
Abstract
Background
Microbial symbioses in marine invertebrates are commonplace. However, characterizations of invertebrate microbiomes are vastly outnumbered by those of vertebrates. Protists and fungi run the gamut of symbiosis, yet eukaryotic microbiome sequencing is rarely undertaken, with much of the focus on bacteria. To explore the importance of microscopic marine invertebrates as potential symbiont reservoirs, we used a phylogenetic-focused approach to analyze the host-associated eukaryotic microbiomes of 220 animal specimens spanning nine different animal phyla.
Results
Our data expanded the traditional host range of several microbial taxa and identified numerous undescribed lineages. A lack of comparable reference sequences resulted in several cryptic clades within the Apicomplexa and Ciliophora and emphasized the potential for microbial invertebrates to harbor novel protistan and fungal diversity.
Conclusions
Microscopic marine invertebrates, spanning a wide range of animal phyla, host various protist and fungal sequences and may therefore serve as a useful resource in the detection and characterization of undescribed symbioses.
Video Abstract
Similar content being viewed by others


Background
The ubiquity of single-celled protists and fungi in various environments, alongside their ecological and metabolic diversity, has facilitated their capacity for niche exploitation. Some species support vast ecosystems as photosynthetic primary producers, while others utilize varying forms of heterotrophy, such as phagotrophs and parasites, which link ecological networks and trophic scales [1]. Indeed, all eukaryotic groups contain parasites that have evolved to exploit a host [2, 3]. The Apicomplexa, for example, form an exclusively symbiotic phylum (many being harmful parasites), characterized by the presence of a morphological structure called the apical complex, which aids host cell penetration and the initiation of infection [4]. However, relationships between protists, fungi, and metazoan hosts span the entire range of symbiosis. At the other end of the spectrum, photosynthetic dinoflagellates of the Symbiodiniaceae are well-known mutualists in coral [5], lignocellulose-degrading metamonads have facilitated niche expansion and the subsequent success of termites [6], while leucocoprineous fungi form a (typically) vertically transmitted association with fungus-growing attine ants [7].
Nevertheless, microbiome research has most commonly focused exclusively on bacterial communities, despite the understanding that microbial eukaryotes, along with archaea and viruses, are likely contributing to the pool of interactions that the term defines [3]. The lack of eukaryotic data in microbiome marker gene surveys is mostly due to methodological limitations rather than a genuine absence of protists. The co-amplification of host DNA when targeting symbiotic microbial eukaryotes can often dwarf non-metazoan reads and nullify attempts to fully characterize the eukaryome. However, various approaches have been developed to mitigate this problem, and sequence characterization of the eukaryotic microbiome is a possibility [8, 9].
The majority of microbiome studies focus on vertebrate hosts which ultimately represent a minute proportion of animal diversity [10, 11]. The Arthropoda alone makes up ~80% of all known animal species [11]. Microscopic marine invertebrates remain highly underrepresented in even bacterial microbiome literature [12]. As a consequence, eukaryotic microbiomes of these animals are almost completely unknown, and even for larger, commercially relevant marine invertebrates, the data are scarce [13].
Most invertebrate phyla include species smaller than 1–2 mm and belong to either planktonic or meiofaunal communities [14]; their abundance and diversity raise the possibility that microscopic marine invertebrates may interact with microbial eukaryotes in various ways. Many stable, symbiotic, microeukaryote-invertebrate associations are well documented [15,16,17], but protists can also be found inside/associated with these animals because they are consumed in their diet [18]. Therefore, the classification of microbial eukaryotes as true symbionts or components of a host-associated microbiome may be difficult with marker gene analysis alone.
Here, in an attempt to characterize protistan and fungal diversity in over 200 microscopic marine invertebrates, we rely on phylogenetic reconstruction to identify taxa that fall within typically host-associated clades, mitigating potential overemphasis and misidentification of microorganisms in the diet as symbionts. We expected that these minute animals could either be too small to host microbial eukaryotes, in which case we would not find sequence variants that could be reliably identified as symbiotic (i.e., fall within our target taxa), or simply understudied as viable hosts, which would result in the detection of a large proportion of unidentified lineages.
Methods
Specimen collection
Microscopic invertebrate specimens were taken from a larger cohort of animals collected for bacterial microbiome analysis [12]. All specimens were isolated from one of three locations in British Columbia, Canada (Calvert Island, Quadra Island, and Vancouver) or Curaçao in the Dutch Caribbean, from July 2017 to January 2019. The majority of specimens were collected from either sediment, with a meiobenthic dredge (subtidal), or shovel (intertidal and subtidal); water column via horizontal and vertical plankton tows with a 64-μm mesh; or macroalgae, picked from rock pools. A small number of animals were sampled extemporaneously from other habitats, referred to as “other” in Fig. 1. The samples were taken back to the laboratory and stored at 4 °C. Animals were extracted from sediment and macroalgal samples with MgCl2 treatment [19] or the “bubble and blot” protocol [20], and specimens were isolated with Irwin Loops [21] under a dissecting microscope (Zeiss Stemi 508) within 24 h of arrival. All tools were sterilized with 10% bleach and 70% ethanol before use.

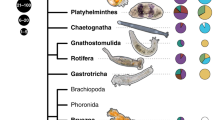
Overview of more than 200 specimens and visual evidence of symbiosis. a Sankey diagram showing the distribution of specimen locations, habitats, and phyla, with the number of specimens accompanying each factor. b Photos of representative specimens from each invertebrate phylum. c Unknown Apicomplexa shown in a Platyhelminthes specimen belonging to the family Koinocystididae. d Epibiont ciliates of the genus Rhabdostyla (black arrows) on an unknown species of a Syllis polychaete (Annelida). e Potential fungal structures on the dorsal side of a harpacticoid copepod. Image darkened to aid visibility
Prior to the preservation, specimens were transferred to droplets of sterile marine water, imaged on either a Zeiss Axioscope A1 or Leica DMIL microscope (British Columbia and Curaçao locations, respectively) with Axiocam 503 color or Sony a6000 cameras. Specimens were then assigned taxonomic groups following the World Register of Marine Species (WoRMS, https://www.marinespecies.org/index.php) and given a unique alphanumeric code. Recorded specimens were then washed in at least three successive transfers of sterile water and immediately frozen in 20 μL of sterile water at −20 °C until DNA extraction.
