
Abstract
The renin-angiotensin system (RAS) was classically considered a circulating hormonal system that regulates blood pressure. However, different tissues and organs, including the brain, have a local paracrine RAS. Mutual regulation between the dopaminergic system and RAS has been observed in several tissues. Dysregulation of these interactions leads to renal and cardiovascular diseases, as well as progression of dopaminergic neuron degeneration in a major brain center of dopamine/angiotensin interaction such as the nigrostriatal system. A decrease in the dopaminergic function induces upregulation of the angiotensin type-1 (AT1) receptor activity, leading to recovery of dopamine levels. However, AT1 receptor overactivity in dopaminergic neurons and microglial cells upregulates the cellular NADPH-oxidase-superoxide axis and Ca2+ release, which mediate several key events in oxidative stress, neuroinflammation, and α-synuclein aggregation, involved in Parkinson's disease (PD) pathogenesis. An intraneuronal antioxidative/anti-inflammatory RAS counteracts the effects of the pro-oxidative AT1 receptor overactivity. Consistent with this, an imbalance in RAS activity towards the pro-oxidative/pro-inflammatory AT1 receptor axis has been observed in the substantia nigra and striatum of several animal models of high vulnerability to dopaminergic degeneration. Interestingly, autoantibodies against angiotensin-converting enzyme 2 and AT1 receptors are increased in PD models and PD patients and contribute to blood–brain barrier (BBB) dysregulation and nigrostriatal pro-inflammatory RAS upregulation. Therapeutic strategies addressed to the modulation of brain RAS, by AT1 receptor blockers (ARBs) and/or activation of the antioxidative axis (AT2, Mas receptors), may be neuroprotective for individuals with a high risk of developing PD or in prodromal stages of PD to reduce progression of the disease.
Similar content being viewed by others

Background
The renin-angiotensin system (RAS) was identified for the first time by Tigerstedt and Bergman (1898) [1] in the rabbit’s kidney. Phylogenetically, the RAS is a very old hormone system, present in primitive vertebrates such as the lamprey, playing a key role in the adaptation from aquatic to terrestrial life [2, 3] and the continuation of life in little salt ecosystems [4]. This was related to the major role of RAS, as a circulating hormonal system, in the regulation of blood pressure and sodium and water homeostasis, which was the classical function associated with RAS for decades. Then, local or paracrine RAS were observed in different tissues and organs, including the brain. In peripheral tissues, although both the circulating RAS and the local tissue RAS may act together, the circulating RAS appears less important than the local RAS in the RAS tissue effects [5].
Dopamine was synthesized in 1910 [6], and dopamine deficits in patients with Parkinson's disease (PD) were initially observed by Ehringer and Hornykiewicz (1960) [7]. Later, the role of dopamine in peripheral organs, particularly in the renal and cardiovascular systems, was identified [8]. More recently, different functions of dopamine in peripheral organs and tissues have been revealed, including regulation of blood pressure, sodium and water homeostasis, gut motility, respiration, and immune responses [9, 10]. In addition to brain cells, dopamine D1-like and D2-like receptor subtypes have been observed in peripheral tissues such as the blood vessels, kidney, heart, adrenal gland, gastrointestinal tract sympathetic nerve terminals, and almost all immune cell subpopulations [11, 12].
Interestingly, important functional interactions between the local dopaminergic and angiotensin systems have been observed in several peripheral organs. Dysregulation of the interactions between both systems, such as dysregulation of the expression or dimerization between dopamine receptors and angiotensin receptors, results in renal degenerative diseases and hypertension [13, 14]. Over the last decades, similar interactions between the dopaminergic system and the local RAS have been observed in the brain. Several studies from our research group and others have revealed a major role of the dysregulation of dopamine-RAS interactions in brain diseases, particularly in dopaminergic degeneration in PD, as detailed in the following sections.
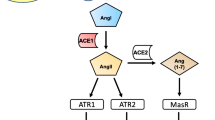
Angiotensin II (AngII) is the main RAS effector peptide, produced from the precursor protein angiotensinogen through sequential cleavages by the enzymes renin and angiotensin-converting enzyme (ACE, ACE1) (Fig. 1). AngII binds two major G-protein coupled receptors (GPCR) named AngII type 1 (AT1) and 2 (AT2) receptors. The AT1 receptors are related to most of the classical RAS peripheral effects, such as vasoconstriction and kidney water and salt retention. The human AT1 receptor gene is localized on chromosome 3q, coding for a 40–42 kDa protein (359 amino acids). AT1 receptor activation promotes hydrolysis of membrane phosphatidylinositol-4,5-bisphosphate, which produces inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 binds IP3 receptors, which are ligand-gated Ca2+ release channels located in intracellular Ca2+ store sites (such as the endoplasmic reticulum), inducing mobilization of intracellular Ca2+ stores [15, 16]. DAG activates protein kinase C, which promotes the activation of the NADPH oxidase complex [17, 18], the second major source of superoxide after mitochondria [19, 20]. NADPH-oxidase-derived superoxide and superoxide-derived reactive oxygen species (ROS) are major factors responsible for the pro-oxidative and pro-inflammatory effects of AT1 receptor activation [21, 22].

The renin-angiotensin system (RAS) is organized into two opposite arms that must be properly balanced: a pro-oxidative/pro-inflammatory axis (in red), mainly formed by Angiotensin II that binds AT1 receptors (AT1R), and an antioxidative/anti-inflammatory axis (in green), mainly formed by Angiotensin II-binding AT2 receptors and Angiotensin 1–7-binding Mas receptors (MasR) or Mas-related G protein-coupled receptors. The enzyme prorenin/renin acting on the precursor protein angiotensinogen produces Angiotensin I, which is converted to Angiotensin II by the angiotensin-converting enzyme (ACE or ACE1). Renin and its precursor prorenin (PR) can also bind specific pro-oxidative PR receptors (PRR). Angiotensin-converting enzyme 2 (ACE2; also known as the major entry receptor for the SARS-COV viruses) plays a major role in balancing both RAS arms, as ACE2 (together with other peptidases such as Neprilysin, NEP) transforms peptides of the pro-inflammatory axis (Angiotensin I and, particularly, Angiotensin II) into peptides of the anti-inflammatory axis (Angiotensin 1–9 and, particularly Angiotensin 1–7)
The AT2 receptor is a protein of 363 amino acids [23, 24]. The human AT2 gene is located in the X chromosome [25], particularly in the Xq23 region [26]. AT2 receptor effects are normally opposed to those induced by AT1 receptors. AT2 receptor activation decreases the NADPH oxidase activity and superoxide production, and inhibits NFκB and ERK1/2 phosphorylation, with nitric oxide being a key second messenger for AT2 signaling [27,28,29]. Sex differences in AT2 expression have been observed, which may be related to both hormonal and sex chromosome complement effects [30].
Beyond the classical AT1 and AT2 receptors, additional angiotensin peptides and receptors modulate the RAS function, which is overall organized into a pro-oxidative and pro-inflammatory arm and a protective or counter-regulatory anti-oxidative and anti-inflammatory arm (Fig. 1). Both arms must be correctly balanced in physiological conditions. The pro-oxidative/pro-inflammatory axis is mainly constituted by AngII/AT1 receptor activation, which upregulates the function of the NADPH-oxidase complex and induces Ca2+ release, as described above. In addition, renin and its precursor prorenin also act on the corresponding receptors (Fig. 1). Binding of prorenin to its receptor provides prorenin with catalytic properties similar to those of renin. Furthermore, the prorenin receptor induces a signaling pathway resulting in pro-oxidative effects as those induced by AT1 receptor activation [31]. The anti-oxidative/anti-inflammatory axis is mainly constituted by the AngII/AT2 receptor component, and by the activation of the G-protein-coupled receptor Mas [32, 33] and the Mas-related GPCR members (Mrg), such as MrgD [34] and MrgE [35] by Ang(1–7) and alamandine (Fig. 1). The angiotensin-converting enzyme 2 (ACE2) plays a key role in the balance between the two RAS arms because ACE2 converts peptides of the pro-oxidative arm (AngI and AngII) into components of the anti-oxidative arm such as Ang1-9 and, particularly, Ang1-7 [36,37,38]. Alamandine is generated by decarboxylation of the Asp residue of angiotensin-(1–7) [34]. The role of other RAS components such as AngA, AngIII, and AngIV is more controversial (see for review [34, 39]).
ACE2 has been intensely studied over the last few years because ACE2 is the primary binding site for SARS-CoV-2 entry into host cells [40, 41]. Many studies have suggested a major role of the tissue RAS in the pathophysiology and severity of COVID-19 [42, 43], as viral binding reduces the ACE2 levels at the cell surface [44], which leads to a shift in the RAS balance toward inflammation and disease severity. This raised the dilemma of increasing ACE2 levels in tissues to inhibit the inflammatory responses or decreasing tissue ACE2 levels to inhibit viral entry and replication. Similarly, the possible beneficial or detrimental effects of therapies with ARBs and ACE inhibitors were highly controversial [45, 46]. Research [47, 48] and clinical studies [49, 50] showed the non-detrimental and possible beneficial effects of these drugs. This has been detailed in our recent review article [51]. Interestingly, a possible increase in the risk of PD related to COVID-19 has been suggested [52, 53]; however, the possible involvement of RAS dysregulation in this link remains to be studied.
In addition to the classic circulating RAS and the tissue or paracrine RAS (Fig. 1), an intracellular or intracrine RAS has been described in several types of cells, including fibroblasts, vascular smooth muscle cells, cardiac cells, kidney cells, and neurons [38, 54, 55]. The existence of a third level of RAS is supported by the intracellular synthesis of AngII and the intracellular location of different RAS components and receptors. Although the role of the intracellular RAS is still unclear, our data in neurons suggest cell protective effects, as described below [35, 38, 56,57,58].
The complexity of the RAS functioning is further increased by the possible formation of receptor complexes. Over the last decades, research on GPCRs revealed that individual receptors can interact to form heteroreceptor complexes, leading to new functional units providing cell responses that may differ from those of the individually acting receptors [59]. Regarding RAS GPCRs, receptor heteromers have been observed both between different RAS receptors and between RAS receptors and receptors of different systems such as dopaminergic, adenosine, cannabinoid, bradykinin, and β-adrenergic receptors [60,61,62]. The AT1 receptor dimerizes with itself and forms AT1-AT1 homodimers [63]. AT1-AT2 receptor association leads to AT1 signal inhibition [64], and administration of an antagonist of one receptor releases the inhibition of the partner receptor activity [65]. In AT1-CB1 heteromers, the CB1 receptor can modulate the AT1-mediated signaling [66]. We have also observed that Mas receptor can interact with the AT1 receptor and/or AT2 receptor to form heterodimers and heterotrimers in microglia and neurons [61]. In the striatum, we observed that the AT1 receptor forms heteromers with dopamine D2 receptors and that AT1 agonist and antagonist drugs can selectively alter the functional responses of the D2 receptors [67].
The existence of a local brain RAS. RAS in the dopaminergic nigrostriatal system
In the brain, the RAS was initially associated with blood pressure regulation through the circumventricular organs [68], as AngII cannot cross a healthy BBB [69]. However, brain levels of AngII are higher than the circulating levels [70], suggesting the presence of a brain paracrine RAS. Astrocytes are the major source of the precursor protein (angiotensinogen) for the brain RAS paracrine system [70,71,72], with minor contributions from other cells such as neurons [38, 73, 74]. Initially, some authors were unable to detect renin in the brain and suggested that the brain AngII may be uptaken from circulating AngII, thus questioning an independent brain RAS [75]. Different studies detected low levels of renin [76, 77] and, essentially, high levels of prorenin and prorenin receptors in the brain. Prorenin activation by the prorenin receptors confers a catalytic function of prorenin like that of renin [31, 78, 79]. In addition, more recent studies suggest that, although angiotensin peptides cannot cross a healthy BBB, pathological upregulation of peripheral RAS components may modify the BBB integrity [80, 81].
Initially, several studies detected the presence of RAS components in the basal ganglia, particularly in the nigrostriatal system [82,83,84]. More recently, a paracrine local RAS was observed in the substantia nigra (SN) and striatum of rodents [22, 79, 85], as well as non-human primates and humans [86, 87]. In dopaminergic neurons and glial cells, the presence and functional effects of different components of the pro-oxidative/pro-inflammatory arm have been shown, including the AngII/AT1 receptor axis [22, 85, 88] and the pro-renin signaling pathway [78, 79]. The presence and functional effects of the anti-oxidative/anti-inflammatory arm have also been shown, including the AngII/AT2 axis [30, 89] and the Ang1-7/ Mas receptor pathway [56]. Consistent with this, a recent study using single‐cell genomic profiling of human dopaminergic neurons revealed high expression of the AT1 receptor gene as a marker of most vulnerable dopaminergic neurons in humans, including PD patients, which are located in the ventral tier of the SN pars compacta (SNpc) [90], further supporting the potential role of the pro-oxidative AngII/AT1 receptor axis in dopaminergic degeneration, as detailed below.
