admixture history of East Asians have increased significantly in the last two decades, the genetic diversity of East Asian populations has been studied with fewer representative populations compared to European populations. Human genomic studies have promoted a better understanding of the benefit of personal genomics and precision medicine [49, 50]. Under-representation and European bias in human genetic studies have hindered the healthy equality of ethnolinguistically diverse populations [51]. Thus, large-scale population genomic studies in under-studied regions are important and emergency [51]. As early as 2010, scholars have illuminated the patterns of the diversity of ethnicity, language, culture, and colonial history, and they also discovered the complex migration events between Southeast Asia and North Asia and the genetic relationships and various social events among East Asian hunters based on multiple evidence [1]. For a long time, scholars had studied the genetic diversity of East Asian populations through low-density genetic markers (STRs, SNPs, and Indels) and high-density genetic markers, including genome-wide SNPs and whole-genome sequencing, which have provided initial explorations of the population structure and mixing history of East Asian populations [5, 52,53,54,55,56]. Genetic relationships among various East Asian ethnic groups have been studied mainly using STRs, Indels, and Y-chromosomal markers, which have identified the north–south stratification and north–south genetic mixing patterns in China [57, 58]. Previous genetic studies also suggested that the distribution of genetic diversity of different ethnic groups in China was closely related to their linguistic and geographical divisions [59, 60], such as the identified genetic differentiation between highlanders and lowlanders [61]. Liu et al. genotyped forensic-related genetic markers of Tibetans on the Tibetan Plateau and combined low-density/high-density genome-wide data of modern and ancient high-altitude East Asians, and they demonstrated the close relationship between the Tibetan Plateau and Yellow River Basin populations [61].
To comprehensively characterize the genetic diversity of East Asians and develop a next-step sampling plan in the genomic cohorts in the 10K_CPGDP (10,000 Chinese Person Genomic Diversity Project) and GSRD100KWCH (Genome sequencing of Rare Diseases of 100,000 people in China), we are conducting comprehensive genetic screening for the basic background of ethnolinguistically diverse populations. Here, we collected 67 Miao samples from four regions in Guizhou. Our analysis revealed genetic characteristics unique to the HM populations in Guizhou, including the genetic affinity and genetic structure of the studied HM populations with ancient and modern East Asian people as determined by PCA, East Asian-scale ADMIXTURE, and Fst. It showed that our studied HM populations were influenced by north–south gene flow. The tight clustering pattern of HM-speaking populations and the relationship between HM and Gaohuahua people were confirmed by f4-statistics, out-group f3-statistics, shared IBD fragments, and fineSTRUCTURE, and the genetic substructure among studied HM populations was determined by f4-statistics and TreeMix phylogeny.
We defined candidate genes associated with several different biological functions (locomotion, signaling, localization, regulation of biological process, and developmental process) in the HM populations (CDK18, SMYD1, KCNJ3, PALLD, MUC22, PSORS1C1|PSORS1C2, PSORS1C1, CCHCR1, TCF19, PSORS1C3, KHDRBS2, CDSN|PSORS1C1, DLC1, ZNF25, ZNF248, ZNF33BP1|ZNF248, ZNF33A, and ABCC (ZNF33A, ABCC8, SLC15A1, NIPA1)). More Denisovan ancient adaptive infiltration signals associated with immune function [62] have been confirmed in Island Southeast Asian and Oceanian populations. Interestingly, we observed natural selection signals associated with immune function in the AGER gene on chromosome 6 (iHS score: 3.8638 in rs2071288, 3.7444 in rs2070600), and the RHOH gene on chromosome 4 (3.833187 in rs73808637). Rachel et al. showed that OCA2 and BNC2 are genes that affect the skin pigmentation phenotype [63], but no corresponding natural selection signal was observed in the Guizhou HM populations. And we did not observe a natural selection signal for the lactase (LCT) [64] gene in the Guizhou HM-speaking populations. ALDH2 and ADH1B are strongly associated with alcohol metabolism [65], and we only observed that the ADH1B gene showed a strong selectivity, reaching a maximum XPEHH score at position rs1229984 on chromosome 4 for 2.7829. The genomic selection processes observed in HM-speaking populations are related to the unique demographic history of Southwest China and the specific living environment of the Yungui Plateau. Therefore, comprehensive sampling and sequencing of the various ethnic groups in Southwest China will help us gain new insights into the genetic landscape of Southwest China and the history of migratory and adaptive changes in HM populations.