DNA extraction and library preparation
DNA was extracted using the DNeasy PowerSoil Kit (QIAGEN Gmbh) according to the recommended protocol and quantified with the dsDNA HS Assay Kit (Life Technologies) and a Qubit Fluorometer. Amplicon libraries were generated using a nested PCR comprising an initial amplification with non-metazoan specific (UNon-Met-PCR) primers (18S-EUK581-F: 5’-GTGCCAGCAGCCGCG-3’, 18S-EUK1134-R: 5’-TTTAAGTTTCAGCCTTGCG-3’) [22]. UNon-Met-PCR primers not only significantly reduce metazoan reads but perform as well or better than common universal primers when amplifying the V4 region from a range of microeukaryotic taxa [23]. PCRs were performed in total volumes of 20 μL using Phusion High-Fidelity PCR Master Mix (New England BioLabs) and 2–4 μL of template DNA. Thermal cycler settings were as follows: initial denaturation, 98 °C (30 s); 35 cycles at 98 °C (10 s), 51 °C (30 s), and 72 °C (60 s); final extension, 72 °C (10 min). Amplicons were purified with the QIAquick PCR Purification Kit (QIAGEN) and quantified with a Qubit fluorometer before being sent to CGEB – Integrated Microbiome Resource for the subsequent PCR using “fusion primers” (Illumina adaptors + indices + specific regions) targeting the V4 region of the 18S rRNA gene (E572F: 5’-CYGCGGTAATTCCAGCTC-3’, E1009R: 5’-AYGGTATCTRATCRTCTTYG-3’) [24]. Fusion primer PCRs were performed in duplicate with 2 μL of the initial UNon-Met-PCR reaction, and one reaction using a 1/10th template dilution (as detailed on https://github.com/LangilleLab/microbiome_helper/wiki/Microbiome-Amplicon-Sequencing-Workflow). Duplicate libraries were then pooled, cleaned, and normalized with the Just-a-Plate 96-well Normalization Kit (Charm Biotech), and sequenced using 2 x 300 bp reads and Illumina Miseq v3 chemistry.
Amplicon sequence variant (ASV) generation
Raw reads were initially trimmed with Cutadapt (v3.4) [25] to remove primers, before being processed in R [26] using the DADA2 package (v.1.14.1) [27]. Following the standard pipeline, trimmed reads were truncated based on quality profiles and filtered using the default parameters (maxN=0 and max EE=c(2,2)). Error rates were modeled on the first 100 million bases and “pseudo” pooling was implemented during sample inference to allow singletons, providing they appear in more than one library. Paired-end reads were then merged before chimera detection and taxonomic classification with the RDP Naïve Bayesian Classifier against the PR2 database (v.4.12.0) [28].
Preprocessing and filtering were done with the phyloseq package [29]. Libraries were removed if their relative abundance of metazoan reads was greater than 70%. Subsequently, all metazoan reads were removed from the remaining samples, as were non-eukaryotic sequences. Libraries with less than 1000 reads were discarded and any ASVs left with a read count of zero were also removed. To account for sequence variation across multiple copies of the 18S rRNA gene in individual protists, a phylogeny was reconstructed for all reads using IQTree (v.1.6.12) [30] and ASV sequences were subsequently grouped based on phyloseq’s tip_glom function (using Agnes hierarchical clustering and a tree height (h) of 0.05) [29]. The Sankey diagram, detailing specimen metadata of final library selection, was produced with the ggaluvial package in R [31].
Phylogenetics
To reconstruct potential symbiont phylogenies, ASVs above a minimum relative abundance threshold of 0.1% in any one library were selected according to broad taxa assigned by the ASV pipeline (e.g., Apicomplexa, Ciliophora, Fungi, etc.). Unassigned sequences were also included if BLAST results showed similarities to the taxon in question. A selection of diverse, near full-length, reference sequences was added to structure each phylogeny including the top five BLAST hits (for each ASV) in the NCBI nt database (blastn, e value threshold of 1e−25). All sequences were trimmed to a maximum length of 2000 bp prior to aligning. The multiple sequence alignment was produced using the mafft EINSi iterative alignment algorithm [32] and masked with trimal to remove sites with gaps in more than 90% of sequences or with a similarity score of less than 0.001 [33]. A maximum-likelihood phylogeny was reconstructed with IQtree, using a GTR+F+R7 substitution model and 1000 ultrafast bootstraps [34]. The unaligned fasta files were trimmed to remove sequences that were irrelevant and/or represented minor strain variations from the same studies, and alignment, masking and phylogenies were repeated.
To visualize phylogenies, IQtree output trees were imported in R, rooted with the treeio package [35], and plotted using ggtree [36] and ggtreeExtra [37]. Branch lengths were removed to improve visualization of topologies, and bar charts displaying ASV prevalence measures in specimens were produced with ggplot2 [38] and later added using Adobe Illustrator.
Results and discussion
Specimen overview
The final dataset contains 220 isolated specimens: 56.4% of those were isolated from Quadra Island in British Columbia, Canada, and just under half of all specimens were isolated from the sediment (49.5%) (Fig. 1a). These animals span nine invertebrate phyla: Annelida, Mollusca, Platyhelminthes, Chaetognatha, Kinorhyncha, Arthropoda, Nematoda, Xenacoelomorpha, and Cnidaria (Fig. 1a, b). The most highly represented phylum was Arthropoda (n=82), followed by Annelida (n=37), and Nematoda (n=28) (Fig. 1a).
Apicomplexa
A total of 52 ASVs were characterized as apicomplexans (Fig. 2, SFig. 1). The proportion of Apicomplexa-positive invertebrate specimens varied across phyla, ranging from just 12.5% of cnidarians (1/8) to 77.8% of chaetognaths (7/9) (Fig. 2a, STable 1). Notably, visual evidence of infection was observed in several platyhelminthes (which are typically less opaque than other animals in our dataset) (Fig. 1c); 42.3% of all platyhelminthes (11/26) contained at least one apicomplexan ASV (Fig. 2a, STable 1).

Apicomplexa prevalence and diversity. Maximum-likelihood phylogeny of all Apicomplexa ASVs, reference sequences, and BLAST hits using the GTR+F+R7 substitution model. Individual ASVs are indicated by white rectangles in the gray ring. Accompanying dots reflect the presence in each host phylum (colored accordingly). Black bars in the outer ring reflect the number of specimens associated with each ASV (on a log scale). Nodes are labeled to show UltraFast bootstrap support and taxonomic clades are annotated by color. a Percentage of individuals with at least one ASV in the tree. b Absolute number of distinct ASVs. c–g Highlighted lineages discussed in the text
The Annelida contained the largest number of distinct apicomplexan sequences (Fig. 2b, STable 1); indeed, it is assumed that annelids are the ancestral hosts of gregarines (a subgroup of the Apicomplexa), before these parasites spread to other marine invertebrates [39]. Forty of the 52 ASVs were found in association with only a single host phylum, suggesting that many apicomplexans may have a high degree of host specificity. Although no single ASV was detected in all host phyla, six ASVs were found in four or more phyla. The majority of ASVs (n=19) were spread across known Eugregarinorida diversity, which is also true of other amplicon surveys [40].