Angiotensin-dopamine interactions in the nigrostriatal system
Dopamine is involved in cardiovascular, renal, endocrine, gastrointestinal, and immune functions [11, 12, 91,92,93], and different dopamine D1-like and D2-like receptors are located in peripheral tissues [11, 12]. In these tissues, a functional interaction between the local RAS and dopamine has been shown. This has been particularly studied in kidney regulation of sodium and water homeostasis and cardiovascular regulation, observing that the dopaminergic system and RAS counter-regulate each other [94, 95]. Furthermore, dimerization between receptors of both systems has been observed in peripheral cells [60]. Interestingly, dysregulation of interactions between the two systems, including the imbalance between dopamine and angiotensin receptor expression [13] or alterations in dopamine or angiotensin levels [14], leads to pathological processes such as renal degenerative diseases and hypertension.
In the brain, an interaction between dopamine and AngII was initially shown by microdialysis, which revealed that acute striatal administration of AngII induced a release of dopamine in the striatum that could be blocked by AT1 receptor antagonists [96, 97]. These results suggest that AngII, by activating AT1 receptors, induces dopamine release and that a decrease in dopamine levels may induce a compensatory increase in AngII/AT1 receptor activity to restore the dopaminergic function. AngII has also been shown to modulate the expression and trafficking of key enzymes for catecholamine biosynthesis such as tyrosine hydroxylase and dopamine β-hydroxylase, thus regulating the synthesis of norepinephrine and dopamine [98]. Processes of counter-regulation between angiotensin and dopamine receptors have been shown in the SN and striatum in several studies [99, 100]. Dopamine depletion, in 6-hydroxydopamine (6-OHDA) or reserpine models, upregulates the AT1/NADPH-oxidase activity, which can be reversed after restoring dopamine levels by L-DOPA administration [100]. Furthermore, both D1 and D2 KO mice showed upregulation of AT1 receptor expression [99]. Dimerization between receptors of the two systems has also been observed in the nigrostriatal system [61, 67]. As AT1 activation induces dopamine release, and D2 autoreceptors in dopaminergic neurons modulate dopamine release, the formation of AT1/D2 heteromers appears particularly interesting. It is also interesting to note that the striatal dopamine depletion (e.g., using 6-OHDA) and treatment with L-dopa modify the levels of different types of RAS-receptor heteromers [61, 101].
Dysregulation of RAS-dopamine interactions and dopaminergic degeneration
As previously reported in cardiovascular and renal tissues, dysregulation of the RAS/dopamine interactions in the nigrostriatal system promotes neuroinflammation and dopaminergic neurodegeneration [99, 102] (Fig. 2). The possible deleterious effect of RAS/dopamine dysregulation has been studied in several in vivo and in vitro models of PD. In animal models, the dopaminergic lesion induces a loss of dopamine and upregulation of the pro-oxidative/pro-inflammatory AngII/AT1 receptor axis, enhancing the progression of dopaminergic degeneration. This has been observed in our studies and reports from different laboratories using the main in vivo PD models including neurotoxic models such as the 6-OHDA [22, 103] and the MPTP models [21, 85, 104, 105] and, more recently, PD models based on over-expression of α-synuclein using adeno-associated viral vectors [106]. The dopaminergic lesion is significantly decreased by ACE inhibitors [104, 107, 108] and, more specifically, by the blockage of AT1 receptors [22, 85, 103, 106]. In vitro models, such as primary mesencephalic cultures treated with low doses of the neurotoxin 6-OHDA or MPP+ revealed similar results, showing that the neurotoxin-induced dopaminergic neuron death is significantly increased by administration of AngII and inhibited by treatment with AT1 receptor blockers [22, 85].

The brain RAS plays a role in the progression of dopaminergic neuron degeneration. Different pathogenic factors may trigger molecular and cellular changes that lead to an initial dysregulation of the brain RAS, or dysregulation of the dopaminergic neuron function leading to decreased dopamine production, which affects the dopamine/RAS interaction in neurons and glial cells. In neurons, a decrease in dopamine level upregulates the angiotensin type-1 (AT1) receptor activity, leading to the recovery of dopamine levels together with overactivation of the NADPH-oxidase-superoxide-mitochondria axis and Ca2+ release, which mediate several key events such as oxidative stress, α-synuclein aggregation, and neuroinflammation involved in the progression of Parkinson's disease (PD). An intraneuronal antioxidative/anti-inflammatory RAS counteracts the effects of the pro-oxidative AT1 receptor overactivation (detailed in Fig. 3). In microglial cells, AT1 receptor upregulation activates the NADPH-oxidase complex, increasing the release of ROS to the extracellular space and the inflammatory response. In astrocytes, a decrease in dopamine level induces an increase in paracrine angiotensinogen/AngII production that can act on neurons and microglial cells. AngII, angiotensin II; AT1, angiotensin type I; ROS, reactive oxygen species
Over the last decade, a series of clinical observations also supported that the overactivation of the AngII/AT1 pro-inflammatory axis contributes to the progression of PD. Early studies reported increased ACE activity in the cerebrospinal fluid (CSF) of PD patients [109], associations between ACE-genetic polymorphisms and PD [110], and beneficial effects of ACE inhibitors in PD patients [111]. The effects of antihypertensive drugs on the risk of PD have been studied in several cohort studies [112, 113]. However, the conclusive value of these studies was limited by the low number of patients or other confounding factors such as short periods of exposure to the analyzed drug or the inclusion of different anti-hypertensive drugs or different ARBs and ACE inhibitors in the same group of patients [114]. More recent studies also suggest the potential clinical effects of ARBs [115, 116], including studies using artificial intelligence [117], which support the neuroprotective effects of AT1 receptor blockers on PD risk. Recent retrospective cohort studies involving a large number of patients are particularly interesting and show that ARBs may be an effective neuroprotective strategy to reduce PD risk and progression [118, 119]. ARB treatment is associated with a marked reduction of PD risk in patients with recently diagnosed hypertension [119]. Furthermore, ARBs with BBB-penetrating properties and a high cumulative duration of treatment are particularly effective [118].
The development of prodromal clinical trials, using ARBs that can cross the BBB or other molecules able to inhibit the effects of AT1 receptor over-activity, is further supported by recent key data from Kamath et al. [90]. Using single-nucleus RNA sequencing and unbiased clustering analysis of human SN dopaminergic neurons, they identified one neuron population showing high levels of SOX6 and AT1 receptor gene (AGTR1), specifically localized in the most vulnerable region (i.e., the ventral tier) of the SNpc. Consistent with this, the highest loss of neurons in PD patients and patients with Lewy body dementia relative to controls was observed in the SOX6_AGTR1 subpopulation, and the levels of AGTR1 expression correlated with the susceptibility to neurodegeneration in neurons. Similar results were found in the SNpc of other mammalian species [90, 120]. Interestingly, compared to more resistant neurons in other regions of the SN and other brain regions, these most vulnerable neurons in the ventral tier of the SNpc also show lower levels of buffer mechanisms, such as low calbindin levels, to counteract rising intracellular calcium [121, 122]. As described above, the major consequences of AT1 receptor overactivation are the NADPH-oxidase-derived oxidative stress and intracellular calcium mobilization (Fig. 2).
Mechanisms of the increase in dopaminergic degeneration induced by brain RAS dysregulation
In both peripheral tissues and the brain, NADPH-oxidase-derived oxidative stress, intracellular calcium dysregulation, and enhanced inflammatory responses appear to mediate the deleterious effects of overactivation of the pro-oxidative/pro-inflammatory axis of the tissue RAS (Fig. 2; Table 1). It is known that oxidative stress and neuroinflammation are early components of dopaminergic degeneration, and both factors, probably acting synergistically with additional factors, lead to the progression of PD [123,124,125]. Consistent with the role of NADPH-oxidase activation and NADPH-oxidase-derived ROS in the exacerbation of dopaminergic neurodegeneration induced by AT1 receptor overactivation, neurodegeneration is inhibited by blockers of NADPH-oxidase activation in animal models [22, 85, 103]. Furthermore, both angiotensin receptors and NADPH-oxidase components have been observed in dopaminergic neurons and glial cells in the SN of different mammals, including humans [22, 85, 87]. As previously observed in peripheral processes, particularly in kidney and cardiovascular tissues, AngII acts both on tissue-resident cells (neurons in the brain) and inflammatory cells (glia, particularly microglial cells in the brain) (Fig. 2). Activation of AT1 receptors in dopaminergic neurons induces upregulation of NADPH-oxidase-derived superoxide and intracellular ROS levels (Fig. 2). As observed in other types of cells [126, 127], the NADPH oxidase-derived ROS interact with the mitochondria in dopaminergic neurons [105, 128, 129], via mitochondrial ATP-sensitive potassium channels, leading to a further increase in mitochondrial ROS production; inversely, the mitochondrial-derived ROS further increase NADPH-oxidase activation and ROS generation, leading to a vicious cycle [20, 105]. In addition, the NADPH-oxidase-derived ROS can act as a second messenger in cellular signaling pathways, including those triggering the inflammatory response and the migration of inflammatory glial cells into the affected region [19, 20, 130]. Furthermore, we have already indicated that AT1 receptor activation, via IP3, induces intracellular calcium mobilization. Therefore, overactivation of AT1 receptors can induce dysregulation of cell calcium homeostasis, which has been involved in PD pathogenesis, as physiological calcium levels must be correctly balanced to prevent neuronal death [131,132,133].
RAS receptors were also observed in glial cells (Fig. 2), including microglia, indicating that AngII can act directly on their receptors to modulate the neuroinflammatory response. In inflammatory cells, such as microglia, NADPH oxidase activation can induce high levels of ROS that are released to the extracellular space, exacerbating oxidative stress in neurons. In microglia, the activation of NADPH-oxidase also induces low levels of intracellular ROS to act as a second messenger to modulate the inflammatory response [19, 20, 160]. Over the last decade, the major role of the RAS in the modulation of microglial inflammatory response has been shown in a considerable number of studies. In microglia, activation of the RhoA/ROCK pathway [88, 161, 162], release of TNF-α [163], and altered iron homeostasis [164] are involved in the enhancement of microglial response and dopaminergic degeneration by AngII/AT1 receptor activation (Fig. 2; Table 1). Up-regulation of NLRP3 inflammasome is also mediated by AT1 receptor activation [140]. Other RAS components also modulate the microglial response, such as AT2 receptors that promote the expression of anti-inflammatory responses [30, 89], and prorenin receptors that enhance inflammation and its damaging effects on dopaminergic neurons [78].
In addition to oxidative stress and neuroinflammation, a major factor involved in PD progression is α-synuclein aggregation and cell-to-cell transmission. However, it was unknown whether AngII/AT1 receptor overactivation affects α-synuclein aggregation and transmission in neurons and glial cells. In recent experiments [156], we observed that AngII/AT1 receptor overactivation promotes α-synuclein expression, aggregation, and glial transmission (Fig. 2; Table 1). This further supports the role of local RAS dysregulation in PD progression, and suggests that AT1 receptor blockers or RAS modulation, by enhancing the activity of the counterregulatory RAS receptors such as AT2 or Ang1-7/Mas receptors, is a promising therapeutic target for PD.
Potential mechanisms and processes triggering the angiotensin/dopamine dysregulation
Functional decline of the dopaminergic system induces RAS dysregulation
Several major processes such as oxidative stress, neuroinflammation, and accumulation of α-synuclein aggregates are involved in the dopaminergic neurodegeneration in PD. However, their order of appearance is not clear and it remains unknown which of them acts first. It is frequently suggested that α-synuclein aggregation and Lewy body pathology are the initial processes triggering neurodegeneration. However, molecular and cellular changes originating from different genetic and/or environmental triggers may occur before α-synuclein aggregation, particularly affecting the most vulnerable neurons [177, 178]. An initial impairment in dopamine metabolism [179], a decrease in tyrosine hydroxylase [180], oxidative damage [181], and early neuroinflammation [182] have been suggested as initial triggers of the disease. Dysregulation of the brain RAS, and particularly, over-activation of the AngII/AT1 receptor axis are involved in all these potential triggering processes, and may be an early mechanism mediating the progression of dopaminergic neuron degeneration. However, the next question could be what the cause for the RAS dysregulation is (Table 1).