Interestingly, we found that genes closely associated with coronary heart disease (CETP, LDLR, APOE, and ABCA1) were selected in HM populations. Cholesteryl ester transfer protein (CETP) (iHS score of rs12597002:1.6131, and rs291044: 1.6484) is a liver-synthesized glycoprotein, and some studies have shown that CETP inhibitors can be used for the prevention and treatment of coronary heart disease and atherosclerosis [66]. Low-density lipoprotein receptor (LDLR) was selected when using the population from southern East Asia as a reference (rs2738456\rs2738458\rs2569538\rs2569537\rs14158\rs143309, XPEHH score > 2); LDLR was selected in the HM populations when using the northern East Asian population as a reference (rs1433099, rs14158, rs2569537, rs5929, XPEHH score > 2), and LDLR achieves modulation of atherosclerosis by regulating cholesterol homeostasis (Li et al. 2021; Go and Mani 2012). ATP-binding cassette subfamily A member 1 (ABCA1) was selected in the HM populations when southern East Asians were used as a reference (rs2472377, rs2472508, rs10991415, rs4149310, rs4149311, rs2487052, XPEHH score > 1.5, and rs62566032, iHS score = 2.6302), and ABCA1 prevents cardiovascular disease by promoting the efflux of intracellular cholesterol and phospholipids, controlling the rate-limiting step of reverse cholesterol transport and by inhibiting inflammation and maintaining lipid homeostasis.
Conclusion
We provided genome-wide SNP data for HM-speaking Miaos in Guizhou Province and identified a distinct genetic branch of HM populations. Our results revealed a mixed pattern of HM people. Han-related genetic materials influenced Guizhou Miao people to varying degrees, and our studied Miaos were all influenced by a smaller stream of exotic genetic material than those in previous studies. Our results suggested the existence of genetic substructures among our studied Miaos, in which Han-related genes influenced the DYM and CJM significantly. At the same time, the LLM and NYM received more gene flow from the ancient people of Guangxi. Our results showed that NYM is more representative of the ancestry of the HM populations than Gaohuahua. We provided genetic evidence for a north–south East Asian mixing pattern in the HM populations and ALDER‐based admixture times further inferred the onset of north–south admixture in East Asians since Neolithic period and the timing of large-scale population mixing in the south occurred between 1100 and 2800 years ago. Finally, we identified hundreds of loci involved in the immune function disorders located on chromosome 6 and ADH1B, CETP, LDLR, and ABCA1 associated with alcohol and coronary heart disease.
Methods and materials
Sample collection, genotyping, and quality control
We collected blood samples from 67 unrelated individuals from four counties of Guizhou Province in southwestern China (CJM, NYM, DYM, and LLM, Fig. 1a). These samples were randomly selected, and written informed consent was provided before participating in this study. Their parents and grandparents were indigenous residents for at least three generations, and they had no consanguineous marriages. This study was reviewed and approved by the Medical Ethics Committee of Guizhou Medical University and North Sichuan Medical College. Besides, the procedure followed the recommendations of the Helsinki Declaration as revised in 2000 [67]. We genotyped these samples using the Affymetrix array containing approximately 699,896 SNPs. Quality control was performed by Plink 1.9 [68] with the option “-missing 0.05 -HWE 0.0001” to calculate the SNP call rate. SNPs that failed to reach the missing threshold of 0.05 and an alpha level of 0.0001 for HWE testing were removed.
Relatedness analysis
Before merging data, we initially performed KING2 [34] to check individual relationships by calculating kinship coefficients [32]. All unrelated samples were kept for the following analyses.
Data merging
We merged our newly generated data of 67 samples with the previously published Human HO and the 1240K datasets from the Allen Ancient DNA Resource (AADR) via the mergeit tool in the EIGENSOFT [69] with the strandcheck parameter setting of Yes to remove the AT/CG SNPs (https://reich.hms.harvard.edu/datasets). We generated three combined datasets that was used in subsequent analysis. The merged HO-based dataset with more present-day reference populations covered 95,342 SNPs variants, and the merged 1240K-based dataset covered 245,627 SNPs variants. We also merged other recently published ancient East Asian people in our analysis [8, 70, 71]. To obtain one high-density dataset, we combined our data with whole-genome sequencing data from Oceania, Asia, Europe, and Africa to form a standard merged WGS) dataset [62, 72] and used it to explore complex admixture models. The combined WGS dataset covered 679,602 SNPs. We used low-density SNP data in Fst, PCA and ADMIXTURE analyses and the high-density SNP data in f3-statistics and f4-statistics.