Our data also provide evidence for wider host ranges of many known apicomplexan clades. For instance, one cluster of ASVs (spread across all nine host phyla) was found within the insect-infecting Neogregarinorida (Fig. 2c, Sfig.1), branching sister to a clade containing Syncystis mirabilis isolated from a “water scorpion” (Nepa cincera, Insecta), but also found in dragonflies, and Quadruspinospora mexicana from the Mexican lubber grasshopper (Taeniopoda centurio) [41]. Typically, the Neogregarinorida are known for infecting terrestrial hosts [42,43,44,45], and they are often found in amplicon surveys of soils and marine sediment [40]. Our phylogeny does include BLAST hits of environmental sequences isolated from soil (and sediment) within this cluster; therefore, we should not discount the idea that Neogregarinorida sequences found in our marine invertebrates could be derived from ingested cysts in terrestrial run-off. However, only 14 of the 46 occurrences of Neogregarinorida ASVs were from animals isolated from the sediment.
Gregarines, in general, are thought to be mostly monoxenous, meaning their life cycle involves just one host organism. Although two sequences of the Neogregarina were found in multiple host phyla (four and eight phyla respectively), most gregarine ASVs (20/27) were phylum-specific, with the remaining five ASVs found in just two host phyla (Fig. 2). Notably, two of these dixenous gregarines were detected in nematodes; six apicomplexan ASVs were detected in eleven individual nematodes in total (Fig. 2a, STable 1), despite no prior record of nematode-infecting Apicomplexa in the literature.
In the Marosporida, two ASVs found in various host phyla branched with a group of mollusc parasites as sisters to the Rhytidocystidae. ASV_713, found in a single kinorhynch, mollusc, and chaetognath, is the sister group of the remainder of this mollusc-infecting clade, whereas ASV_168 (one of our most widespread apicomplexan lineages and found in all host phyla except Cnidaria and Xenacoelomorpha) was identical to Margolisiella islandica, a heart-infecting parasite of the Islandic scallop Chlamys islandica [45] (Fig. 2d, SFig. 1). We also found several ASVs in the Rhytidocystidae—some of which were isolated from molluscs, platyhelminthes, and arthropods and therefore found outside of their typically associated hosts (annelids) [46].
The most abundant ASV is a sister lineage of the blastogregarine Siedleckia cf. nematoides, a parasite of the bristle worm (Scoloplos armiger), but only shares 87.32% sequence identity (Fig. 2e). This lineage was found in all phyla with the exception of Cnidaria. Another of the more abundant ASV branches as a novel clade within Coccidia (the subclass to which Corallicolida, Adeleorina, and Eimeriorina belong; Fig. 2). This position within the coccidia has low support (Fig. 2f, SFig. 1). Again, these cryptic sequences share low sequence similarity scores to GenBank accessions (< 85% to environmental sequences), and there appears to be no specific host and/or environmental trait consistent across all associated specimens in our dataset.
Finally, we detected lineages closely related to fish-infecting Goussia (Fig. 2g), supporting the hypothesis that small invertebrates may serve as paratenic hosts for some species. Three distinct sequences were found in two annelids, three chaetognaths, and two molluscs, respectively. The sequence found in annelids shares over 96% identity to Goussia ameliae, which was isolated from the pyloric caecum of landlocked alewives (Alosa pseudoharengus) and is not known to infect other hosts [47]. The chaetognath isolate is slightly more dissimilar (94.0% sequence identity) to the highest scoring reference sequence (Goussia washuti from wild bluegill, Lepomis macrochirus) [48] and likely represents an undescribed species. Finally, the molluscan sequence is closest to that of Goussia pannonica (99.2% sequence identity) from the blue bream (Abramis syn. Ballerus sapa) [49].
Ciliophora
Contrary to apicomplexans, which are entirely host-restricted, most described ciliates are free-living. Consequently, the distribution of ciliate ASVs found in association with microscopic invertebrates does not match the taxonomic diversity and relative abundance predicted by environmental surveys of the group as a whole, but it does reflect what is known about lineages that are predominantly parasitic. We detected relatively few Spirotrichea (mostly oligotrichs and choreotrichs) and Litostomatea, which alone typically make up 70–90% of free-living ciliates in the marine environment [50]. ASVs belonging to these common ciliate groups are almost exclusively found in arthropods—animals at the largest end of the size range investigated here—and are most likely food. Indeed, nearly all ciliate ASVs were detected in arthropod specimens, 41 were detected in arthropods alone, and 50% (41/82) of all arthropod specimens contained at least one ciliate ASV (Fig. 3a, STable 1), resulting in the largest number of distinct ciliate ASVs compared to the other host phyla (Fig. 3b, STable 1). Half of all Cnidaria (4/8) also contained more than one ciliate ASV (although the total number of specimens analyzed was considerably lower). Conversely, ciliate ASVs were found in just 14.3% of Kinorhyncha and Nematoda (1/7 and 4/28, respectively) (Fig. 3a, STable 1).
The majority of ciliate ASVs in our dataset clearly belong to clades of known ecto- and endosymbionts, with a marked overrepresentation (compared to their relatively lower known diversity) of taxa from Suctoria and especially Apostomatia (epibiotic and parasitic subgroups of the Phyllopharyngea and Oligohymenophorea, respectively). Although most of these taxa are already known symbionts of marine invertebrates, they are generally documented in much larger specimens: adult echinoderms [51, 52], large cephalopods [53] and other molluscs [54], hydroids [55], and crustaceans [56]. Notably, associations between the suctorian genus, Ephelota, and small crustaceans (copepods) have also been reported [57, 58]. ASV_013, found in 18 host specimens across six phyla, formed a small cluster with other ASVs appearing as a sister group to species of Ephelota (Fig. 3c, SFig. 2). ASV_016 appears to be a member of the Rhabdostyla genus (Fig. 3d, SFig. 2), a well-known invertebrate epibiont and noted for their symbiotic relationship with annelids of the Salvatoria genus [59, 60]. We observed the same ciliate genus on a specimen of the Syllis polychaete (Fig. 1d). These epibionts are noted to sometimes result in the misidentification of some annelid species, given their morphological similarity to papillae [59].

Environmental and host-associated ciliate lineages. Maximum-likelihood phylogeny of all Ciliophora ASVs, reference sequences and best BLAST hits using the GTR+F+R7 substitution model. Individual ASVs indicated by white rectangles in a gray ring. Accompanying dots reflect the presence in each host phylum (colored accordingly). Black bars in the outer ring reflect the number of specimens associated with each ASV (on a log scale). Nodes are labeled to show UltraFast bootstrap support and taxonomic clades are annotated by color. Outer red clade labels show host-associated taxa (single line) and epibiotic symbionts (double line). a Percentage of individuals with at least one ASV in the tree. b Absolute number of distinct ASVs. c–e Highlighted lineages discussed in the text
Many of our ciliate ASVs were notably dissimilar from known reference sequences and often formed uninformative clusters. The most prevalent lineage (ASV_072) appeared in 24 animal specimens across seven of the nine phyla investigated. It branched in a weakly supported cluster with two, more spurious, ASVs and uncultured sequences from various marine environments, within the usually host-associated Oligohymenophorea (Fig. 3e, SFig. 2).