First, an initial decrease in dopamine levels (Fig. 2), due to dopamine homeostasis dysregulation, may lead to a compensatory increase in AngII/AT1 receptor activity to normalize dopamine levels. This may occur in the very initial stages of PD, aging, and other situations that have been associated with an increased risk of developing PD (see below). AngII/AT1 receptor overactivity may lead to a transitory upregulation of dopamine levels [67, 99, 100, 102, 134]. However, the AngII/AT1 receptor axis upregulation may simultaneously promote oxidative stress, neuroinflammation, and α-synuclein aggregation through increased NADPH-oxidase activity and alteration of intracellular calcium homeostasis. Furthermore, it is well known that dopamine is an immunomodulatory molecule, that dopamine receptors are present in immune effector cells [135, 136], and that dopamine inhibits NLRP3 inflammasome via dopamine receptors in several cell types [137, 138]. Therefore, an initial decrease in dopamine or dopaminergic function may enhance the inflammatory response, and upregulation of the pro-inflammatory RAS axis appears to be involved in this process (Fig. 2). Consistent with this, we have recently shown that dopamine regulates RAS activity in glial cells, modulating AngII release by astrocytes and the expression of angiotensin receptors in microglia [102]. Dopaminergic neurotoxins such as MPP+ can increase the release of angiotensinogen and AngII from astrocytes. However, dopamine, via type-2 (D2) receptors, down-regulates the production of angiotensinogen and the expression of AT1 receptors while increasing the expression of AT2 receptors in astrocytes. In microglia, dopamine administration induces downregulation of the AT1/AT2 ratio and inhibits the inflammatory response [102]. Furthermore, recent studies showed that AT1 receptors mediate microglial inflammasome complex activation in dopamine-depleted models [140].
Consistent with the role of RAS dysregulation in PD progression, overactivity of the pro-oxidative/pro-inflammatory AngII/AT1 receptor axis has been observed in several processes described below, which are known to increase the risk of PD.
Aging-related RAS dysregulation and PD
Different studies have shown that aging increases the vulnerability of dopaminergic neurons to degeneration. Advanced age is the first risk factor for PD and other neurodegenerative diseases. It has been shown that the pro-inflammatory and pro-oxidative state associated with aging (inflammaging) [183, 184] may favor the development of degenerative diseases [185, 186]. As RAS is involved in the neuroinflammatory response, the AngII/AT1 receptor upregulation in the nigrostriatal system may be a factor for increased dopaminergic vulnerability with aging. In aged male rats (see below for aged females), we observed higher levels of neuroinflammatory and oxidative markers in the SN and striatum, and an increase in the dopaminergic neuron death triggered by dopaminergic neurotoxins, which are downregulated by the AT1 receptor antagonist candesartan [99, 141]. AT1 receptor inhibition also ameliorates the upregulation of the NLRP3 inflammasome observed in aged rats [140, 187]. Aging-related upregulation of the pro-inflammatory axis components such as AT1 receptors and prorenin receptors has also been observed [89, 99, 142]. In dopaminergic neurons, this may be mediated by an age-related decrease in dopaminergic activity and the already mentioned RAS compensatory changes. Consistent with this, several studies have shown a loss of striatal D2 and D1 receptors in aged animals and humans [143, 144], and that the dopaminergic system is altered during normal aging [145, 146]. However, other non-dopaminergic factors appear also involved, as the age-related upregulation of the pro-oxidative/pro-inflammatory RAS has also been observed in other tissues apparently unrelated with the dopaminergic systems [147,148,149]. Interestingly, aged animals also show a decrease in the expression of components of the RAS anti-inflammatory axis, such as AT2 or Mas receptor expression, not only at the level of the cell membrane but also in the intracellular RAS system detailed below [35, 56, 57], revealing a decrease in the compensatory response of the RAS anti-inflammatory axis that contributes to the aging-related RAS imbalance.
Menopause, RAS dysregulation and PD
Menopause is also a risk factor for PD, as the incidence and the prevalence of the disease are higher in men and postmenopausal women than in premenopausal women of similar age [188, 189]. Over the last few decades, different experimental studies have reported the beneficial effects of estrogen against dopaminergic neurodegeneration [190, 191], and shown that the anti-inflammatory effects of estrogen are responsible for the neuroprotective effects [192, 193]. The beneficial effects of estrogen replacement therapies are more controversial [194, 195], with age and the period without estrogen before receiving the replacement treatment as underlying factors for the discrepancies. As estrogen-induced regulation of the RAS has been suggested to mediate the beneficial effects of estrogen in several peripheral tissues [150, 151], we studied the effects of the lack of estrogen on the nigrostriatal RAS in female rats with early surgical menopause (young ovariectomized rats) and natural menopause (aged rats) [152,153,154,155]. The activity of the RAS pro-oxidative/pro-inflammatory arm was increased in both groups of menopausal rats. Interestingly, treatment with the AT1 receptor blocker candesartan reduced the loss of neurons induced by low doses of dopaminergic neurotoxins in both groups of menopausal rats; however, the neuroprotection of the replacement therapy was effective only in the young rat groups [154]. We observed that there is a critical period for the neuroprotection with estrogen against dopaminergic neurodegeneration and that the local RAS plays a major role, as treatment with the AT1 receptor antagonist candesartan provided significant neuroprotection beyond the critical period for estrogen, [152]. The brain RAS system may be an efficient therapeutic target for the treatment or prevention of PD in estrogen-deficient women, together with or substituting estrogen replacement therapies [152].
Interestingly, recent studies have shown that levels of estrogen are not the only factor responsible for the reduced activity of the RAS pro-inflammatory axis in the nigrostriatal system of females relative to males and that the expression of the anti-inflammatory AT2 receptors is higher in females, independently of circulating levels of estrogen and probably related with the sex chromosome complement effects, which contribute to attenuate the inflammatory response [30].
Chronic brain hypoperfusion, RAS dysregulation and PD
Chronic brain hypoperfusion has also been related to an increased risk of neurodegeneration and PD [157, 158, 196]. This is consistent with previous studies in animal models showing that brain hypoperfusion enhances the neuronal loss induced by dopaminergic neurotoxins, together with the nigrostriatal expression of inflammatory markers such as IL-1β and increased oxidative stress such as increased NADPH-oxidase activity [159]. Interestingly, the hypoperfusion-induced changes are accompanied by upregulation of the AngII/AT1 receptor activity in the SN, and these changes are downregulated by chronic treatment with the AT1 receptor blocker candesartan [159].
RAS-related autoimmunity and PD
Autoimmune processes have also been involved in the triggering and progression of degenerative diseases, including PD [197,198,199]. We have recently observed an increase in the serum levels of autoantibodies for AT1 receptors (AT1 receptor agonistic autoantibodies, AT1-AA) and ACE2 autoantibodies (ACE2 antagonists) in PD patients compared to non-PD controls. We also found both autoantibodies in the CSF samples from some PD patients [80]. Furthermore, there was a significant correlation between serum levels of AT1-AA and serum inflammatory cytokines in PD patients but not in controls. In parallel experiments, using in vivo and in vitro PD models, we confirmed that these autoantibodies are associated with the neurodegenerative process and the accompanying neuroinflammatory changes, and led to further progression of neurodegeneration by increasing the activity of AT1 receptors and decreasing the activity of ACE2-related anti-inflammatory RAS axis, respectively. Consistent with the findings in PD patients, we observed a significant increase of these RAS-related autoantibodies in both serum and CSF of rats lesioned with the dopaminergic neurotoxin 6-OHDA. Furthermore, we confirmed in cultures that administration of AT1-AA increased the loss of dopaminergic neurons, which was inhibited by treatment with the AT1 receptor blocker candesartan [80]. Altogether, these findings suggest that the generation of RAS autoantibodies during early degenerative stages contributes to RAS dysregulation towards the proinflammatory axis and to the increased progression of dopaminergic degeneration and PD. ARBs or treatments that inhibit the generation of these autoantibodies may inhibit these effects.
Metabolic syndrome (MetS), RAS dysregulation and PD
In addition to the already mentioned risk factors, several peripheral diseases related to chronic inflammation, appear to increase the risk of neurodegenerative diseases including PD [200, 201]. MetS has been associated with chronic peripheral inflammation and increased risk of PD [202, 203]. As in the case of neurodegenerative diseases, MetS can be currently considered a silent epidemic disease. The definition of MetS consists of the presence of obesity and at least 2 of the following conditions: hypertension, hypertriglyceridemia, low HDL cholesterolemia, and type-2 diabetes/hyperglycemia [204]. In a rat model, we showed that MetS leads to the upregulation of the pro-oxidative/pro-inflammatory RAS axis in the SN, together with increases in oxidative stress, neuroinflammatory markers, and dopaminergic neurodegeneration, and all these changes were decreased by treatment with ARBs [81]. In rats, MetS also increases the circulating levels of major pro-inflammatory cytokines and 27-hydroxycholesterol. Interestingly, serum levels of pro-inflammatory AT1 and ACE2 autoantibodies are increased and correlated with several MetS parameters. AT1 and ACE2 autoantibodies are also present in the CSF of these rats. Osmotic minipump infusions of AT1 receptor autoantibodies disrupt BBB and affect the brain, leading to upregulation of the pro-inflammatory RAS activity in the SN and a significant increase in dopaminergic neurodegeneration in two different rat PD models [81]. Activation of AT1 endothelial receptors by the circulating agonistic AT1 receptor autoantibodies appears as a major mechanism of BBB disruption. This is consistent with several previous studies showing that stimulation of AT1 receptors in endothelial cells and perivascular macrophages by circulating AngII, but not hypertension itself, is a major mechanism of the BBB disruption observed in hypertension. Consistently, the disruption can be blocked by AT1 receptor blockers (ARBs) and not by other anti-hypertensive drugs [205,206,207,208,209]. In MetS patients, previous studies have also observed increases in the levels of pro-inflammatory cytokines [210, 211] and BBB permeability [212, 213], which were attributed to the increase in circulating cytokines. However, the increase in circulating agonistic AT1 receptor autoantibodies may also play a major role in BBB disruption, as patients with MetS show significantly higher levels of AT1 receptor autoantibodies, which lead to dysregulation of the SN RAS as observed in the MetS rat model [81]. In addition, circulating levels of AT1 receptor autoantibodies are significantly higher in non-Parkinsonian patients with MetS than in non-Parkinsonian patients without MetS. However, no significant difference has been observed between Parkinsonian patients with and without MetS, both showing higher levels of AT1 receptor autoantibodies than normal controls. This may be because dopaminergic degeneration and neuroinflammation in PD patients (without MetS) also lead to an increase in circulating autoantibodies [80], as detailed above. In MetS patients, both processes may trigger a vicious cycle that accelerates PD progression, which may be blocked by strategies against generation of these autoantibodies or by AT1 receptor blockers.
Additional mechanisms may be involved in the association between MetS and PD. Recent results from rat models of MetS suggest involvement of circulating extracellular vesicles (EVs) and their RAS cargo in the link between MetS and PD [81]. EV cargo shows the molecular state of their cells of origin [214], including the cellular level of RAS components [215]. Circulating EVs can cross the BBB and serve as inflammatory mediators [216, 217] and carriers of oxidative stress signals [218]. In the adipose tissues from obese animals, increases of angiotensinogen [219, 220], AT1 receptors [221],and prorenin receptors [222] have been reported. Consistent with this, we have recently observed that in rat models of MetS, EVs are highly increased in the serum and show increased pro-oxidative/pro-inflammatory and decreased anti-oxidative/anti-inflammatory RAS components, as well as increased inflammatory and oxidative stress markers. Interestingly, the increase of serum EVs, the RAS dysregulation and the increases of inflammatory and oxidative stress markers in the EV cargo are inhibited by chronical treatment with theAT1 blocker candesartan in MetS rats [165]. In vitro, administration of EVs isolated from the serum of MetS rats increases dopaminergic cell death and regulates the astrocytic function, leading to the upregulation of neuroinflammation and oxidative stress markers. The effects of treatment with EVs are inhibited by pre-treatment of cultures with the AT1 blocker candesartan [165]. Altogether, in MetS, circulating EVs may contribute, via RAS dysregulation, to the progression of neuroinflammation and dopaminergic cell death. This mechanism can be inhibited by treatment with ARBs such as candesartan.
Gastrointestinal processes, RAS dysregulation and PD
Many recent studies have suggested the association of gastrointestinal diseases with a higher risk of PD, and revealed the presence of a gut-brain axis. Gut dysmotility is a PD component, although the mechanisms are still unclear. Conversely, the role of gut diseases in PD remains to be clarified. Braak´s hypothesis suggested that PD may be caused by pathogens that act on the gastrointestinal tract, which induce gastrointestinal inflammation and oxidative stress, leading to α-synuclein deposition that is retrogradely transported to the brain [223, 224]. However, the gut-brain axis also regulates the nigrostriatal dopamine homeostasis, via the vagus nerve, as a caloric intake regulatory system [225, 226]. Consistent with this, a decrease in nigrostriatal dopamine level leads to changes in colonic expression of dopamine receptors as well as dopamine and acetylcholine levels in rodents, and experimental gut inflammation leads to changes in the nigrostriatal dopaminergic homeostasis [92]. Those results suggest that the nigrostriatal dopaminergic system and the gastrointestinal system interact bidirectionally and that both brain dopaminergic lesions and gastrointestinal lesions can lead to dysregulation of the functional interaction. This may explain the gastrointestinal alterations observed in PD patients, and the higher vulnerability of central dopaminergic neurons after gastrointestinal inflammation [92]. Several recent studies in rodents and humans support this proposition [227,228,229], and brain-first and body-first subtypes of PD have been proposed [228].