Principal component analysis
We performed three levels of PCA analyses, focusing on the relationship between East and Southeast Asian populations and the HM populations. We used the smartpca program of the EIGENSOFT package [69] with default parameters of numoutlieriter: 0 and lsqproject: YES to conduct PCA analysis and projected ancient samples onto the PCA axes generated by present-day samples. The PCA at the East and Southeast Asian level includes 46 individuals from 5 Tungusic and Mongolic-speaking populations in northern China, 35 individuals from 4 Tibeto-Burman-speaking populations, 9 Dai individuals from TK-speaking populations, and 33 Han Chinese from South China, 27 individuals from Japan, 19 individuals from 2 AN-speaking populations, 40 Ayta and Agta individuals from Southeast Asia, and 87 individuals from 6 HM-speaking populations [62, 72]. PCA at the Chinese level includes 410 individuals from 51 populations from ancient Northwest China, Northeast China, Middle Yellow River, Southeast China coastal and island, modern Altaic, ST, and HM-speaking populations [8, 70, 71].
Model-based ADMIXTURE analysis
We performed ADMIXTURE [73] based on the model-based maximum likelihood (ML) clustering algorithm to estimate the ancestry of individuals and determine the population structure. We trimmed SNPs of strong linkage disequilibrium with the parameters “-indep-pairwise 200 25 0.4” using Plink [68] before analyzing the admixture. We re-ran ADMIXTURE 100 times with default tenfold cross-validation (− CV = 10), with the number of ancestral populations from K = 2 to K = 10 in bootstrap sequences with different random seeds. We selected the best run based on the highest log-likelihood and used CV values to identify an “optimal” number of clusters. We then used AncestryPainter [74] to visualize the individual and population ancestry.
Admixture f 3-statistics and out-group f 3-statistics
We used the qp3Pop (version 435) package in ADMIXTOOLS software [75] to evaluate the possible admixture signatures in Guizhou Miao via the admixture-f3-statistics in the form f3(X, Y; studied Miao). Target populations with negative f3-values and Z-scores less than − 3 were regarded as mixed populations of two ancestral people related to X and Y. We then evaluated the genetic affinity between studied Miao and other reference populations via the out-group f3-statistics in the form f3(Reference, studied Miao; Mbuti).
f 4-Statistics
We employed the qpDstat program in ADMIXTOOLS to obtain f4-statistics with default parameters [75]. We calculated the f4-statistics in the form f4(Reference1, studied Miao; Reference2, Mbuti) to test whether the studied Miao shared more gene flow with the reference1 population compared with the reference2 population or shared more alleles with Reference2 compared with Reference1. A significant negative Z-score smaller than − 3 indicated that the studied Miao shared more alleles with reference2 populations compared with reference1 populations. The f4-statistics in the form f4(studied Miao1, studied Miao2; Reference, Mbuti) were used to test the genetic difference among the studied geographically different Miao.
MALDER
To explore the admixture signatures and fit the potential admixture model with admixture times, we used MALDER to estimate the generations since the admixture events occurred [76] with the default parameters.
qpAdm
We used ancient northern East Asians as the northern surrogate of the ancestral source and used southern East Asians as the southern surrogate of the ancestral source to model the formation of studied Miao people via qpAdm [77]. We used the following populations as out-groups: Mbuti, Iran_GanjDareh_N, Italy_North_Villabruna_HG, Mixe, Papuan, Onge, and Agta_Philippines.
FineSTRUCTURE analysis based on the sharing haplotypes
We phased 1049 genomes from 47 populations with the default parameters using the SHAPEIT software [78] as well as computed the sharing haplotypes using the ChromoPainterv2 software [79]. Fine-scale population structures were explored using fineSTRUCTURE (version 4.0) according to the co-ancestry matrix [79].
Haplotype-based admixture sources, proportions, and dates
We used ChromoPainter v2 [79] to estimate the copied vector, paint all targeted individuals with different ancestral surrogates, and used fastGLOBETROTTER to identify, describe, and date the admixture events with the recommended parameters [80].
Maximum likelihood tree
We generated the ML admixture graph, with the migration events from 1 to 10, through TreeMix to further explore the relationship between the newly studied Miao and modern southern Chinese and Southeast Asian populations [81].
IBD estimation and effective population size
We used the refined IBD [82] to estimate the pairwise IBD among individuals. We then calculated the average total IBD among populations based on the length of a single IBD less than or equal to one, ranging from one to five, five to ten, and over ten. We also used Plink to estimate the individual-level ROH) to show the status of the interbreeding coefficient [68]. IBDNe was used to estimate the effective population size among geographically diverse Miao people [83].