The detection of two Colpoda-like ASVs is unusual (Fig. 3, SFig. 2), given that the genus Colpoda is quintessentially terrestrial. Despite an old report (based on morphology) of a Colpoda commensal of the sea urchin (Toxopneustes variegatus) [61], and the existence of marine species within the class Colpodea [62, 63], these signals could also be soil-derived cysts ingested by the animals.
Fungi
We detected a large diversity of fungal ASVs associated with marine invertebrates (Fig. 4, SFig. 3) and observed fungal-like structures emanating from some specimens (Fig. 1e). Many putatively marine fungi are assigned to species that are also found in terrestrial habitats—this is particularly true of the Ascomycota and Basidiomycota [64, 65] which make up the majority of species in our dataset (Fig. 4, SFig. 3). This may be indicative of terrestrial contamination, for instance, if marine invertebrates ingested spores, but fungal phylogenies often show putatively marine fungi nested within clades of typically terrestrial lineages [65]. This led to a hypothesis that most marine fungi diversified before animals transitioned to a terrestrial lifestyle [65], but it has also been proposed that many truly marine isolates recently evolved from terrestrial ancestors [64, 66]. Some fungi are capable of tolerating vastly different habitats [67], so may inhabit both marine and terrestrial environments. Our data does, however, support the idea that habitat can influence species localization [65, 68, 69]. Eighty-two of the 121 unique ASVs were from specimens localized to a single habitat; 49 were from sediment.

Evidence of fungal ASVs in marine invertebrates. Maximum-likelihood phylogeny of all Fungi ASVs, reference sequences and best BLAST hits using the GTR+F+R7 substitution model. Individual ASVs indicated by white rectangles in a gray ring. Accompanying dots reflect the presence in each host phylum (colored accordingly). Black bars in the outer ring reflect the number of specimens associated with each ASV (on a log scale). Nodes are labeled to show UltraFast bootstrap support and taxonomic clades are annotated by color. a Percentage of individuals with at least one ASV in the tree. b Absolute number of distinct ASVs. c–e Highlighted lineages discussed in the text
The Ascomycota and Basidiomycota represented 56 and 54 ASVs, respectively. By comparison, we found just ten ASVs belonging to the Chytridiomycota, which typically dominate other nearshore and sediment samples [64]. Fungal ASVs were found in the majority of specimens in all phyla except Cnidaria (where they were found in only 37.5% of specimens; Fig. 4a, STable 1). Furthermore, all Platyhelminthes, Chaetognatha, and Kinorhyncha contain at least one fungal ASV (Fig. 4a, STable 1). Despite this, there were relatively few unique fungal ASVs in both Chaetognatha and Kinorhyncha (Fig. 4b, STable 1). Of the total 121 unique fungal ASVs, 74 were found to be specific to just one host phylum, 25 of these host-phylum-specific sequences were found in arthropods and 19 in platyhelminthes. Although there is ample evidence of coevolution between fungal species and plant hosts [70], each host phylum-specific lineage in our dataset only ever occurred in one or two specimens. In contrast, two fungal sequences were found in more than 50 specimens and previous reports have shown how a single fungal species can engage with multiple ant genera [71]. ASV_117, found in six different host phyla, branches sister to a sequence from the Chaetothyriales (Fig. 4c, SFig. 3): often referred to as “black yeasts” and sometimes implicated as potentially pathogenic [72, 73].
ASV_019 is identical to several Aspergillus and Penicillium spp. (Fig. 4d, SFig. 3), which are often co-isolated from marine samples. Aspergillus spp. infect a wide range of vertebrate hosts, including cetaceans [74], and can produce metabolites detrimental to the photophysiological performance of the coral symbiont, Symbiodinium [75]. Both fungal genera have been isolated from diseased coral and sponges [76, 77]. Phylogenetic and microsatellite-based analyses have been unable to distinguish between aquatic and terrestrial strains of some species [78], but marine sequences are common [79] and ASV_019 was found in all host phyla except Xenacoelomorpha (Fig. 4d).
Our most common fungal sequence was found in all phyla except in the Cnidaria and appears to be related to Cladosporium spp., along with several uncultured sequences obtained from the marine environment (Fig. 4e, SFig. 3). Notably, Cladosporium produces an enzyme that digests phytoplankton-derived organic matter, and its abundance has been linked to diatoms in the ocean [80], which are likely ingested by our hosts. Some fungi, like the Cryptomycota (of which we detected one ASV), are indeed parasites of protists and other fungi [80].
Other potential symbionts
Syndiniales
Marine alveolates (MALVs), or Syndiniales, are thought to be exclusively parasitic lineages that form a paraphyletic group outside of the core dinoflagellate clade [81]. Despite often being the most dominant microbial eukaryote in environmental marker gene surveys [82, 83], the vast majority of Syndiniales are still uncultured, their hosts are unknown, and they are represented only by environmental sequences [84]. There are currently only five characterized species spread across three of the five recognized SSU rRNA clades (Groups I, II, and IV); Groups III and V are inferred only from environmental sequencing and are yet to be observed. We found Syndiniales in all invertebrate phyla except Xenacoelomorpha, with most ASVs being found in arthropods and molluscs (Fig. 5a, SFig. 4). This reflects our current understanding of these protists: Syndiniales are thought to be small flagellates that dominate seawater samples and would therefore be found in filter feeders like molluscs, and two of the five known Syndiniales genera typically infect arthropods.

Other invertebrate symbiont taxa. Maximum-likelihood phylogeny of a marine alveolates (MALVs) and Perkinsea and d Stramenopiles ASVs, reference sequences and best BLAST hits using the GTR+F+R7 substitution model. Individual ASVs indicated by white rectangles in a gray ring. Accompanying dots reflect the presence in each host phylum (colored accordingly). Black bars in the outer ring reflect the number of specimens associated with each ASV (on a log scale). Nodes are labeled to show UltraFast bootstrap support and taxonomic clades are annotated by color. b, c, e, f Highlight lineages discussed in the text
Group II, in which the genus Amoebophrya is described, represents eight of our ASVs. Amoebophrya has been found in a wide range of dinoflagellate hosts and was recently estimated to represent eight different species [85]. Most of our Group II ASVs branched outside of the Amoebophrya clade. Of the further eight ASVs that fall within the Group IV Syndiniales, all but one were found in, but are not exclusive to, arthropods. Three of these sequences form orphan lineages that appear to have diverged prior to the clade containing both known Group IV genera: Syndinium (found in copepods and radiolarians [86]) and Hematodinium (found in crustaceans [87]). The most frequently detected Syndiniales sequence in our dataset (ASV_198) belongs to Group I (Fig. 5b, SFig. 4). However, it appears distinct from the two described genera within Group I: Ichthyodinium and Euduboscquella (syn. Dubosquella). Ichthyodinium spp. infect fish eggs [88] whereas Euduboscquella spp. are found in tintinnid ciliates [89]. ASV_198, found in the Annelida, Mollusca, Platyhelminthes, Kinorhyncha, Arthropoda, and Nematoda, branches sister to an environmental sequence from the Northwest Pacific Ocean. We did, however, find ASVs that shared much more recent ancestors with both Group I type species. Furthermore, most of our Syndiniales likely fall in Group I, which is also noted to be the dominant group in other zooplanktonic hosts [90].