Interestingly, the gastrointestinal tract has a local RAS that is involved in major functional processes such as motility, absorption, and gastrointestinal inflammation [166,167,168]. The gastrointestinal RAS also plays a role in gut diseases such as inflammatory bowel disease and gastrointestinal motility disorders [167, 168]. As in the case of the nigrostriatal dopaminergic system, mutual regulation between the gastrointestinal dopaminergic and the angiotensin systems has been observed, which may be dysregulated with aging and by different processes, leading to increased vulnerability to gastrointestinal inflammatory diseases [91, 93]. Furthermore, nigrostriatal dopaminergic depletion also leads to upregulation of the pro-inflammatory axis of the gastrointestinal RAS, together with increased gut levels of oxidative stress and pro-inflammatory markers [92]. Conversely, experimental gastrointestinal inflammation leads to changes in dopaminergic homeostasis and upregulation of the pro-inflammatory RAS in the SN, which may contribute to increases in neuroinflammation, dopaminergic neuron vulnerability, and progression towards PD [92, 169].
Microbiota, RAS dysregulation, and PD
Recent studies suggest a connection of gut microbiota with neuroinflammatory and neurodegenerative disorders such as Alzheimer's disease and PD, and that intervention on microbiota may provide a novel strategy for treating and preventing neurodegeneration [230]. Although the exact mechanism remains to be clarified, gut microbiota and its metabolites may contribute to PD pathophysiology through modulation of gut inflammation (see above). However, additional mechanisms including the release of short-chain fatty acids or compounds affecting the BBB may also be involved [230].
Interestingly, the RAS, including systemic and gastrointestinal RAS, has emerged as a major mediator of microbiota-derived effects [170, 171]. The microbiota and its metabolites may modulate gastrointestinal and systemic RAS, and RAS alterations may modify microbiota composition and metabolism. In animal models, treatment with microbiota metabolites such as trimethylamine-oxide results in altered expression of RAS receptors in the heart and kidney [170, 172], ACE inhibitory peptides are produced during the bacterial fermentation processes [173], and chronic losartan treatment reduces gut dysbiosis [174]. Consistent with this, the microbiome, acting via RAS regulation, has been related to diabetic-induced kidney injury, hypertension, and organ damage related to hypertension [171, 175]. Interestingly, gut microbiota, via RAS, has been involved in obesity and the development of MetS [176]. As described in a previous section, MetS is related to a higher risk of PD [202, 203], and we have recently shown several potential mechanisms connecting MetS and PD [81, 165].
Given the above-mentioned major role of RAS dysfunction in the inflammatory changes involved in COVID-19, a role of the microbiota/RAS interaction in the pathogenesis and progression of COVID-19 has been suggested [231, 232]. The RAS is suggested to play a pivotal role in inflammatory processes that affect microbiome dysregulation, COVID-19 and the development of PD [233]. As a consequence, prebiotics and probiotics have been suggested for the treatment of RAS-related diseases. Recent studies have shown that probiotics can activate the ACE2/MAS receptor axis [234], and that oral delivery of Ang(1–7)-expressing Lactobacillus paracasei led to significant modification of microbiota and decreased expression of neuroinflammatory genes in the cortex [235]. However, the microbiome-related effects require further clarification in future studies.
RAS-related compensatory mechanisms protecting dopaminergic neuron. The intraneuronal RAS
It has been assumed that peptides of the antioxidative/anti-inflammatory RAS, acting on their corresponding cell surface receptors (AT2, Mas, and Mas-related receptors), counteract the effects of the pro-oxidative AngII/AT1 receptor axis (Fig. 3). However, we have observed that at least in dopaminergic neurons, the intracellular RAS plays a major role in cell protection (Fig. 3). Several studies in peripheral cells have suggested that the intracellular RAS may increase the effects of the pro-oxidative AngII/AT1 receptor axis of the paracrine or tissue RAS [236, 237]. As in several peripheral cells, different RAS receptors are observed in intracellular components of dopaminergic neurons, such as mitochondria and neuronal nuclei [35, 56,57,58]. However, our studies showed that the nuclear and the mitochondrial RAS constitute a protective mechanism to buffer or counteract excessive pro-oxidative effects of the cell membrane AngII/AT1/NADPH-oxidase (Nox2) effects (Fig. 3), as we reviewed in detail in [38].

The intraneuronal RAS compensates (green lines: neuroprotective mechanisms) for the pro-oxidative effects of plasma membrane AT1 receptor activation by paracrine AngII (red lines: pro-neurodegenerative mechanisms). Internalization of the AT1/Ang II complex to the nucleus and activation of nuclear and mitochondrial receptors by intracellular AngII and Ang 1–7, trigger several mechanisms that protect neurons against AT1-induced oxidative stress during normal cell function. Antioxidative AT2, Mas, and MrgE receptors are more abundant in the mitochondria. In the nucleus, activation of AT1 receptors triggers several compensatory mechanisms, including increased mRNA expression of antioxidative RAS receptors, angiotensinogen, IGF1, and PGC1α. However, an excess of cell membrane AngII/AT1 receptor activity to compensate for dopamine decrease or other pathogenic factors may overwhelm the buffering mechanisms, leading to the progression of dopaminergic degeneration. AngII, angiotensin II; Ang1-7, angiotensin 1–7; AT1, angiotensin type 1; AT2, angiotensin type 2; MAS, Mas receptors; MrgE, Mas-related receptor MrgE; Nox4, NADPH-oxidase 4; ROS, reactive oxygen species
In the mitochondria (Fig. 3), different angiotensin receptors modulate oxidative phosphorylation. AT1 receptors, by activating mitochondrial NADPH-oxidase 4 (Nox4), contribute to superoxide production and increase respiration. However, the receptors of the antioxidative system are much more abundant than the AT1 receptors in the mitochondria. We initially observed AT2 and Mas receptors, which induce, via nitric oxide, a downregulation in mitochondrial respiration and modulate oxidative phosphorylation [56, 57]. Interestingly, we observed high levels of mitochondrial ACE2 and its product Ang1-7, which may act on mitochondrial Mas receptors. However, we surprisingly found high mitochondrial levels of the Mas-related receptor E (MrgE), which appear as the most abundant RAS receptor in the mitochondria of dopaminergic neurons [35]. In peripheral cells, ROS-mediated crosstalk between the cell membrane Nox2 and mitochondria has been shown, during which superoxide (and superoxide-derived ROS) produced by Nox2 induces mitoKATP (mitochondrial ATP-sensitive potassium channel) opening, increasing the generation of mitochondrial ROS [127, 139, 238]. We have shown this mechanism also in dopaminergic neurons treated with AngII [128, 129]. This mechanism, triggered by the activation of plasma membrane AT1 receptors, is counteracted by the mitochondrial AT2, Mas, and, particularly, MrgE receptors (Fig. 3).
As described above, activation of AT1 receptors at the cell membrane activates the membrane Nox2, which produces intracellular superoxide that may lead to oxidative stress. However, it is known that activation of AT1 receptors induces a simultaneous internalization of the AngII/AT1 receptor complex towards the cell nucleus, where AngII activates nuclear AT1 receptors, inducing upregulation of nuclear superoxide/H2O2 (by activating nuclear Nox4) and Ca2+ levels (by activating nuclear IP3). This results in regulation of gene expression to trigger several compensatory mechanisms that protect cells against oxidative stress induced by the activation of surface membrane AT1/Nox2 [38, 58] (Fig. 3). These protective mechanisms include (i) increased production of protective AT2 and Mas receptors that traffic to the cell membrane and, particularly, to the mitochondria, (ii) increased production of intracellular angiotensin, particularly Ang1-7 that can act on mitochondrial AT2 and, particularly, MrgE and Mas receptors, respectively, and (iii) upregulation of mRNA expression of cell protective components such as IGF-1 and PGC-1α [38, 58].
In summary, the intracellular RAS may compensate for the deleterious effects of cell membrane AT1 receptor activation. Internalization of the AngII/AT1 complex activates nuclear AT1 receptors, which triggers protective mechanisms against cell membrane AT1-induced oxidative stress. This is possibly effective within physiological levels of AT1 receptor activation. However, excessive AngII/AT1 receptor activation at the cell surface membrane or an increased membrane AT1/AT2-Mas receptor ratio, under RAS deregulatory conditions as those described in the previous sections, may overwhelm the buffering mechanisms, leading to cell oxidative stress and progression of the disease. Interestingly, aging leads to the downregulation of mitochondrial AT2 and Mas/MrgE receptors [35, 56, 57].
In addition to the specific RAS compensatory mechanisms, other neuronal antioxidant systems and protective mechanisms against intracellular calcium dysregulation also protect neurons from the paracrine AngII/AT1 receptor overactivation. Consistent with this, we have shown that AngII also activates the nuclear factor erythroid 2-related factor 2 (NRF2) signaling pathway in dopaminergic neurons [239], which is a key regulator of cell antioxidant mechanisms and redox homeostasis.
Perspectives, limitations and conclusions
Dopamine/RAS interactions have been observed in several tissues, and dysregulation of these interactions leads to renal, cardiovascular, and other peripheral diseases. In the nigrostriatal system, local RAS dysregulation is involved in major processes responsible for the initiation and progression of dopaminergic neuron degeneration and PD, including oxidative stress, neuroinflammation, and α-synuclein aggregation and transmission. Consistent with this, an imbalance in RAS activity towards the pro-oxidative/pro-inflammatory RAS axis has been observed in the SN and striatum of models exposed to factors associated with dopaminergic degeneration, including aging, menopause, chronic brain hypoperfusion, MetS, gut inflammation, and microbiome dysregulation. Autoantibodies against ACE2 and AT1 receptors are increased in PD models and PD patients and contribute to BBB dysregulation and pro-inflammatory RAS enhancement. Circulating EVs with dysregulated RAS cargo, as observed in MetS models, may also promote neuroinflammation and dopaminergic degeneration. Although the lack of more detailed knowledge of all mechanisms involved in the development and progression of PD limits the development of neuroprotective therapies, current data on the effects of RAS dysregulation, as summarized here, suggest that regulating the brain RAS may be an effective neuroprotective strategy for individuals with a high risk of developing PD or in prodromal stages of PD.
Availability of data and materials
Not applicable.
Abbreviations
- 6-OHDA:
-
6-Hydroxy dopamine
- ACE or ACE1:
-
Angiotensin-converting enzyme
- ACE2:
-
Angiotensin-converting enzyme 2
- AngII:
-
Angiotensin II
- ARBs:
-
AT1 receptor blockers
- AT1:
-
Angiotensin type 1
- AT1R:
-
AT1 receptors
- AT2:
-
Angiotensin type 2
- BBB:
-
Blood–brain barrier
- CSF:
-
Cerebrospinal fluid
- DAG:
-
Diacylglycerol
- EVs:
-
Extracellular vesicles
- GPCR:
-
G-protein coupled receptors.
- IP3:
-
Inositol trisphosphate
- MasR:
-
Mas receptors
- MetS:
-
Metabolic syndrome
- MrgE:
-
Mas-related receptor E
- Nox2:
-
NADPH-oxidase
- Nox4:
-
NADPH-oxidase 4
- PD:
-
Parkinson's disease
- RAS:
-
Renin-angiotensin system
- ROS:
-
Reactive oxygen species
- SN:
-
Substantia nigra
- SNpc:
-
Substantia nigra pars compacta
References
Tigerstedt R, Bergman PQ. Niere und kreislauf. Skand Arch Physiol. 1898;8:223–71.
Nishimura H. Renin-angiotensin system in vertebrates: phylogenetic view of structure and function. Anat Sci Int. 2017;92:215–47.
Wong MKS, Takei Y. Molecular and evolutionary perspectives of the renin-angiotensin system from lamprey. Gen Comp Endocrinol. 2018;257:137–42.
Lev-Ran A, Porta M. Salt and hypertension: a phylogenetic perspective. Diabetes Metab Res Rev. 2005;21:118–31.
Ganong WF. Origin of the angiotensin II secreted by cells. Proc Soc Exp Biol Med. 1994;205:213–9.