Natural selection indexes of XPEHH and iHS estimation
We used the R package of REHH [84] to compute the iHS and XPEHH. Both northern Han Chinese and southern Han Chinese were used as references in the estimation of XPEHH.
Availability of data and materials
The genome-wide variation data were collected from the public dataset of Allen Ancient DNA Resource (AADR) (https://reich.hms.harvard.edu/allen-ancient-dna-resource-aadr-downloadable-genotypes-present-day-and-ancient-dna-data). The new-generated allele frequency data have been submitted to the public database. (The link will be available when this work is accepted.)
Change history
30 March 2023
A Correction to this paper has been published: https://doi.org/10.1186/s40246-023-00477-5
References
Stoneking M, Delfin F. The human genetic history of East Asia: weaving a complex tapestry. Curr Biol. 2010;20(4):R188-193.
Tian JY, Li YC, Kong QP, Zhang YP. The origin and evolution history of East Asian populations from genetic perspectives. Yi Chuan. 2018;40(10):814–24.
Pinhasi R, Douka K. Before and after farming: the genetic structure of South China and Southeast Asia. Cell. 2021;184(14):3597–8.
Kutanan W, Liu D, Kampuansai J, Srikummool M, Srithawong S, Shoocongdej R, Sangkhano S, Ruangchai S, Pittayaporn P, Arias L, et al. Reconstructing the human genetic history of Mainland Southeast Asia: insights from genome-wide data from Thailand and Laos. Mol Biol Evol. 2021;38(8):3459–77.
Wang CC, Yeh HY, Popov AN, Zhang HQ, Matsumura H, Sirak K, Cheronet O, Kovalev A, Rohland N, Kim AM, et al. Genomic insights into the formation of human populations in East Asia. Nature. 2021;591(7850):413–9.
Chen P, He G, Xing H, Gao H, Wang M, Zhao M, Luo L, Wu J, Yu J, Han Y. Forensic characteristics and phylogenetic analysis of 23 Y-STR loci in the Miao population from Guizhou province, southwest China. Ann Hum Biol. 2019;46(1):84–7.
Chen J, He G, Ren Z, Wang Q, Liu Y, Zhang H, Yang M, Zhang H, Ji J, Zhao J, et al. Fine-scale population admixture landscape of Tai–Kadai-Speaking Maonan in Southwest China inferred from genome-wide SNP data. Front Genet. 2022;13: 815285.
Wang T, Wang W, Xie G, Li Z, Fan X, Yang Q, Wu X, Cao P, Liu Y, Yang R, et al. Human population history at the crossroads of East and Southeast Asia since 11,000 years ago. Cell. 2021;184(14):3829–41e3821.
Wu D, Dou J, Chai X, Bellis C, Wilm A, Shih CC, Soon WWJ, Bertin N, Lin CB, Khor CC, et al. Large-scale whole-genome sequencing of three diverse Asian populations in Singapore. Cell. 2019;179(3):736–49e715.
Leipe C, Long T, Sergusheva EA, Wagner M, Tarasov PE. Discontinuous spread of millet agriculture in eastern Asia and prehistoric population dynamics. Sci Adv. 2019;5(9):eaax6225.
Xia Z-Y, Yan S, Wang C-C, Zheng H-X, Zhang F, Liu Y-C, Yu G, Yu B-X, Shu L-L, Jin L. Inland-coastal bifurcation of southern East Asians revealed by Hmong–Mien genomic history; 2019.
Huang X, Xia Z-Y, Bin X, He G, Guo J, Lin C, Yin L, Zhao J, Ma Z, Ma F, et al. Genomic insights into the demographic history of Southern Chinese. Front Ecol Evol. 2020;6:66.
Zhang H, He G, Guo J, Ren Z, Zhang H, Wang Q, Ji J, Yang M, Huang J, Wang C-C. Genetic diversity, structure and forensic characteristics of Hmong–Mien-speaking Miao revealed by autosomal insertion/deletion markers. Mol Genet Genomics. 2019;294(6):1487–98.
Li H, Huang Y, Mustavich LF, Zhang F, Tan JZ, Wang LE, Qian J, Gao MH, Jin L. Y chromosomes of prehistoric people along the Yangtze River. Hum Genet. 2007;122(3–4):383–8.
Wen S-Q, Tong X-Z, Li H. Y-chromosome-based genetic pattern in East Asia affected by Neolithic transition. Quatern Int. 2016;426:50–5.