Perkinsea
Perkinsea are alveolate parasites that can cause mass mortality events in fish, molluscs, and amphibians [91]. We detected a single perkinsid ASV in seven microscopic invertebrate specimens (two annelids, two molluscs, and three arthropods) (Fig. 5c, SFig. 4). Notably, this sequence is nearly 96% identical to that of Perkinsus qugwadi, a species that has caused sporadic mass mortality events in Yesso scallop (Patinopecten yessoensis) stocks in British Columbia [92]. Given that P. qugwadi shares ~96% sequence identity with some other Perkinsus species, it is likely we have detected a novel but related species. Although, notably, all of our associated specimens were isolated from the same location as previous P. qugwadi outbreaks (Quadra Island) [92, 93].
Stramenopiles
We generally found lower proportions of specimens with potentially host-associated stramenopile ASVs—ranging from zero kinorhynchs and cnidarians to 26.7% (4/15) of molluscs (Fig. 5d, SFig. 4). The most prevalent sequence was found in just seven individual hosts and appears to be related to the Labyrinthula genus (Fig. 5e, SFig. 4), a well-known pathogen of various seagrass species and also noted for its association with other algae and phytoplankton [94]. These seven specimens were not however isolated for macroalgae, but rather sediment.
Several studies describe pathologies caused by thraustochytrids in large molluscs [95], and it has been suggested that a specific pathological association may exist. However, most of our sequences within the Thraustochytrida were isolated from host phyla other than Mollusca; ASV_430 was found in one molluscan specimen (Solenogastres), but not exclusively (Fig. 5f, SFig. 4). Sequences from both of these Labyrinthulomycetes orders (Labyrinthulida, to which the abovementioned Labyrinthula belongs, and Thraustochytrida) could represent saprotrophic organisms; however, several invertebrate associations exist [96, 97]. The same can be said of the oomycetes, which are notable for their wide host and geographic range [98,99,100].
Conclusions
Our sampling uncovers a role for microscopic invertebrates in the ecology of microbial eukaryotes. We detected a wide range of diverse organisms, often expanding the host range of previously characterized microbes, and several clades that we could not identify using presently archived reference sequences. Thus, our data support the hypothesis that, despite their size, microscopic marine invertebrates still harbor protist and fungal symbionts—many of which are currently uncharacterized.
It should be noted that short regions of the 18S SSU gene alone limit our ability to distinguish unique species; even distinct protistan species can have almost identical 18S genes [101]. Utilizing the full length of the 18S gene sequence would be the next step in improving the taxonomic resolution of our potential symbionts [102].
Although we acknowledge the potential for terrestrial run-off, these works support the notion that protists and fungi should be included in analyses of invertebrate microbiomes, and highlight host taxa that could warrant further exploration.
Availability of data and materials
All sequence data are deposited in the NCBI Short Read Archive under the BioProject accession number PRJNA746569.
References
Weisse T, Anderson R, Arndt H, Calbet A, Hansen PJ, Montagnes DJS. Functional ecology of aquatic phagotrophic protists – concepts, limitations, and perspectives. Eur J Protistol. 2016;55:50–74.
Bass D, Stentiford GD, Littlewood DTJ, Hartikainen H. Diverse applications of environmental DNA methods in parasitology. Trends Parasitol. 2015;31:499–513.
del Campo J, Bass D, Keeling PJ. The eukaryome: diversity and role of microeukaryotic organisms associated with animal hosts. Funct Ecol. 2020;34:2045–54.
Dos Santos PN, Tosetti N, Koreny L, Waller RF, Soldati-Favre D. Evolution, composition, assembly, and function of the conoid in Apicomplexa. Trends Parasitol. 2020;36:688–704.
LaJeunesse TC, Parkinson JE, Gabrielson PW, Jeong HJ, Reimer JD, Voolstra CR, et al. Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr Biol. 2018;28:2570–80 e6.
Brune A. Symbiotic digestion of lignocellulose in termite guts. Nat Rev Microbiol. 2014;12:168–80.
Currie CR. A community of ants, fungi, and bacteria: a multilateral approach to studying symbiosis. Annu Rev Microbiol. 2001;55:357–80.
Minardi D, Ryder D, del Campo J, Garcia Fonseca V, Kerr R, Mortensen S, et al. Improved high throughput protocol for targeting eukaryotic symbionts in metazoan and eDNA samples. Mol Ecol Resour. 2022;22:664–78.
Zhong KX, Cho A, Deeg CM, Chan AM, Suttle CA. Revealing the composition of the eukaryotic microbiome of oyster spat by CRISPR-Cas Selective Amplicon Sequencing (CCSAS). Microbiome. 2021;9:230.
Petersen JM, Osvatic J. Microbiomes in natura: importance of invertebrates in understanding the natural variety of animal-microbe interactions. Msystems. 2018;3:e00179–17.
Zhang Z-Q. Animal biodiversity: an introduction to higher-level classification and taxonomic richness. Zootaxa. 2011;3148:39–55.
Boscaro V, Holt CC, Van Steenkiste NWL, Herranz M, Irwin NAT, Àlvarez-Campos P, et al. Microbiomes of microscopic marine invertebrates do not reveal signatures of phylosymbiosis. Nat Microbiol. 2022;7:810–9.
Holt CC, Bass D, Stentiford GD, van der Giezen M. Understanding the role of the shrimp gut microbiome in health and disease. J Invertebr Pathol. 2021;186:107387.
Giere O. Meiobenthology: the microscopic motile fauna of aquatic sediments. 2nd rev. and extended ed. Berlin: Springer; 2009.
Van Steenkiste NWL, Stephenson I, Herranz M, Husnik F, Keeling PJ, Leander BS. A new case of kleptoplasty in animals: marine flatworms steal functional plastids from diatoms. Sci Adv. 2019;5:eaaw4337.
Yarden O. Fungal association with sessile marine invertebrates. Front Microbiol. 2014;5:228.
Yellowlees D, Rees TAV, Leggat W. Metabolic interactions between algal symbionts and invertebrate hosts. Plant Cell Environ. 2008;31:679–94.
Majdi N, Schmid-Araya JM, Traunspurger W. Examining the diet of meiofauna: a critical review of methodologies. Hydrobiologia. 2020;847:2737–54.
Hall GS, Lasserre P, Hawksworth DL, CAB International, Unesco, International Union of Biological Sciences. Methods for the examination of organismal diversity in soils and sediments. Wallingford; New York: CAB International in association with United Nations Educational, Scientific, and Cultural Organization and the International Union of Biological Sciences; 1996.