Barger G, Dale HH. Chemical structure and sympathomimetic action of amines. J Physiol. 1910;41:19–59.
Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin Wochenschr. 1960;38:1236–9.
Goldberg LI. Cardiovascular and renal actions of dopamine: potential clinical applications. Pharmacol Rev. 1972;24:1–29.
Channer B, Matt SM, Nickoloff-Bybel EA, Pappa V, Agarwal Y, Wickman J, et al. Dopamine, immunity, and disease. Pharmacol Rev. 2023;75:62–158.
Matt SM, Gaskill PJ. Where is dopamine and how do immune cells see it? Dopamine-mediated immune cell function in health and disease. J Neuroimmune Pharmacol. 2020;15:114–64.
Mackie P, Lebowitz J, Saadatpour L, Nickoloff E, Gaskill P, Khoshbouei H. The dopamine transporter: an unrecognized nexus for dysfunctional peripheral immunity and signaling in Parkinson’s disease. Brain Behav Immun. 2018;70:21–35.
Vidal PM, Pacheco R. The cross-talk between the dopaminergic and the immune system involved in schizophrenia. Front Pharmacol. 2020;11:3.
Chugh G, Lokhandwala MF, Asghar M. Altered functioning of both renal dopamine d1 and angiotensin II type 1 receptors causes hypertension in old rats. Hypertension. 2012;59:1029–36.
Yang S, Yao B, Zhou Y, Yin H, Zhang MZ, Harris RC. Intrarenal dopamine modulates progressive angiotensin II-mediated renal injury. Am J Physiol Renal Physiol. 2012;302:F742–9.
Garcia MI, Boehning D. Cardiac inositol 1,4,5-trisphosphate receptors. Biochim Biophys Acta Mol Cell Res. 2017;1864:907–14.
Simo-Cheyou ER, Tan JJ, Grygorczyk R, Srivastava AK. Stim-1 and orai-1 channel mediate angiotensin-ii-induced expression of egr-1 in vascular smooth muscle cells. J Cell Physiol. 2017;232:3496–509.
Basu U, Case AJ, Liu J, Tian J, Li YL, Zimmerman MC. Redox-sensitive calcium/calmodulin-dependent protein kinase iialpha in angiotensin II intra-neuronal signaling and hypertension. Redox Biol. 2019;27:101230.
Bollag WB. Regulation of aldosterone synthesis and secretion. Compr Physiol. 2014;4:1017–55.
Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–7.
Belarbi K, Cuvelier E, Destee A, Gressier B, Chartier-Harlin MC. NADPH oxidases in Parkinson’s disease: a systematic review. Mol Neurodegener. 2017;12:84.
Grammatopoulos TN, Jones SM, Ahmadi FA, Hoover BR, Snell LD, Skoch J, et al. Angiotensin type 1 receptor antagonist losartan, reduces MPTP-induced degeneration of dopaminergic neurons in substantia nigra. Mol Neurodegener. 2007;2:1.
Rodriguez-Pallares J, Rey P, Parga JA, Munoz A, Guerra MJ, Labandeira-Garcia JL. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ros. Neurobiol Dis. 2008;31:58–73.
Kambayashi Y, Bardhan S, Takahashi K, Tsuzuki S, Inui H, Hamakubo T, et al. Molecular cloning of a novel angiotensin II receptor isoform involved in phosphotyrosine phosphatase inhibition. J Biol Chem. 1993;268:24543–6.
Nakajima M, Mukoyama M, Pratt RE, Horiuchi M, Dzau VJ. Cloning of cdna and analysis of the gene for mouse angiotensin II type 2 receptor. Biochem Biophys Res Commun. 1993;197:393–9.
Gard PR. The role of angiotensin II in cognition and behaviour. Eur J Pharmacol. 2002;438:1–14.
de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. Xxiii. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–72.
Bhat SA, Sood A, Shukla R, Hanif K. At2r activation prevents microglia pro-inflammatory activation in a nox-dependent manner: inhibition of pkc activation and p47(phox) phosphorylation by pp2a. Mol Neurobiol. 2019;56:3005–23.
McCarthy CA, Widdop RE, Denton KM, Jones ES. Update on the angiotensin at(2) receptor. Curr Hypertens Rep. 2013;15:25–30.
Patel SN, Fatima N, Ali R, Hussain T. Emerging role of angiotensin at2 receptor in anti-inflammation: an update. Curr Pharm Des. 2020;26:492–500.
Garrido-Gil P, Pedrosa MA, Garcia-Garrote M, Pequeno-Valtierra A, Rodriguez-Castro J, Garcia-Souto D, et al. Microglial angiotensin type 2 receptors mediate sex-specific expression of inflammatory cytokines independently of circulating estrogen. Glia. 2022;70:2348–60.
Nguyen G, Contrepas A. The (pro)renin receptors. J Mol Med (Berl). 2008;86:643–6.
Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, Sexton PM, et al. G-protein-coupled receptor mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation. 2005;111:1806–13.
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ace2/angiotensin-(1–7)/mas axis of the renin-angiotensin system: Focus on angiotensin-(1–7). Physiol Rev. 2018;98:505–53.
Hrenak J, Paulis L, Simko F. Angiotensin a/alamandine/mrgd axis: another clue to understanding cardiovascular pathophysiology. Int J Mol Sci. 2016;17:1098.
Valenzuela R, Rodriguez-Perez AI, Costa-Besada MA, Rivas-Santisteban R, Garrido-Gil P, Lopez-Lopez A, et al. An ace2/mas-related receptor mrge axis in dopaminergic neuron mitochondria. Redox Biol. 2021;46:102078.
Jackson L, Eldahshan W, Fagan SC, Ergul A. Within the brain: the renin angiotensin system. Int J Mol Sci. 2018;19:876.
Labandeira-Garcia JL, Rodriguez-Perez AI, Garrido-Gil P, Rodriguez-Pallares J, Lanciego JL, Guerra MJ. Brain renin-angiotensin system and microglial polarization: implications for aging and neurodegeneration. Front Aging Neurosci. 2017;9:129.
Labandeira-Garcia JL, Valenzuela R, Costa-Besada MA, Villar-Cheda B, Rodriguez-Perez AI. The intracellular renin-angiotensin system: Friend or foe. Some light from the dopaminergic neurons. Prog Neurobiol. 2021;199:101919.
Paz Ocaranza M, Riquelme JA, Garcia L, Jalil JE, Chiong M, Santos RAS, et al. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat Rev Cardiol. 2020;17:116–29.
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. Sars-cov-2 cell entry depends on ace2 and tmprss2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80 e8.
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of sars-cov-2 by full-length human ace2. Science. 2020;367:1444–8.
Brojakowska A, Narula J, Shimony R, Bander J. Clinical implications of sars-cov-2 interaction with renin angiotensin system: JACC review topic of the week. J Am Coll Cardiol. 2020;75:3085–95.
Ferrara F, Vitiello A. The renin-angiotensin system and specifically angiotensin-converting enzyme 2 as a potential therapeutic target in sars-cov-2 infections. Naunyn Schmiedebergs Arch Pharmacol. 2021;394:1589–93.
Wang S, Guo F, Liu K, Wang H, Rao S, Yang P, et al. Endocytosis of the receptor-binding domain of sars-cov spike protein together with virus receptor ace2. Virus Res. 2008;136:8–15.
South AM, Tomlinson L, Edmonston D, Hiremath S, Sparks MA. Controversies of renin-angiotensin system inhibition during the COVID-19 pandemic. Nat Rev Nephrol. 2020;16:305–7.
Wang K, Gheblawi M, Oudit GY. Angiotensin converting enzyme 2: A double-edged sword. Circulation. 2020;142:426–8.
Pedrosa MA, Valenzuela R, Garrido-Gil P, Labandeira CM, Navarro G, Franco R, et al. Experimental data using candesartan and captopril indicate no double-edged sword effect in COVID-19. Clin Sci (Lond). 2021;135:465–81.
Valenzuela R, Pedrosa MA, Garrido-Gil P, Labandeira CM, Navarro G, Franco R, et al. Interactions between ibuprofen, ace2, renin-angiotensin system, and spike protein in the lung. Implications for COVID-19. Clin Transl Med. 2021;11:e371.
Landolfo M, Maino A, Di Salvo E, Fiorini G, Peterlana D, Borghi C. Renin-angiotensin system modulation and outcomes in patients hospitalized for interstitial sars-cov2 pneumonia: a cohort study. Intern Emerg Med. 2022;17:1335–41.
Nunez-Gil IJ, Olier I, Feltes G, Viana-Llamas MC, Maroun-Eid C, Romero R, et al. Renin-angiotensin system inhibitors effect before and during hospitalization in COVID-19 outcomes: Final analysis of the international hope COVID-19 (health outcome predictive evaluation for COVID-19) registry. Am Heart J. 2021;237:104–15.
Labandeira-Garcia JL, Labandeira CM, Valenzuela R, Pedrosa MA, Quijano A, Rodriguez-Perez AI. Drugs modulating renin-angiotensin system in COVID-19 treatment. Biomedicines. 2022;10:502.
Smeyne RJ, Eells JB, Chatterjee D, Byrne M, Akula SM, Sriramula S, et al. COVID-19 infection enhances susceptibility to oxidative stress-induced Parkinsonism. Mov Disord. 2022;37:1394–404.
Tiwari S, Yadav N, Singh S. COVID-19 and Parkinson’s disease: possible links in pathology and therapeutics. Neurotox Res. 2022;40:1586–96.
Li XC, Zhu D, Zheng X, Zhang J, Zhuo JL. Intratubular and intracellular renin-angiotensin system in the kidney: a unifying perspective in blood pressure control. Clin Sci (Lond). 2018;132:1383–401.
Re RN. Role of intracellular angiotensin II. Am J Physiol Heart Circ Physiol. 2018;314:H766–71.
Costa-Besada MA, Valenzuela R, Garrido-Gil P, Villar-Cheda B, Parga JA, Lanciego JL, et al. Paracrine and intracrine angiotensin 1–7/mas receptor axis in the substantia nigra of rodents, monkeys, and humans. Mol Neurobiol. 2018;55:5847–67.
Valenzuela R, Costa-Besada MA, Iglesias-Gonzalez J, Perez-Costas E, Villar-Cheda B, Garrido-Gil P, et al. Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging-related vulnerability to neurodegeneration. Cell Death Dis. 2016;7:e2427.
Villar-Cheda B, Costa-Besada MA, Valenzuela R, Perez-Costas E, Melendez-Ferro M, Labandeira-Garcia JL. The intracellular angiotensin system buffers deleterious effects of the extracellular paracrine system. Cell Death Dis. 2017;8:e3044.
Ferre S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, et al. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5:131–4.
Durdagi S, Erol I, Salmas RE, Aksoydan B, Kantarcioglu I. Oligomerization and cooperativity in GPCRS from the perspective of the angiotensin AT1 and dopamine D2 receptors. Neurosci Lett. 2019;700:30–7.
Rivas-Santisteban R, Lillo J, Munoz A, Rodriguez-Perez AI, Labandeira-Garcia JL, Navarro G, et al. Novel interactions involving the MAS receptor show potential of the renin-angiotensin system in the regulation of microglia activation: altered expression in Parkinsonism and dyskinesia. Neurotherapeutics. 2021;18:998–1016.
Rivas-Santisteban R, Lillo J, Raich I, Munoz A, Lillo A, Rodriguez-Perez AI, et al. The cannabinoid CB(1) receptor interacts with the angiotensin AT(2) receptor. Overexpression of AT(2)-CB(1) receptor heteromers in the striatum of 6-hydroxydopamine hemilesioned rats. Exp Neurol. 2023;362:114319.
Szalai B, Barkai L, Turu G, Szidonya L, Varnai P, Hunyady L. Allosteric interactions within the AT(1) angiotensin receptor homodimer: Role of the conserved dry motif. Biochem Pharmacol. 2012;84:477–85.
AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an at1 receptor antagonist. J Biol Chem. 2001;276:39721–6.
Zeng C, Liu Y, Wang Z, He D, Huang L, Yu P, et al. Activation of d3 dopamine receptor decreases angiotensin II type 1 receptor expression in rat renal proximal tubule cells. Circ Res. 2006;99:494–500.
Rozenfeld R, Gupta A, Gagnidze K, Lim MP, Gomes I, Lee-Ramos D, et al. AT1R-CB(1)R heteromerization reveals a new mechanism for the pathogenic properties of angiotensin II. EMBO J. 2011;30:2350–63.
Martinez-Pinilla E, Rodriguez-Perez AI, Navarro G, Aguinaga D, Moreno E, Lanciego JL, et al. Dopamine D2 and angiotensin II type 1 receptors form functional heteromers in rat striatum. Biochem Pharmacol. 2015;96:131–42.