Laos R. Diaspora and the predicament of origins: Interrogating Hmong postcolonial history and identity. Hmong Stud J. 2007;8:66.
Fei XT. The pattern of diversity in unity of the Chinese nation. Beijing: Central Univ for Nationalities Press; 1999.
Nguanchoo V, Wangpakapattanawong P, Balslev H, Inta A. Exotic plants used by the Hmong in Thailand. Plants. 2019;8(11):66.
Dubost JM, Phakeovilay C, Her C, Bochaton A, Elliott E, Deharo E, Xayvue M, Bouamanivong S, Bourdy G. Hmong herbal medicine and herbalists in Lao PDR: pharmacopeia and knowledge transmission. J Ethnobiol Ethnomed. 2019;15(1):27.
Ali AH, Kang MS, Kaur K, Al Adhami S, Yuvienco CR. Review of Hmong-related health problems: a quick guide for healthcare providers. Cureus. 2020;12(8): e9808.
He G, Wang Z, Zou X, Wang M, Liu J, Wang S, Ye Z, Chen P, Hou Y. Tai–Kadai-speaking Gelao population: forensic features, genetic diversity and population structure. Forensic Sci Int Genet. 2019;40:e231–9.
Fan GY, An YR, Zhou YJ, Liu MN, Xiang J, Ye Y. Phylogenic analysis and forensic genetic characterization of Guizhou Miao tribes from 58 microareas via autosomal STR. Leg Med. 2020;47: 101737.
Tang J, Yang M, Wang X, Wang Q, Wang Q, Zhang H, Qian E, Zhang H, Ji J, Ren Z, et al. Genetic structure and forensic characterisation of 36 Y-chromosomal STR loci in Hmong–Mien-speaking Miao population. Ann Hum Biol. 2020;47(6):541–8.
Wen B, Li H, Gao S, Mao X, Gao Y, Li F, Zhang F, He Y, Dong Y, Zhang Y, et al. Genetic structure of Hmong–Mien speaking populations in East Asia as revealed by mtDNA lineages. Mol Biol Evol. 2005;22(3):725–34.
Han Y, He G, Gong S, Chen J, Jiang Z, Chen P. Genetic diversity and haplotype analysis of Guizhou Miao identified with 19 X-chromosomal short tandem repeats. Int J Legal Med. 2019;133(1):99–101.
Yang CH, Jin XY, Guo YX, Cui W, Chen C, Meng HT, Zhu BF. Genetic distribution analyses and population background explorations of Gansu Yugur and Guizhou Miao groups via InDel markers. J Hum Genet. 2019;64(6):535–43.
Yang M, He G, Ren Z, Wang Q, Liu Y, Zhang H, Zhang H, Chen J, Ji J, Zhao J, et al. Genomic insights into the unique demographic history and genetic structure of five Hmong–Mien-speaking Miao and Yao populations in Southwest China. Front Ecol Evol. 2022;10:66.
Luo T, Wang R, Wang CC. Inferring the population structure and admixture history of three Hmong–Mien-speaking Miao tribes from southwest China based on genome-wide SNP genotyping. Ann Hum Biol. 2021;48(5):418–29.
Wang Y, Zou X, Wang M, Yuan D, Yang L, Zeng Y, Cheng F, Tang R, He G. The genomic history of southwestern Chinese populations demonstrated massive population migration and admixture among proto-Hmong–Mien speakers and incoming migrants. Mol Genet Genomics. 2022;297(1):241–62.
Qian YP, Chu ZT, Dai Q, Wei CD, Chu JY, Tajima A, Horai S. Mitochondrial DNA polymorphisms in Yunnan nationalities in China. J Hum Genet. 2001;46(4):211–20.
Liu Y, Xie J, Wang M, Liu C, Zhu J, Zou X, Li W, Wang L, Leng C, Xu Q, et al. Genomic insights into the population history and biological adaptation of Southwestern Chinese Hmong–Mien people. Front Genet. 2021;12: 815160.
Roman YM, Lor K, Xiong T, Culhane-Pera K, Straka RJ. Gout prevalence in the Hmong: a prime example of health disparity and the role of community-based genetic research. Per Med. 2021;18(3):311–27.
Barone G, Staples CJ, Ganesh A, Patterson KW, Bryne DP, Myers KN, Patil AA, Eyers CE, Maslen S, Skehel JM, et al. Human CDK18 promotes replication stress signaling and genome stability. Nucleic Acids Res. 2016;44(18):8772–85.