Higgins RP, Thiel H. Introduction to the study of meiofauna. Washington, D.C: Smithsonian Institution Press; 1988.
Schram MD. Irwin Loops – a history and method of constructing homemade loops. Trans Kans Acad Sci. 1903;115:35–40.
Bower SM, Carnegie RB, Goh B, Jones SRM, Lowe GJ, Mak MWS. Preferential PCR amplification of parasitic protistan small subunit rDNA from metazoan tissues. J Eukaryot Microbiol. 2004;51:325–32.
del Campo J, Pons MJ, Herranz M, Wakeman KC, del Valle J, Vermeij MJA, et al. Validation of a universal set of primers to study animal-associated microeukaryotic communities. Environ Microbiol. 2019;21:3855–61.
Comeau AM, Li WKW, Tremblay J-É, Carmack EC, Lovejoy C. Arctic Ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS One. 2011;6:e27492.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10.
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2021.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Brunson J. ggalluvial: layered grammar for alluvial plots. J Open Source Softw. 2020;5:2017.
Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–8.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35:518–22.
Wang L-G, Lam TT-Y, Xu S, Dai Z, Zhou L, Feng T, et al. Treeio: An R package for phylogenetic tree input and output with richly annotated and associated data. Mol Biol Evol. 2020;37:599–603.
Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8:28–36.
Xu S, Dai Z, Guo P, Fu X, Liu S, Zhou L, et al. ggtreeExtra: Compact visualization of richly annotated phylogenetic data. Mol Biol Evol. 2021;38:4039–42.
Wickham H. ggplot2: elegant graphics for data analysis. 2nd ed. Cham: Springer International Publishing: Imprint: Springer; 2016.
Cox FEG. The evolutionary expansion of the Sporozoa. Int J Parasitol. 1994;24:1301–16.
del Campo J, Heger TJ, Rodríguez-Martínez R, Worden AZ, Richards TA, Massana R, et al. Assessing the diversity and distribution of apicomplexans in host and free-living environments using high-throughput amplicon data and a phylogenetically informed reference framework. Front Microbiol. 2019;10:2373.
Medina-Durán JH, Mayén-Estrada R, Mariño-Pérez R, Song H. Morphology and phylogenetic position of two new gregarine species (Apicomplexa: Eugregarinorida) parasitizing the lubber grasshopper Taeniopoda centurio (Drury, 1770) (Insecta: Orthoptera: Romaleidae) in Mexico. J Eukaryot Microbiol. 2020;67:4–17.
Alarcón ME, Huang C-G, Tsai Y-S, Chen W-J, Dubey AK, Wu W-J. Life cycle and morphology of Steinina ctenocephali (Ross 1909) comb. nov. (Eugregarinorida: Actinocephalidae), a gregarine of Ctenocephalides felis (Siphonaptera: Pulicidae) in Taiwan. Zool Stud. 2011;50:7663–772.
Field SG, Michiels NK. Parasitism and growth in the earthworm Lumbricus terrestris: fitness costs of the gregarine parasite Monocystis sp. Parasitology. 2005;130:397–403.
Plischuk S, Meeus I, Smagghe G, Lange CE. Apicystis bombi (Apicomplexa: Neogregarinorida) parasitizing Apis mellifera and Bombus terrestris (Hymenoptera: Apidae) in Argentina. Environ Microbiol Rep. 2011;3:565–8.
Kristmundsson Á, Helgason S, Bambir SH, Eydal M, Freeman MA. Margolisiella islandica sp. nov. (Apicomplexa: Eimeridae) infecting Iceland scallop Chlamys islandica (Müller, 1776) in Icelandic waters. J Invertebr Pathol. 2011;108:139–46.
Rueckert S, Leander BS. Phylogenetic position and description of Rhytidocystis cyamus sp. n. (Apicomplexa, Rhytidocystidae): a novel intestinal parasite of the north-eastern Pacific ‘stink worm’ (Polychaeta, Opheliidae, Travisia pupa). Marine Biodiver. 2009;39:227–34.
Lovy J, Friend SE. Intestinal coccidiosis of anadromous and landlocked alewives, Alosa pseudoharengus, caused by Goussia ameliae n. sp. and G. alosii n. sp. (Apicomplexa: Eimeriidae). Int J Parasitol. 2015;4:159–70.
Lovy J, Friend SE, Lewis NL. Seasonal intestinal coccidiosis in wild bluegill Lepomis macrochirus is associated with a spring bacterial epizootic. J Fish Dis. 2019;42:1697–711.
Rosenthal BM, Dunams-Morel D, Ostoros G, Molnár K. Coccidian parasites of fish encompass profound phylogenetic diversity and gave rise to each of the major parasitic groups in terrestrial vertebrates. Infect Genet Evol. 2016;40:219–27.
Boscaro V, Santoferrara LF, Zhang Q, Gentekaki E, Syberg-Olsen MJ, del Campo J, et al. EukRef-Ciliophora: a manually curated, phylogeny-based database of small subunit rRNA gene sequences of ciliates. Environ Microbiol. 2018;20:2218–30.
Lynn DH, Strüder-Kypke M. Scuticociliate endosymbionts of echinoids (phylum Echinodermata): phylogenetic relationships among species in the genera Entodiscus, Plagiopyliella, Thyrophylax, and Entorhipidium (phylum Ciliophora). J Parasitol. 2005;91:1190–9.
Lynn DH, Strüder-Kypke M. Phylogenetic position of Licnophora, Lechriopyla, and Schizocaryum, three unusual ciliates (phylum Ciliophora) endosymbiotic in echinoderms (phylum Echinodermata). J Eukaryot Microbiol. 2002;49:460–8.
Souidenne D, Florent I, Dellinger M, Justine JL, Romdhane MS, Furuya H, et al. Diversity of apostome ciliates, Chromidina spp. (Oligohymenophorea, Opalinopsidae), parasites of cephalopods of the Mediterranean Sea. Parasite. 2016;23:33.
Dias RJP, D’Ávila S, D’Agosto M. First record of epibionts peritrichids and suctorians (Protozoa, Ciliophora) on Pomacea lineata (Spix, 1827). Braz Arch Biol Technol. 2006;49:807–12.
Tazioli S, Di Camillo CG. Ecological and morphological characteristics of Ephelota gemmipara (Ciliophora, Suctoria), epibiontic on Eudendrium racemosum (Cnidaria, Hydrozoa) from the Adriatic Sea. Eur J Protistol. 2013;49:590–9.
Fernandez-Leborans G, Tato-Porto ML. A review of the species of protozoan epibionts on crustaceans. II. Suctorian ciliates. Crustaceana. 2000;73:1205–37.
Purushothaman A, Dovgal I, Francis SV, Padmakumar KB. Observation of a suctorian ciliate Ephelota coronata on the calanoid copepod Pontella spinipes in the southeastern Arabian Sea. Symbiosis. 2020;81:321–7.