Phillips MI, de Oliveira EM. Brain renin angiotensin in disease. J Mol Med (Berl). 2008;86:715–22.
Harding JW, Sullivan MJ, Hanesworth JM, Cushing LL, Wright JW. Inability of [125I]SAR1, ILE8-angiotensin II to move between the blood and cerebrospinal fluid compartments. J Neurochem. 1988;50:554–7.
Hermann K, McDonald W, Unger T, Lang RE, Ganten D. Angiotensin biosynthesis and concentrations in brain of normotensive and hypertensive rats. J Physiol (Paris). 1984;79:471–80.
Milsted A, Barna BP, Ransohoff RM, Brosnihan KB, Ferrario CM. Astrocyte cultures derived from human brain tissue express angiotensinogen mrna. Proc Natl Acad Sci U S A. 1990;87:5720–3.
Stornetta RL, Hawelu-Johnson CL, Guyenet PG, Lynch KR. Astrocytes synthesize angiotensinogen in brain. Science. 1988;242:1444–6.
Kumar A, Rassoli A, Raizada MK. Angiotensinogen gene expression in neuronal and glial cells in primary cultures of rat brain. J Neurosci Res. 1988;19:287–90.
Thomas WG, Greenland KJ, Shinkel TA, Sernia C. Angiotensinogen is secreted by pure rat neuronal cell cultures. Brain Res. 1992;588:191–200.
van Thiel BS, Goes Martini A, Te Riet L, Severs D, Uijl E, Garrelds IM, et al. Brain renin-angiotensin system: Does it exist? Hypertension. 2017;69:1136–44.
Bader M, Ganten D. It’s renin in the brain: transgenic animals elucidate the brain renin angiotensin system. Circ Res. 2002;90:8–10.
Lavoie JL, Cassell MD, Gross KW, Sigmund CD. Localization of renin expressing cells in the brain, by use of a REN-EGFP transgenic model. Physiol Genomics. 2004;16:240–6.
Lopez-Lopez A, Villar-Cheda B, Quijano A, Garrido-Gil P, Garcia-Garrote M, Diaz-Ruiz C, et al. NADPH-oxidase, rho-kinase and autophagy mediate the (pro)renin-induced pro-inflammatory microglial response and enhancement of dopaminergic neuron death. Antioxidants (Basel). 2021;10:1340.
Valenzuela R, Barroso-Chinea P, Villar-Cheda B, Joglar B, Munoz A, Lanciego JL, et al. Location of prorenin receptors in primate substantia nigra: effects on dopaminergic cell death. J Neuropathol Exp Neurol. 2010;69:1130–42.
Labandeira CM, Pedrosa MA, Quijano A, Valenzuela R, Garrido-Gil P, Sanchez-Andrade M, et al. Angiotensin type-1 receptor and ACE2 autoantibodies in Parkinson’S disease. NPJ Parkinsons Dis. 2022;8:76.
Pedrosa MA, Labandeira CM, Valenzuela R, Quijano A, Sanchez-Andrade M, Suarez-Quintanilla JA, et al. AT1 receptor autoantibodies mediate effects of metabolic syndrome on dopaminergic vulnerability. Brain Behav Immun. 2023;108:255–68.
Allen AM, MacGregor DP, Chai SY, Donnan GA, Kaczmarczyk S, Richardson K, et al. Angiotensin II receptor binding associated with nigrostriatal dopaminergic neurons in human basal ganglia. Ann Neurol. 1992;32:339–44.
Quinlan JT, Phillips MI. Immunoreactivity for an angiotensin II-like peptide in the human brain. Brain Res. 1981;205:212–8.
Simonnet G, Giorguieff-Chesselet MF, Carayon A, Bioulac B, Cesselin F, Glowinski J, et al. Angiotensin II and nigostriatal system (author’s transl). J Physiol (Paris). 1981;77:71–9.
Joglar B, Rodriguez-Pallares J, Rodriguez-Perez AI, Rey P, Guerra MJ, Labandeira-Garcia JL. The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: relevance to progression of the disease. J Neurochem. 2009;109:656–69.
Garrido-Gil P, Rodriguez-Perez AI, Fernandez-Rodriguez P, Lanciego JL, Labandeira-Garcia JL. Expression of angiotensinogen and receptors for angiotensin and prorenin in the rat and monkey striatal neurons and glial cells. Brain Struct Funct. 2017;222:2559–71.
Garrido-Gil P, Valenzuela R, Villar-Cheda B, Lanciego JL, Labandeira-Garcia JL. Expression of angiotensinogen and receptors for angiotensin and prorenin in the monkey and human substantia nigra: an intracellular renin-angiotensin system in the nigra. Brain Struct Funct. 2013;218:373–88.
Rodriguez-Perez AI, Borrajo A, Rodriguez-Pallares J, Guerra MJ, Labandeira-Garcia JL. Interaction between NADPH-oxidase and rho-kinase in angiotensin II-induced microglial activation. Glia. 2015;63:466–82.
Rodriguez-Perez AI, Garrido-Gil P, Pedrosa MA, Garcia-Garrote M, Valenzuela R, Navarro G, et al. Angiotensin type 2 receptors: Role in aging and neuroinflammation in the substantia nigra. Brain Behav Immun. 2020;87:256–71.
Kamath T, Abdulraouf A, Burris SJ, Langlieb J, Gazestani V, Nadaf NM, et al. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in Parkinson’s disease. Nat Neurosci. 2022;25:588–95.
Garrido-Gil P, Dominguez-Meijide A, Moratalla R, Guerra MJ, Labandeira-Garcia JL. Aging-related dysregulation in enteric dopamine and angiotensin system interactions: Implications for gastrointestinal dysfunction in the elderly. Oncotarget. 2018;9:10834–46.
Garrido-Gil P, Rodriguez-Perez AI, Dominguez-Meijide A, Guerra MJ, Labandeira-Garcia JL. Bidirectional neural interaction between central dopaminergic and gut lesions in Parkinson’s disease models. Mol Neurobiol. 2018;55:7297–316.
Garrido-Gil P, Rodriguez-Perez AI, Lage L, Labandeira-Garcia JL. Estrogen deficiency and colonic function: surgical menopause and sex differences in angiotensin and dopamine receptor interaction. J Gerontol A Biol Sci Med Sci. 2021;76:1533–41.
Gildea JJ. Dopamine and angiotensin as renal counterregulatory systems controlling sodium balance. Curr Opin Nephrol Hypertens. 2009;18:28–32.
Gildea JJ, Xu P, Kemp BA, Carey RM, Jose PA, Felder RA. The dopamine D(1) receptor and angiotensin II type-2 receptor are required for inhibition of sodium transport through a protein phosphatase 2A pathway. Hypertension. 2019;73:1258–65.
Brown DC, Steward LJ, Ge J, Barnes NM. Ability of angiotensin II to modulate striatal dopamine release via the AT1 receptor in vitro and in vivo. Br J Pharmacol. 1996;118:414–20.
Mendelsohn FA, Jenkins TA, Berkovic SF. Effects of angiotensin II on dopamine and serotonin turnover in the striatum of conscious rats. Brain Res. 1993;613:221–9.
Aschrafi A, Berndt A, Kowalak JA, Gale JR, Gioio AE, Kaplan BB. Angiotensin II mediates the axonal trafficking of tyrosine hydroxylase and dopamine beta-hydroxylase mrnas and enhances norepinephrine synthesis in primary sympathetic neurons. J Neurochem. 2019;150:666–77.
Villar-Cheda B, Dominguez-Meijide A, Valenzuela R, Granado N, Moratalla R, Labandeira-Garcia JL. Aging-related dysregulation of dopamine and angiotensin receptor interaction. Neurobiol Aging. 2014;35:1726–38.
Villar-Cheda B, Rodriguez-Pallares J, Valenzuela R, Munoz A, Guerra MJ, Baltatu OC, et al. Nigral and striatal regulation of angiotensin receptor expression by dopamine and angiotensin in rodents: implications for progression of Parkinson’s disease. Eur J Neurosci. 2010;32:1695–706.
Rivas-Santisteban R, Rodriguez-Perez AI, Munoz A, Reyes-Resina I, Labandeira-Garcia JL, Navarro G, et al. Angiotensin AT(1) and AT(2) receptor heteromer expression in the hemilesioned rat model of Parkinson’s disease that increases with levodopa-induced dyskinesia. J Neuroinflammation. 2020;17:243.
Dominguez-Meijide A, Rodriguez-Perez AI, Diaz-Ruiz C, Guerra MJ, Labandeira-Garcia JL. Dopamine modulates astroglial and microglial activity via glial renin-angiotensin system in cultures. Brain Behav Immun. 2017;62:277–90.
Rey P, Lopez-Real A, Sanchez-Iglesias S, Munoz A, Soto-Otero R, Labandeira-Garcia JL. Angiotensin type-1-receptor antagonists reduce 6-hydroxydopamine toxicity for dopaminergic neurons. Neurobiol Aging. 2007;28:555–67.
Sonsalla PK, Coleman C, Wong LY, Harris SL, Richardson JR, Gadad BS, et al. The angiotensin converting enzyme inhibitor captopril protects nigrostriatal dopamine neurons in animal models of Parkinsonism. Exp Neurol. 2013;250:376–83.
Zawada WM, Banninger GP, Thornton J, Marriott B, Cantu D, Rachubinski AL, et al. Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. J Neuroinflammation. 2011;8:129.
Rodriguez-Perez AI, Sucunza D, Pedrosa MA, Garrido-Gil P, Kulisevsky J, Lanciego JL, et al. Angiotensin type 1 receptor antagonists protect against alpha-synuclein-induced neuroinflammation and dopaminergic neuron death. Neurotherapeutics. 2018;15:1063–81.
Lopez-Real A, Rey P, Soto-Otero R, Mendez-Alvarez E, Labandeira-Garcia JL. Angiotensin-converting enzyme inhibition reduces oxidative stress and protects dopaminergic neurons in a 6-hydroxydopamine rat model of Parkinsonism. J Neurosci Res. 2005;81:865–73.
Munoz A, Rey P, Guerra MJ, Mendez-Alvarez E, Soto-Otero R, Labandeira-Garcia JL. Reduction of dopaminergic degeneration and oxidative stress by inhibition of angiotensin converting enzyme in a MPTP model of Parkinsonism. Neuropharmacology. 2006;51:112–20.
Konings CH, Kuiper MA, Bergmans PL, Grijpma AM, van Kamp GJ, Wolters EC. Increased angiotensin-converting enzyme activity in cerebrospinal fluid of treated patients with Parkinson’s disease. Clin Chim Acta. 1994;231:101–6.
Lin JJ, Yueh KC, Chang DC, Lin SZ. Association between genetic polymorphism of angiotensin-converting enzyme gene and Parkinson’s disease. J Neurol Sci. 2002;199:25–9.
Reardon KA, Mendelsohn FA, Chai SY, Horne MK. The angiotensin converting enzyme (ace) inhibitor, perindopril, modifies the clinical features of Parkinson’s disease. Aust N Z J Med. 2000;30:48–53.
Becker C, Jick SS, Meier CR. Use of antihypertensives and the risk of Parkinson disease. Neurology. 2008;70:1438–44.
Lee YC, Lin CH, Wu RM, Lin JW, Chang CH, Lai MS. Antihypertensive agents and risk of Parkinson’s disease: a nationwide cohort study. PLoS ONE. 2014;9:e98961.
Ascherio A, Tanner CM. Use of antihypertensives and the risk of Parkinson disease. Neurology. 2009;72:578–9.
Kulisevsky J, Martinez-Horta S, Campolongo A, Pascual-Sedano B, Marin-Lahoz J, Bejr-Kasem H, et al. A randomized clinical trial of candesartan for cognitive impairment in Parkinson’s disease. Parkinsonism Relat Disord. 2023;110:105367.
Udovin L, Otero-Losada M, Bordet S, Chevalier G, Quarracino C, Capani F, et al. Effects of angiotensin type 1 receptor antagonists on Parkinson’s disease progression: An exploratory study in the PPMI database. Parkinsonism Relat Disord. 2021;86:34–7.
Visanji NP, Madan P, Lacoste AMB, Buleje I, Han Y, Spangler S, et al. Using artificial intelligence to identify anti-hypertensives as possible disease modifying agents in Parkinson’s disease. Pharmacoepidemiol Drug Saf. 2021;30:201–9.
Jo Y, Kim S, Ye BS, Lee E, Yu YM. Protective effect of renin-angiotensin system inhibitors on Parkinson’s disease: a nationwide cohort study. Front Pharmacol. 2022;13:837890.
Lin HC, Tseng YF, Shen AL, Chao JC, Hsu CY, Lin HL. Association of angiotensin receptor blockers with incident Parkinson disease in patients with hypertension: a retrospective cohort study. Am J Med. 2022;135:1001–7.