Fan LL, Ding DB, Huang H, Chen YQ, Jin JY, Xia K, Xiang R. A de novo mutation of SMYD1 (p.F272L) is responsible for hypertrophic cardiomyopathy in a Chinese patient. Clin Chem Lab Med. 2019;57(4):532–9.
Liotta L, Lange S, Maurer HC, Olive KP, Braren R, Pfarr N, Burger S, Muckenhuber A, Jesinghaus M, Steiger K, et al. PALLD mutation in a European family conveys a stromal predisposition for familial pancreatic cancer. JCI Insight. 2021;6(8):66.
Sun H, Xu B, Meng Q, Yang Q, Chang X. PSORS1C1/CDSN is associated with ankylosing spondylitis. Joint Bone Spine. 2014;81(3):268–72.
Abbas Zadeh S, Mlitz V, Lachner J, Golabi B, Mildner M, Pammer J, Tschachler E, Eckhart L. Phylogenetic profiling and gene expression studies implicate a primary role of PSORS1C2 in terminal differentiation of keratinocytes. Exp Dermatol. 2017;26(4):352–8.
Conigliaro P, Triggianese P, De Martino E, Fonti GL, Chimenti MS, Sunzini F, Viola A, Canofari C, Perricone R. Challenges in the treatment of Rheumatoid Arthritis. Autoimmun Rev. 2019;18(7):706–13.
Lin S, Tian C, Li J, Liu B, Ma T, Chen K, Gong W, Wang JM, Huang J. Differential MUC22 expression by epigenetic alterations in human lung squamous cell carcinoma and adenocarcinoma. Oncol Rep. 2021;45(5):66.
Chen JB, Zhang J, Hu HZ, Xue M, Jin YJ. Polymorphisms of TGFB1, TLE4 and MUC22 are associated with childhood asthma in Chinese population. Allergol Immunopathol. 2017;45(5):432–8.
Mondal P, Gadad SS, Adhikari S, Ramos EI, Sen S, Prasad P, Das C. TCF19 and p53 regulate transcription of TIGAR and SCO2 in HCC for mitochondrial energy metabolism and stress adaptation. Faseb J. 2021;35(9): e21814.
Abd El-Aziz MM, Patel RJ, El-Ashry MF, Barragan I, Marcos I, Borrego S, Antiñolo G, Bhattacharya SS. Exclusion of four candidate genes, KHDRBS2, PTP4A1, KIAA1411 and OGFRL1, as causative of autosomal recessive retinitis pigmentosa. Ophthalmic Res. 2006;38(1):19–23.
Liu Q, Liu X, Zhao D, Ruan X, Su R, Shang X, Wang D, Yang C, Xue Y. Pseudogene ACTBP2 increases blood–brain barrier permeability by promoting KHDRBS2 transcription through recruitment of KMT2D/WDR5 in Abeta1-42 microenvironment. Cell Death Discov. 2021;7(1):142.
Zhang Y, Li G. A tumor suppressor DLC1: the functions and signal pathways. J Cell Physiol. 2020;235(6):4999–5007.
Guy J, Hearn T, Crosier M, Mudge J, Viggiano L, Koczan D, Thiesen HJ, Bailey JA, Horvath JE, Eichler EE, et al. Genomic sequence and transcriptional profile of the boundary between pericentromeric satellites and genes on human chromosome arm 10p. Genome Res. 2003;13(2):159–72.
De Franco E, Saint-Martin C, Brusgaard K, Knight Johnson AE, Aguilar-Bryan L, Bowman P, Arnoux JB, Larsen AR, Sanyoura M, Greeley SAW, et al. Update of variants identified in the pancreatic beta-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum Mutat. 2020;41(5):884–905.
Liu L, Liu H, Ning L, Li F. Rabbit SLC15A1, SLC7A1 and SLC1A1 genes are affected by site of digestion, stage of development and dietary protein content. Animal. 2019;13(2):326–32.
Tazelaar GHP, Dekker AM, van Vugt J, van der Spek RA, Westeneng HJ, Kool L, Kenna KP, van Rheenen W, Pulit SL, McLaughlin RL, et al. Association of NIPA1 repeat expansions with amyotrophic lateral sclerosis in a large international cohort. Neurobiol Aging. 2019;74:234e239–234e215.
Collins RL, Brand H, Karczewski KJ, Zhao X, Alfoldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, et al. A structural variation reference for medical and population genetics. Nature. 2020;581(7809):444–51.