Purushothaman J, Bhowal A, Siddique A, Francis SV, Raghunathan C. A report on epibionts and new record of two ciliates Ephelota plana and Ephelota gigantea in the coastal waters of Bay of Bengal, Northern Indian Ocean. Symbiosis. 2020;80:217–30.
Álvarez-Campos P, Fernández-Leborans G, Verdes A, San Martín G, Martin D, Riesgo A. The tag-along friendship: epibiotic protozoans and syllid polychaetes. Implications for the taxonomy of Syllidae (Annelida), and description of three new species of Rhabdostyla and Cothurnia (Ciliophora, Peritrichia). Zool J Linnean Soc. 2014;172:265–81.
Lu B, Shen Z, Zhang Q, Hu X, Warren A, Song W. Morphology and molecular analyses of four epibiotic peritrichs on crustacean and polychaete hosts, including descriptions of two new species (Ciliophora, Peritrichia). Eur J Protistol. 2020;73:125670.
Powers PBA. Studies on the ciliates from sea urchins: I. General taxonomy. Biol Bull. 1933;65:106–21.
Dunthorn M, Eppinger M, Schwarz MVJ, Schweikert M, Boenigk J, Katz LA, et al. Phylogenetic placement of the Cyrtolophosididae Stokes, 1888 (Ciliophora; Colpodea) and neotypification of Aristerostoma marinum Kahl, 1931. Int J Syst Evol Microbiol. 2009;59:167–80.
Dunthorn M, Otto J, Berger SA, Stamatakis A, Mahé F, Romac S, et al. Placing environmental next-generation sequencing amplicons from microbial eukaryotes into a phylogenetic context. Mol Biol Evol. 2014;31:993–1009.
Amend A, Burgaud G, Cunliffe M, Edgcomb VP, Ettinger CL, Gutiérrez MH, et al. Fungi in the marine environment: open questions and unsolved problems. mBio. 2019;10:e01189–18.
Gladfelter AS, James TY, Amend AS. Marine fungi. Curr Biol. 2019;29:R191–5.
Schoch CL, Crous PW, Groenewald JZ, Boehm EWA, Burgess TI, de Gruyter J, et al. A class-wide phylogenetic assessment of Dothideomycetes. Stud Mycol. 2009;64:1–15.
Velez P, Alejandri-Ramírez ND, González MC, Estrada KJ, Sanchez-Flores A, Dinkova TD. Comparative transcriptome analysis of the cosmopolitan marine fungus Corollospora maritima under two physiological conditions. G3 Genes, Genomes, Genetics. 2015;5:1805–14.
Panzer K, Yilmaz P, Weiß M, Reich L, Richter M, Wiese J, et al. Identification of habitat-specific biomes of aquatic fungal communities using a comprehensive nearly full-length 18S rRNA dataset enriched with contextual data. PLoS One. 2015;10:e0134377.
Wurzbacher C, Warthmann N, Bourne E, Attermeyer K, Allgaier M, Powell JR, et al. High habitat-specificity in fungal communities in oligo-mesotrophic, temperate Lake Stechlin (North-East Germany). MycoKeys. 2016;16:17–44.
Lim SJ, Bordenstein SR. An introduction to phylosymbiosis. Proc Biol Sci. 2020;287:20192900.
Mikheyev AS, Mueller UG, Abbot P. Cryptic sex and many-to-one coevolution in the fungus-growing ant symbiosis. Proc Natl Acad Sci U S A. 2006;103:10702–6.
Teixeira MM, Moreno LF, Stielow BJ, Muszewska A, Hainaut M, Gonzaga L, et al. Exploring the genomic diversity of black yeasts and relatives (Chaetothyriales, Ascomycota). Stud Mycol. 2017;86:1–28.
Van Dover CL, Ward ME, Scott JL, Underdown J, Anderson B, Gustafson C, et al. A fungal epizootic in mussels at a deep-sea hydrothermal vent. Mar Ecol. 2007;28:54–62.
Seyedmousavi S, Guillot J, Arné P, de Hoog GS, Mouton JW, Melchers WJG, et al. Aspergillus and aspergilloses in wild and domestic animals: a global health concern with parallels to human disease. Med Mycol. 2015;53:765–97.
Hayashi A, Crombie A, Lacey E, Richardson AJ, Vuong D, Piggott AM, et al. Aspergillus sydowii marine fungal bloom in Australian coastal waters, its metabolites and potential impact on Symbiodinium dinoflagellates. Marine Drugs. 2016;14:59.
Greco G, Di Piazza S, Gallus L, Amaroli A, Pozzolini M, Ferrando S, et al. First identification of a fatal fungal infection of the marine sponge Chondrosia reniformis by Aspergillus tubingensis. Dis Aquat Org. 2019;135:227–39.
Soler-Hurtado MM, Sandoval-Sierra JV, Machordom A, Diéguez-Uribeondo J. Aspergillus sydowii and other potential fungal pathogens in gorgonian octocorals of the Ecuadorian Pacific. PLoS One. 2016;11:e0165992.
Zuluaga-Montero A, Ramírez-Camejo L, Rauscher J, Bayman P. Marine isolates of Aspergillus flavus: Denizens of the deep or lost at sea? Fungal Ecol. 2010;3:386–91.
Richards TA, Jones MDM, Leonard G, Bass D. Marine fungi: their ecology and molecular diversity. Annu Rev Mar Sci. 2012;4:495–522.
Letcher PM, Powell MJ. A taxonomic summary and revision of Rozella (Cryptomycota). IMA Fungus. 2018;9:383–99.
Strassert JFH, Karnkowska A, Hehenberger E, del Campo J, Kolisko M, Okamoto N, et al. Single cell genomics of uncultured marine alveolates shows paraphyly of basal dinoflagellates. ISME J. 2018;12:304–8.
Clarke LJ, Bestley S, Bissett A, Deagle BE. A globally distributed Syndiniales parasite dominates the Southern Ocean micro-eukaryote community near the sea-ice edge. ISME J. 2019;13:734–7.
de Vargas C, Audic S, Henry N, Decelle J, Mahé F, Logares R, et al. Eukaryotic plankton diversity in the sunlit ocean. Science. 2015;348:1261605.
Guillou L, Viprey M, Chambouvet A, Welsh RM, Kirkham AR, Massana R, et al. Widespread occurrence and genetic diversity of marine parasitoids belonging to Syndiniales (Alveolata). Environ Microbiol. 2008;10:3349–65.
Cai R, Kayal E, Alves-de-Souza C, Bigeard E, Corre E, Jeanthon C, et al. Cryptic species in the parasitic Amoebophrya species complex revealed by a polyphasic approach. Sci Rep. 2020;10:2531.
Skovgaard A, Massana R, Balagué V, Saiz E. Phylogenetic position of the copepod-infesting parasite Syndinium turbo (Dinoflagellata, Syndinea). Protist. 2005;156:413–23.