Labandeira-Garcia JL, Parga JA. Nigral neurons degenerating in Parkinson’s disease express the angiotensin receptor type 1 gene. Mov Disord. 2022;37:1610–1.
Yamada T, McGeer PL, Baimbridge KG, McGeer EG. Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-d28k. Brain Res. 1990;526:303–7.
Zampese E, Surmeier DJ. Calcium, bioenergetics, and Parkinson’s disease. Cells. 2020;9:2045.
Gao HM, Liu B, Zhang W, Hong JS. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. FASEB J. 2003;17:1954–6.
Rodriguez-Pallares J, Parga JA, Munoz A, Rey P, Guerra MJ, Labandeira-Garcia JL. Mechanism of 6-hydroxydopamine neurotoxicity: The role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J Neurochem. 2007;103:145–56.
Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, et al. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2003;100:6145–50.
Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–96.
Wosniak J Jr, Santos CX, Kowaltowski AJ, Laurindo FR. Cross-talk between mitochondria and NADPH oxidase: Effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal. 2009;11:1265–78.
Rodriguez-Pallares J, Parga JA, Joglar B, Guerra MJ, Labandeira-Garcia JL. The mitochondrial ATP-sensitive potassium channel blocker 5-hydroxydecanoate inhibits toxicity of 6-hydroxydopamine on dopaminergic neurons. Neurotox Res. 2009;15:82–95.
Rodriguez-Pallares J, Parga JA, Joglar B, Guerra MJ, Labandeira-Garcia JL. Mitochondrial ATP-sensitive potassium channels enhance angiotensin-induced oxidative damage and dopaminergic neuron degeneration. Relevance for aging-associated susceptibility to Parkinson’s disease. Age (Dordr). 2012;34:863–80.
Ruiz-Ortega M, Lorenzo O, Suzuki Y, Ruperez M, Egido J. Proinflammatory actions of angiotensins. Curr Opin Nephrol Hypertens. 2001;10:321–9.
Liu Y, Zhu X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl Neurodegener. 2017;6:21.
Michel PP, Hirsch EC, Hunot S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron. 2016;90:675–91.
Rittenhouse AR, Zigmond RE. Role of n- and l-type calcium channels in depolarization-induced activation of tyrosine hydroxylase and release of norepinephrine by sympathetic cell bodies and nerve terminals. J Neurobiol. 1999;40:137–48.
Kobiec T, Otero-Losada M, Chevalier G, Udovin L, Bordet S, Menendez-Maissonave C, et al. The renin-angiotensin system modulates dopaminergic neurotransmission: a new player on the scene. Front Synaptic Neurosci. 2021;13:638519.
Arreola R, Alvarez-Herrera S, Perez-Sanchez G, Becerril-Villanueva E, Cruz-Fuentes C, Flores-Gutierrez EO, et al. Immunomodulatory effects mediated by dopamine. J Immunol Res. 2016;2016:3160486.
Sarkar C, Basu B, Chakroborty D, Dasgupta PS, Basu S. The immunoregulatory role of dopamine: an update. Brain Behav Immun. 2010;24:525–8.
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160:62–73.
Zhu J, Hu Z, Han X, Wang D, Jiang Q, Ding J, et al. Dopamine d2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of beta-arrestin2 and NLRP3. Cell Death Differ. 2018;25:2037–49.
Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797:897–906.
Quijano A, Diaz-Ruiz C, Lopez-Lopez A, Villar-Cheda B, Munoz A, Rodriguez-Perez AI, et al. Angiotensin type-1 receptor inhibition reduces nlrp3 inflammasome upregulation induced by aging and neurodegeneration in the substantia nigra of male rodents and primary mesencephalic cultures. Antioxidants (Basel). 2022;11:329.
Villar-Cheda B, Valenzuela R, Rodriguez-Perez AI, Guerra MJ, Labandeira-Garcia JL. Aging-related changes in the nigral angiotensin system enhances proinflammatory and pro-oxidative markers and 6-OHDA-induced dopaminergic degeneration. Neurobiol Aging. 2012;33(204):e1-11.
Diaz-Ruiz C, Villar-Cheda B, Dominguez-Meijide A, Garrido-Gil P, Guerra MJ, Labandeira-Garcia JL. Aging-related overactivity of the angiotensin/at1 axis decreases sirtuin 3 levels in the substantia nigra, which induces vulnerability to oxidative stress and neurodegeneration. J Gerontol A Biol Sci Med Sci. 2020;75:416–24.
Ishibashi K, Ishii K, Oda K, Kawasaki K, Mizusawa H, Ishiwata K. Regional analysis of age-related decline in dopamine transporters and dopamine D2-like receptors in human striatum. Synapse. 2009;63:282–90.
Wang Y, Chan GL, Holden JE, Dobko T, Mak E, Schulzer M, et al. Age-dependent decline of dopamine d1 receptors in human brain: a pet study. Synapse. 1998;30:56–61.
Collier TJ, Lipton J, Daley BF, Palfi S, Chu Y, Sortwell C, et al. Aging-related changes in the nigrostriatal dopamine system and the response to mptp in nonhuman primates: Diminished compensatory mechanisms as a prelude to Parkinsonism. Neurobiol Dis. 2007;26:56–65.
Kubis N, Faucheux BA, Ransmayr G, Damier P, Duyckaerts C, Henin D, et al. Preservation of midbrain catecholaminergic neurons in very old human subjects. Brain. 2000;123(Pt 2):366–73.
Cassis P, Conti S, Remuzzi G, Benigni A. Angiotensin receptors as determinants of life span. Pflugers Arch. 2010;459:325–32.
Min LJ, Mogi M, Iwai M, Horiuchi M. Signaling mechanisms of angiotensin II in regulating vascular senescence. Ageing Res Rev. 2009;8:113–21.
Thompson MM, Oyama TT, Kelly FJ, Kennefick TM, Anderson S. Activity and responsiveness of the renin-angiotensin system in the aging rat. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1787–94.
Chen J, Yang S, Hu S, Choudhry MA, Bland KI, Chaudry IH. Estrogen prevents intestinal inflammation after trauma-hemorrhage via downregulation of angiotensin II and angiotensin II subtype I receptor. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1131–7.
Dean SA, Tan J, O’Brien ER, Leenen FH. 17beta-estradiol downregulates tissue angiotensin-converting enzyme and Ang II type 1 receptor in female rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R759–66.
Rodriguez-Perez AI, Borrajo A, Valenzuela R, Lanciego JL, Labandeira-Garcia JL. Critical period for dopaminergic neuroprotection by hormonal replacement in menopausal rats. Neurobiol Aging. 2015;36:1194–208.
Rodriguez-Perez AI, Valenzuela R, Joglar B, Garrido-Gil P, Guerra MJ, Labandeira-Garcia JL. Renin angiotensin system and gender differences in dopaminergic degeneration. Mol Neurodegener. 2011;6:58.
Rodriguez-Perez AI, Valenzuela R, Villar-Cheda B, Guerra MJ, Labandeira-Garcia JL. Dopaminergic neuroprotection of hormonal replacement therapy in young and aged menopausal rats: role of the brain angiotensin system. Brain. 2012;135:124–38.
Rodriguez-Perez AI, Valenzuela R, Villar-Cheda B, Guerra MJ, Lanciego JL, Labandeira-Garcia JL. Estrogen and angiotensin interaction in the substantia nigra. Relevance to postmenopausal Parkinson’s disease. Exp Neurol. 2010;224:517–26.
Lage L, Rodriguez-Perez AI, Villar-Cheda B, Labandeira-Garcia JL, Dominguez-Meijide A. Angiotensin type 1 receptor activation promotes neuronal and glial alpha-synuclein aggregation and transmission. NPJ Parkinsons Dis. 2024;10(1):37.
Kitazaki Y, Ikawa M, Yamaguchi T, Enomoto S, Shirafuji N, Yamamura O, et al. Regional cortical hypoperfusion and atrophy correlate with striatal dopaminergic loss in Parkinson’s disease: a study using arterial spin labeling mr perfusion. Neuroradiology. 2023;65:569–77.
Papapetropoulos S, Ellul J, Argyriou AA, Talelli P, Chroni E, Papapetropoulos T. The effect of vascular disease on late onset Parkinson’s disease. Eur J Neurol. 2004;11:231–5.
Rodriguez-Perez AI, Dominguez-Meijide A, Lanciego JL, Guerra MJ, Labandeira-Garcia JL. Dopaminergic degeneration is enhanced by chronic brain hypoperfusion and inhibited by angiotensin receptor blockage. Age (Dordr). 2013;35:1675–90.
Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–21.
Borrajo A, Rodriguez-Perez AI, Villar-Cheda B, Guerra MJ, Labandeira-Garcia JL. Inhibition of the microglial response is essential for the neuroprotective effects of rho-kinase inhibitors on MPTP-induced dopaminergic cell death. Neuropharmacology. 2014;85:1–8.
Villar-Cheda B, Dominguez-Meijide A, Joglar B, Rodriguez-Perez AI, Guerra MJ, Labandeira-Garcia JL. Involvement of microglial rhoa/rho-kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol Dis. 2012;47:268–79.
Borrajo A, Rodriguez-Perez AI, Diaz-Ruiz C, Guerra MJ, Labandeira-Garcia JL. Microglial TNF-alpha mediates enhancement of dopaminergic degeneration by brain angiotensin. Glia. 2014;62:145–57.
Garrido-Gil P, Rodriguez-Pallares J, Dominguez-Meijide A, Guerra MJ, Labandeira-Garcia JL. Brain angiotensin regulates iron homeostasis in dopaminergic neurons and microglial cells. Exp Neurol. 2013;250:384–96.
Pedrosa MA, Labandeira CM, Lago-Baameiro N, Valenzuela R, Pardo M, Labandeira-Garcia JL, et al. Extracellular vesicles and their renin-angiotensin cargo as a link between metabolic syndrome and Parkinson’s disease. Antioxidants (Basel). 2023;12:2045.
Fandriks L. The renin-angiotensin system and the gastrointestinal mucosa. Acta Physiol (Oxf). 2011;201:157–67.
Garg M, Angus PW, Burrell LM, Herath C, Gibson PR, Lubel JS. Review article: the pathophysiological roles of the renin-angiotensin system in the gastrointestinal tract. Aliment Pharmacol Ther. 2012;35:414–28.
Shi Y, Liu T, He L, Dougherty U, Chen L, Adhikari S, et al. Activation of the renin-angiotensin system promotes colitis development. Sci Rep. 2016;6:27552.
Brudek T. Inflammatory bowel diseases and Parkinson’s disease. J Parkinsons Dis. 2019;9:S331–44.
Jaworska K, Koper M, Ufnal M. Gut microbiota and renin-angiotensin system: a complex interplay at local and systemic levels. Am J Physiol Gastrointest Liver Physiol. 2021;321:G355–66.
Mizoguchi R, Karashima S, Miyajima Y, Ogura K, Kometani M, Aono D, et al. Impact of gut microbiome on the renin-aldosterone system: shika-machi super preventive health examination results. Hypertens Res. 2023;46:2280–92.
Gawrys-Kopczynska M, Konop M, Maksymiuk K, Kraszewska K, Derzsi L, Sozanski K, et al. TMAO, a seafood-derived molecule, produces diuresis and reduces mortality in heart failure rats. Elife. 2020;9:e57028.
Dave LA, Hayes M, Montoya CA, Rutherfurd SM, Moughan PJ. Human gut endogenous proteins as a potential source of angiotensin-i-converting enzyme (ace-i)-, renin inhibitory and antioxidant peptides. Peptides. 2016;76:30–44.
Robles-Vera I, Toral M, de la Visitacion N, Sanchez M, Gomez-Guzman M, Munoz R, et al. Changes to the gut microbiota induced by losartan contributes to its antihypertensive effects. Br J Pharmacol. 2020;177:2006–23.
Karbach SH, Schonfelder T, Brandao I, Wilms E, Hormann N, Jackel S, et al. Gut microbiota promote angiotensin II-induced arterial hypertension and vascular dysfunction. J Am Heart Assoc. 2016;5:e003698.
Guimaraes VHD, Marinho BM, Motta-Santos D, Mendes G, Santos SHS. Nutritional implications in the mechanistic link between the intestinal microbiome, renin-angiotensin system, and the development of obesity and metabolic syndrome. J Nutr Biochem. 2023;113:109252.
Koeglsperger T, Rumpf SL, Schliesser P, Struebing FL, Brendel M, Levin J, et al. Neuropathology of incidental Lewy body & prodromal Parkinson’s disease. Mol Neurodegener. 2023;18:32.
Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, et al. Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology. 2012;79:2307–14.
Masato A, Plotegher N, Boassa D, Bubacco L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol Neurodegener. 2019;14:35.