Wang Q, Dhindsa RS, Carss K, Harper AR, Nag A, Tachmazidou I, Vitsios D, Deevi SVV, Mackay A, Muthas D, et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature. 2021;597(7877):527–32.
McQuillan MA, Zhang C, Tishkoff SA, Platt A. The importance of including ethnically diverse populations in studies of quantitative trait evolution. Curr Opin Genet Dev. 2020;62:30–5.
Chen J, Zheng H, Bei JX, Sun L, Jia WH, Li T, Zhang F, Seielstad M, Zeng YX, Zhang X, et al. Genetic structure of the Han Chinese population revealed by genome-wide SNP variation. Am J Hum Genet. 2009;85(6):775–85.
Pan Z, Xu S. Population genomics of East Asian ethnic groups. Hereditas. 2020;157(1):49.
Qian EF, Deng P, Huang MS, Ma Q, Zhao H, Li CX, Huang J, Jiang L. Genetic polymorphism of Y chromosome haplogroup D-M174 in East Asian populations. Fa Yi Xue Za Zhi. 2019;35(3):308–13.
Yao H, Wang M, Zou X, Li Y, Yang X, Li A, Yeh HY, Wang P, Wang Z, Bai J, et al. New insights into the fine-scale history of western-eastern admixture of the northwestern Chinese population in the Hexi Corridor via genome-wide genetic legacy. Mol Genet Genomics. 2021;296(3):631–51.
Zhou B, Wen S, Sun H, Zhang H, Shi R. Genetic affinity between Ningxia Hui and eastern Asian populations revealed by a set of InDel loci. R Soc Open Sci. 2020;7(1): 190358.
Xie M, Song F, Li J, Lang M, Luo H, Wang Z, Wu J, Li C, Tian C, Wang W, et al. Genetic substructure and forensic characteristics of Chinese Hui populations using 157 Y-SNPs and 27 Y-STRs. Forensic Sci Int Genet. 2019;41:11–8.
Song M, Wang Z, Zhang Y, Zhao C, Lang M, Xie M, Qian X, Wang M, Hou Y. Forensic characteristics and phylogenetic analysis of both Y-STR and Y-SNP in the Li and Han ethnic groups from Hainan Island of China. Forensic Sci Int Genet. 2019;39:e14–20.
Li L, Zou X, Zhang G, Wang H, Su Y, Wang M, He G. Population genetic analysis of Shaanxi male Han Chinese population reveals genetic differentiation and homogenization of East Asians. Mol Genet Genomic Med. 2020;8(5): e1209.
Consortium HP-AS, Abdulla MA, Ahmed I, Assawamakin A, Bhak J, Brahmachari SK, Calacal GC, Chaurasia A, Chen CH, Chen J, et al. Mapping human genetic diversity in Asia. Science. 2009;326(5959):1541–5.
Liu Y, Wang M, Chen P, Wang Z, Liu J, Yao L, Wang F, Tang R, Zou X, He G. Combined low-/high-density modern and ancient genome-wide data document genomic admixture history of high-altitude East Asians. Front Genet. 2021;12: 582357.
Choin J, Mendoza-Revilla J, Arauna LR, Cuadros-Espinoza S, Cassar O, Larena M, Ko AM, Harmant C, Laurent R, Verdu P, et al. Genomic insights into population history and biological adaptation in Oceania. Nature. 2021;592(7855):583–9.
Gittelman RM, Schraiber JG, Vernot B, Mikacenic C, Wurfel MM, Akey JM. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr Biol. 2016;26(24):3375–82.
Segurel L, Bon C. On the evolution of lactase persistence in humans. Annu Rev Genomics Hum Genet. 2017;18:297–319.
Eng MY, Luczak SE, Wall TL. ALDH2, ADH1B, and ADH1C genotypes in Asians: a literature review. Alcohol Res Health. 2007;30(1):22–7.
Nurmohamed NS, Ditmarsch M, Kastelein JJP. Cholesteryl ester transfer protein inhibitors: from high-density lipoprotein cholesterol to low-density lipoprotein cholesterol lowering agents? Cardiovasc Res. 2022;118(14):2919–31.
World Medical Association I: Declaration of Helsinki. Ethical principles for medical research involving human subjects. J Indian Med Assoc. 2009;107(6):403–5.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12): e190.
McColl H, Racimo F, Vinner L, Demeter F, Gakuhari T, Moreno-Mayar JV, van Driem G, Gram Wilken U, Seguin-Orlando A, de la Fuente CC, et al. The prehistoric peopling of Southeast Asia. Science. 2018;361(6397):88–92.