Small HJ. Advances in our understanding of the global diversity and distribution of Hematodinium spp. – significant pathogens of commercially exploited crustaceans. J Invertebr Pathol. 2012;110:234–46.
Gleason FH, Nagarkar M, Chambouvet A, Guillou L. A review of the characteristics of the dinoflagellate parasite Ichthyodinium chabelardi and its potential effect on fin fish populations. Mar Freshw Res. 2019;70:1307.
Coats DW, Bachvaroff TR, Delwiche CF. Revision of the family Duboscquellidae with description of Euduboscquella crenulata n. gen., n. sp. (Dinoflagellata, Syndinea), an intracellular parasite of the ciliate Favella panamensis Kofoid & Campbell. J Eukaryot Microbiol. 2012;59:1–11.
Zamora-Terol S, Novotny A, Winder M. Molecular evidence of host-parasite interactions between zooplankton and Syndiniales. Aquat Ecol. 2021;55:125–34.
Itoïz S, Metz S, Derelle E, Reñé A, Garcés E, Bass D, et al. Emerging parasitic protists: the case of Perkinsea. Front Microbiol. 2022;12:735815.
Itoh N, Meyer GR, Tabata A, Lowe G, Abbott CL, Johnson SC. Rediscovery of the Yesso scallop pathogen Perkinsus qugwadi in Canada, and development of PCR tests. Dis Aquat Org. 2013;104:83–91.
Bower SM, Blackbourn J, Meyer GR. Distribution, prevalence, and pathogenicity of the protozoan Perkinsus qugwadi in Japanese scallops, Patinopecten yessoensis, cultured in British Columbia, Canada. Can J Zool. 1998;76:954–9.
Sullivan BK, Sherman TD, Damare VS, Lilje O, Gleason FH. Potential roles of Labyrinthula spp. in global seagrass population declines. Fungal Ecol. 2013;6:328–38.
Raghukumar S. Ecology of the marine protists, the Labyrinthulomycetes (Thraustochytrids and Labyrinthulids). Eur J Protistol. 2002;38:127–45.
Siboni N, Rasoulouniriana D, Ben-Dov E, Kramarsky-Winter E, Sivan A, Loya Y, et al. Stramenopile microorganisms associated with the massive coral Favia sp. J Eukaryot Microbiol. 2010;57:236–44.
Stokes NA, Ragone Calvo LM, Reece KS, Burreson EM. Molecular diagnostics, field validation, and phylogenetic analysis of Quahog Parasite Unknown (QPX), a pathogen of the hard clam Mercenaria mercenaria. Dis Aquat Org. 2002;52:233–47.
Holt C, Foster R, Daniels CL, van der Giezen M, Feist SW, Stentiford GD, et al. Halioticida noduliformans infection in eggs of lobster (Homarus gammarus) reveals its generalist parasitic strategy in marine invertebrates. J Invertebr Pathol. 2018;154:109–16.
Matthews E, Ellison A, Cable J. Saprolegnia parasitica zoospore activity and host survival indicates isolate variation in host preference. Fungal Biol. 2021;125:260–8.
Vilela R, Taylor JW, Walker ED, Mendoza L. Lagenidium giganteum pathogenicity in mammals. Emerg Infect Dis. 2015;21:290–7.
Boisard J, Duvernois-Berthet E, Duval L, Schrével J, Guillou L, Labat A, et al. Marine gregarine genomes reveal the breadth of apicomplexan diversity with a partially conserved glideosome machinery. BMC Genomics. 2021;23:485.
Jamy M, Foster R, Barbera P, Czech L, Kozlov A, Stamatakis A, et al. Long-read metabarcoding of the eukaryotic rDNA operon to phylogenetically and taxonomically resolve environmental diversity. Mol Ecol Resour. 2020;20:429–43.
Acknowledgements
The authors wish to thank the Hakai Institute and the Caribbean Research and Management of Biodiversity Institute (CARMABI) and their helpful staff (in particular Rebecca Piercey, Neha Acharya-Patel, Carly Janusson, and Carolyn Prentice from Hakai for assistance with DNA extractions).
Funding
The project was funded by the Hakai Institute (Tula Foundation) Project Grant (recipients: PJK & BSL) and the Natural Sciences and Engineering Research Council (NSERC) Canadian Graduate Scholarship (recipient: NATI).
Author information
Authors and Affiliations
Contributions
The project was conceived and planned by CCH, VB, NWLVS, MH, BSL, and PJK. BSL and PJK are the senior authors and provided supervision and funding. VB, NWLVS, MH, and NATI performed most of the fieldwork. VM and GB conducted most of the laboratory work. NWLVS and MH identified the animals. CCH conducted the analyses and wrote the original draft. The authors edited and commented on the final manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Number of Apicomplexa, Ciliophora and Fungi ASVs in each host phylum. n.pos = number of specimens positive for microeukaryote in question, spec.total = total number of specimens, prop.pos = proportion of positive specimens (%), n.uniq.ASVs = number of unique ASVs per host phylum.
Additional file 2: Supplementary Figure 1.
Apicomplexa phylogeny with sequence labels and UltraFast bootstrap support values. Maximum-likelihood phylogeny of all Apicomplexa ASVs, reference sequences, and BLAST hits using the GTR+F+R7 substitution model. Individual ASVs indicated by bold tip labels and white rectangles in grey ring. Accompanying dots reflect presence in each host phylum (coloured accordingly).
Additional file 3: Supplementary Figure 2.
Ciliophora phylogeny with sequence labels and UltraFast bootstrap support values. Maximum-likelihood phylogeny of all Ciliophora ASVs, reference sequences, and BLAST hits using the GTR+F+R7 substitution model. Individual ASVs indicated by bold tip labels and white rectangles in grey ring. Accompanying dots reflect presence in each host phylum (coloured accordingly).
Additional file 4: Supplementary Figure 3.
Fungi phylogeny with sequence labels and UltraFast bootstrap support values. Maximum-likelihood phylogeny of all Fungi ASVs, reference sequences, and BLAST hits using the GTR+F+R7 substitution model. Individual ASVs indicated by bold tip labels and white rectangles in grey ring. Accompanying dots reflect presence in each host phylum (coloured accordingly).
Additional file 5: Supplementary Figure 4.
Marine alveolate (top) and Stramenopiles (bottom) phylogenies with sequence labels and UltraFast bootstrap support values. Maximum-likelihood phylogeny of all MALV and host-associated Stramenopiles ASVs, reference sequences, and BLAST hits using the GTR+F+R7 substitution model. Individual ASVs indicated by bold tip labels and white rectangles in grey ring. Accompanying dots reflect presence in each host phylum (coloured accordingly).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Holt, C.C., Boscaro, V., Van Steenkiste, N.W.L. et al. Microscopic marine invertebrates are reservoirs for cryptic and diverse protists and fungi. Microbiome 10, 161 (2022). https://doi.org/10.1186/s40168-022-01363-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-022-01363-3