Beach TG, Adler CH, Sue LI, Peirce JB, Bachalakuri J, Dalsing-Hernandez JE, et al. Reduced striatal tyrosine hydroxylase in incidental Lewy body disease. Acta Neuropathol. 2008;115:445–51.
Dalfo E, Ferrer I. Early alpha-synuclein lipoxidation in neocortex in lewy body diseases. Neurobiol Aging. 2008;29:408–17.
Doorn KJ, Moors T, Drukarch B, van de Berg W, Lucassen PJ, van Dam AM. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun. 2014;2:90.
Cevenini E, Monti D, Franceschi C. Inflamm-ageing. Curr Opin Clin Nutr Metab Care. 2013;16:14–20.
Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9:586.
Choi DY, Zhang J, Bing G. Aging enhances the neuroinflammatory response and alpha-synuclein nitration in rats. Neurobiol Aging. 2010;31:1649–53.
Ungvari Z, Csiszar A, Kaley G. Vascular inflammation in aging. Herz. 2004;29:733–40.
Panicker N, Kam TI, Wang H, Neifert S, Chou SC, Kumar M, et al. Neuronal NLRP3 is a Parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron. 2022;110:2422-37 e9.
Currie LJ, Harrison MB, Trugman JM, Bennett JP, Wooten GF. Postmenopausal estrogen use affects risk for Parkinson disease. Arch Neurol. 2004;61:886–8.
Ragonese P, D’Amelio M, Callari G, Salemi G, Morgante L, Savettieri G. Age at menopause predicts age at onset of Parkinson’s disease. Mov Disord. 2006;21:2211–4.
Callier S, Le Saux M, Lhiaubet AM, Di Paolo T, Rostene W, Pelaprat D. Evaluation of the protective effect of oestradiol against toxicity induced by 6-hydroxydopamine and 1-methyl-4-phenylpyridinium ion (MPP+) towards dopaminergic mesencephalic neurons in primary culture. J Neurochem. 2002;80:307–16.
Leranth C, Roth RH, Elsworth JD, Naftolin F, Horvath TL, Redmond DE Jr. Estrogen is essential for maintaining nigrostriatal dopamine neurons in primates: Implications for Parkinson’s disease and memory. J Neurosci. 2000;20:8604–9.
Suzuki S, Brown CM, Dela Cruz CD, Yang E, Bridwell DA, Wise PM. Timing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions. Proc Natl Acad Sci U S A. 2007;104:6013–8.
Vegeto E, Benedusi V, Maggi A. Estrogen anti-inflammatory activity in brain: a therapeutic opportunity for menopause and neurodegenerative diseases. Front Neuroendocrinol. 2008;29:507–19.
Popat RA, Van Den Eeden SK, Tanner CM, McGuire V, Bernstein AL, Bloch DA, et al. Effect of reproductive factors and postmenopausal hormone use on the risk of Parkinson disease. Neurology. 2005;65:383–90.
Shulman LM. Is there a connection between estrogen and Parkinson’s disease? Parkinsonism Relat Disord. 2002;8:289–95.
Eskildsen SF, Iranzo A, Stokholm MG, Staer K, Ostergaard K, Serradell M, et al. Impaired cerebral microcirculation in isolated rem sleep behaviour disorder. Brain. 2021;144:1498–508.
Harms AS, Ferreira SA, Romero-Ramos M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021;141:527–45.
Sabatino JJ Jr, Probstel AK, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci. 2019;20:728–45.
Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol. 2022;22:657–73.
Pugazhenthi S, Qin L, Reddy PH. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1037–45.
Sandu RE, Buga AM, Uzoni A, Petcu EB, Popa-Wagner A. Neuroinflammation and comorbidities are frequently ignored factors in cns pathology. Neural Regen Res. 2015;10:1349–55.
Nam GE, Kim SM, Han K, Kim NH, Chung HS, Kim JW, et al. Metabolic syndrome and risk of Parkinson disease: a nationwide cohort study. PLoS Med. 2018;15:e1002640.
Park SH, Nam GE, Han K, Huh Y, Kim W, Lee MK, et al. Association of dynamic changes in metabolic syndrome status with the risk of Parkinson’s disease: a nationwide cohort study. J Parkinsons Dis. 2021;11:1751–9.
Etchegoyen M, Nobile MH, Baez F, Posesorski B, Gonzalez J, Lago N, et al. Metabolic syndrome and neuroprotection. Front Neurosci. 2018;12:196.
Bloch S, Obari D, Girouard H. Angiotensin and neurovascular coupling: beyond hypertension. Microcirculation. 2015;22:159–67.
Faraco G, Sugiyama Y, Lane D, Garcia-Bonilla L, Chang H, Santisteban MM, et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J Clin Invest. 2016;126:4674–89.
Fleegal-DeMotta MA, Doghu S, Banks WA. Angiotensin II modulates BBB permeability via activation of the AT(1) receptor in brain endothelial cells. J Cereb Blood Flow Metab. 2009;29:640–7.
Santisteban MM, Ahn SJ, Lane D, Faraco G, Garcia-Bonilla L, Racchumi G, et al. Endothelium-macrophage crosstalk mediates blood-brain barrier dysfunction in hypertension. Hypertension. 2020;76:795–807.
Setiadi A, Korim WS, Elsaafien K, Yao ST. The role of the blood-brain barrier in hypertension. Exp Physiol. 2018;103:337–42.
Saunders BM, Rudnicka C, Filipovska A, Davies S, Ward N, Hricova J, et al. Shining light on the metabolic role of the cytokine tnfsf14 and the implications on hepatic il-6 production. Immunol Cell Biol. 2018;96:41–53.
Tan X, Hu W, Yang S, Dai H, Xu S, Yang G, et al. Association of metabolic syndrome components with circulating levels of cytokine clusters in young women. Endocr Connect. 2021;10:66–75.
Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, et al. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol. 2006;177:5574–84.
Rochfort KD, Cummins PM. The blood-brain barrier endothelium: a target for pro-inflammatory cytokines. Biochem Soc Trans. 2015;43:702–6.
Cano A, Ettcheto M, Bernuz M, Puerta R, Esteban de Antonio E, Sanchez-Lopez E, et al. Extracellular vesicles, the emerging mirrors of brain physiopathology. Int J Biol Sci. 2023;19:721–43.
Gupte M, Boustany-Kari CM, Bharadwaj K, Police S, Thatcher S, Gong MC, et al. Ace2 is expressed in mouse adipocytes and regulated by a high-fat diet. Am J Physiol Regul Integr Comp Physiol. 2008;295:R781–8.
Li JJ, Wang B, Kodali MC, Chen C, Kim E, Patters BJ, et al. In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J Neuroinflammation. 2018;15:8.
Morales-Prieto DM, Murrieta-Coxca JM, Stojiljkovic M, Diezel C, Streicher PE, Henao-Restrepo JA, et al. Small extracellular vesicles from peripheral blood of aged mice pass the blood-brain barrier and induce glial cell activation. Cells. 2022;11:625.
Chiaradia E, Tancini B, Emiliani C, Delo F, Pellegrino RM, Tognoloni A, et al. Extracellular vesicles under oxidative stress conditions: biological properties and physiological roles. Cells. 2021;10:1763.
Engeli S, Schling P, Gorzelniak K, Boschmann M, Janke J, Ailhaud G, et al. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? Int J Biochem Cell Biol. 2003;35:807–25.
Jones BH, Standridge MK, Taylor JW, Moustaid N. Angiotensinogen gene expression in adipose tissue: analysis of obese models and hormonal and nutritional control. Am J Physiol. 1997;273:R236–42.
Pinheiro TA, Barcala-Jorge AS, Andrade JMO, Pinheiro TA, Ferreira ECN, Crespo TS, et al. Obesity and malnutrition similarly alter the renin-angiotensin system and inflammation in mice and human adipose. J Nutr Biochem. 2017;48:74–82.
Frigolet ME, Torres N, Tovar AR. The renin-angiotensin system in adipose tissue and its metabolic consequences during obesity. J Nutr Biochem. 2013;24:2003–15.
Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396:67–72.
Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Bjorklund T, et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805–20.
de Araujo IE, Ferreira JG, Tellez LA, Ren X, Yeckel CW. The gut-brain dopamine axis: a regulatory system for caloric intake. Physiol Behav. 2012;106:394–9.
Tellez LA, Medina S, Han W, Ferreira JG, Licona-Limon P, Ren X, et al. A gut lipid messenger links excess dietary fat to dopamine deficiency. Science. 2013;341:800–2.
Cui H, Elford JD, Alitalo O, Perez-Pardo P, Tampio J, Huttunen KM, et al. Nigrostriatal 6-hydroxydopamine lesions increase alpha-synuclein levels and permeability in rat colon. Neurobiol Aging. 2023;129:62–71.
Horsager J, Andersen KB, Knudsen K, Skjaerbaek C, Fedorova TD, Okkels N, et al. Brain-first versus body-first Parkinson’s disease: a multimodal imaging case-control study. Brain. 2020;143:3077–88.
O’Donovan SM, Crowley EK, Brown JR, O’Sullivan O, O’Leary OF, Timmons S, et al. Nigral overexpression of alpha-synuclein in a rat Parkinson’s disease model indicates alterations in the enteric nervous system and the gut microbiome. Neurogastroenterol Motil. 2020;32:e13726.
Wang Q, Luo Y, Ray Chaudhuri K, Reynolds R, Tan EK, Pettersson S. The role of gut dysbiosis in Parkinson’s disease: mechanistic insights and therapeutic options. Brain. 2021;144:2571–93.
Ahmadi Badi S, Malek A, Paolini A, Rouhollahi Masoumi M, Seyedi SA, Amanzadeh A, et al. Downregulation of ace, agtr1, and ace2 genes mediating SARS-COV-2 pathogenesis by gut microbiota members and their postbiotics on CACO-2 cells. Microb Pathog. 2022;173:105798.
Yu Z, Yang Z, Wang Y, Zhou F, Li S, Li C, et al. Recent advance of ace2 and microbiota dysfunction in COVID-19 pathogenesis. Heliyon. 2021;7:e07548.
Danilenko V, Devyatkin A, Marsova M, Shibilova M, Ilyasov R, Shmyrev V. Common inflammatory mechanisms in COVID-19 and Parkinson’s diseases: the role of microbiome, pharmabiotics and postbiotics in their prevention. J Inflamm Res. 2021;14:6349–81.
Machado AS, Oliveira JR, Lelis DF, de Paula AMB, Guimaraes ALS, Andrade JMO, et al. Oral probiotic bifidobacterium longum supplementation improves metabolic parameters and alters the expression of the renin-angiotensin system in obese mice liver. Biol Res Nurs. 2021;23:100–8.
Buford TW, Sun Y, Roberts LM, Banerjee A, Peramsetty S, Knighton A, et al. Angiotensin (1–7) delivered orally via probiotic, but not subcutaneously, benefits the gut-brain axis in older rats. Geroscience. 2020;42:1307–21.
Carey RM. Functional intracellular renin-angiotensin systems: potential for pathophysiology of disease. Am J Physiol Regul Integr Comp Physiol. 2012;302:R479–81.
Cook JL, Re RN. Lessons from in vitro studies and a related intracellular angiotensin II transgenic mouse model. Am J Physiol Regul Integr Comp Physiol. 2012;302:R482–93.
Ou Z, Jiang T, Gao Q, Tian YY, Zhou JS, Wu L, et al. Mitochondrial-dependent mechanisms are involved in angiotensin II-induced apoptosis in dopaminergic neurons. J Renin Angiotensin Aldosterone Syst. 2016;17:1470320316672349.
Parga JA, Rodriguez-Perez AI, Garcia-Garrote M, Rodriguez-Pallares J, Labandeira-Garcia JL. Angiotensin II induces oxidative stress and upregulates neuroprotective signaling from the NRF2 and KLF9 pathway in dopaminergic cells. Free Radic Biol Med. 2018;129:394–406.
Acknowledgements
Not applicable.
Funding
This review was supported by Spanish Ministry of Science and Innovation (PID2021-126848NB-I00; PLEC2022-009401), Instituto de Salud Carlos III (RD21/0017/0031; PI20/00345, and CIBERNED), Galician Government (XUGA, ED431C 2022/41), and FEDER (Regional European Development Fund).
Author information
Authors and Affiliations
Contributions
JLL-G and AIR-P conceived, supervised, and wrote the manuscript. AIR-P designed and prepared figures. CLG and MJG contributed to writing the article, provided critical feedback, and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests to declare.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Labandeira-Garcia, J.L., Labandeira, C.M., Guerra, M.J. et al. The role of the brain renin-angiotensin system in Parkinson´s disease. Transl Neurodegener 13, 22 (2024). https://doi.org/10.1186/s40035-024-00410-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-024-00410-3