Ning C, Li T, Wang K, Zhang F, Li T, Wu X, Gao S, Zhang Q, Zhang H, Hudson MJ, et al. Ancient genomes from northern China suggest links between subsistence changes and human migration. Nat Commun. 2020;11(1):2700.
Bergstrom A, McCarthy SA, Hui R, Almarri MA, Ayub Q, Danecek P, Chen Y, Felkel S, Hallast P, Kamm J, et al. Insights into human genetic variation and population history from 929 diverse genomes. Science. 2020;367(6484):66.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64.
Feng Q, Lu D, Xu S. AncestryPainter: a graphic program for displaying ancestry composition of populations and individuals. Genomics Proteom Bioinform. 2018;16(5):382–5.
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D. Ancient admixture in human history. Genetics. 2012;192(3):1065–93.
Loh PR, Lipson M, Patterson N, Moorjani P, Pickrell JK, Reich D, Berger B. Inferring admixture histories of human populations using linkage disequilibrium. Genetics. 2013;193(4):1233–54.
Harney E, Patterson N, Reich D, Wakeley J. Assessing the performance of qpAdm: a statistical tool for studying population admixture. Genetics. 2021;217(4):66.
Browning BL, Tian X, Zhou Y, Browning SR. Fast two-stage phasing of large-scale sequence data. Am J Hum Genet. 2021;108(10):1880–90.
Lawson DJ, Hellenthal G, Myers S, Falush D. Inference of population structure using dense haplotype data. PLoS Genet. 2012;8(1): e1002453.
Hellenthal G, Busby GBJ, Band G, Wilson JF, Capelli C, Falush D, Myers S. A genetic atlas of human admixture history. Science. 2014;343(6172):747–51.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8(11): e1002967.
Browning BL, Browning SR. Improving the accuracy and efficiency of identity-by-descent detection in population data. Genetics. 2013;194(2):459–71.
Browning SR, Browning BL, Daviglus ML, Durazo-Arvizu RA, Schneiderman N, Kaplan RC, Laurie CC. Ancestry-specific recent effective population size in the Americas. PLoS Genet. 2018;14(5): e1007385.
Gautier M, Klassmann A, Vitalis R. rehh 2.0: a reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol Ecol Resour. 2017;17(1):78–90.
Acknowledgements
We thank Prof. Etienne Patin at Institut Pasteur for sharing genome-wide SNP data from the Island Southeast Asian and Oceanian populations. We also thank all volunteers participated this project.
Funding
This study was supported by the Guizhou Scientific Support Project, Qian Science Support (2022) General 280, the National Natural Science Foundation of China (NSFC 82202078), Guizhou Provincial Ordinary Colleges and Universities Young Scientific and Technological Talents Growth Project: Qianjiaohe KY Character [2021]157 and the Doctoral Foundation of Guizhou Medical University [2020]018.
Author information
Authors and Affiliations
Contributions
GH, MW, JW, and JX designed this study. GH, MW, JW, and LY wrote the manuscript. QS, YL, WW, SD, JW, ZW, BY, and DW collected the samples. JW, QS, JW, WW, YL, RT, JY, CL and SD conducted the experiment. JW, MW, GH, LY, and JX analyzed the data. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The authorship has been updated.
Supplementary Information
Additional file 1. Table S1.
Z-scores of admixture-f3 in the form f3(X, Y; Miao_Longli). Table S2. Z-scores of admixture-f3 in the form f3(X, Y; Miao_Duyun). Table S3. Z-scores of admixture-f3 in the form f3(X, Y; Miao_Congjiang). Table S4. Z-scores of admixture-f3 in the form f3(X, Y; Miao_Nayong). Table S5. Results of f4-statistics in the form f4(Northern Han, Four Miao populations; reference populations, Mbuti). Table S6. Results of f4-statistics in the form f4(Han, Four Miao populations; reference populations, Mbuti). Table S7. Results of f4-statistics in the form f4(Miao_Congjiang, Four Miao populations; reference populations, Mbuti). Table S8. Two-way admixture models for the formation of Miao people. Table S9. Three-way admixture models for the formation of Miao people. Table S10. MALDER results showed the admixture times of Miao people.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, G., Wang, J., Yang, L. et al. Genome-wide allele and haplotype-sharing patterns suggested one unique Hmong–Mein-related lineage and biological adaptation history in Southwest China. Hum Genomics 17, 3 (2023). https://doi.org/10.1186/s40246-023-00452-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-023-00452-0