
Abstract
Disruptions of circadian rhythms and sleep cycles are common among neurodegenerative diseases and can occur at multiple levels. Accumulating evidence reveals a bidirectional relationship between disruptions of circadian rhythms and sleep cycles and neurodegenerative diseases. Circadian disruption and sleep disorders aggravate neurodegeneration and neurodegenerative diseases can in turn disrupt circadian rhythms and sleep. Importantly, circadian disruption and various sleep disorders can increase the risk of neurodegenerative diseases. Thus, harnessing the circadian biology findings from preclinical and translational research in neurodegenerative diseases is of importance for reducing risk of neurodegeneration and improving symptoms and quality of life of individuals with neurodegenerative disorders via approaches that normalize circadian in the context of precision medicine. In this review, we discuss the implications of circadian disruption and sleep disorders in neurodegenerative diseases by summarizing evidence from both human and animal studies, focusing on the bidirectional links of sleep and circadian rhythms with prevalent forms of neurodegeneration. These findings provide valuable insights into the pathogenesis of neurodegenerative diseases and suggest a promising role of circadian-based interventions.
Similar content being viewed by others
Background
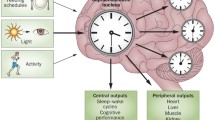
Circadian rhythms are physiological and behavioral oscillations that manifest at every level of tissue from gene expression to interorgan functional coordination, which are regulated by an endogenous process with a periodicity of ~ 24 h that persists in the absence of environmental cues [1]. The circadian system in the brain influences many important functions including sleep-wake cycle, temperature, eating, and social interaction, and is believed to be paramount for maintaining synchrony between internal physiology, behavior, and the stimulus from the environment. The importance of such system lies in several aspects of metabolic, cognitive, immunological and oncogenic processes in the brain. Sleep-wake behavior is the most established and widely recognized sign of circadian system [2]. According to the different manifestations of electroencephalography (EEG), eyes movements and electromyography (EMG), sleep is divided into two different phases: rapid eye movement (REM) and non-rapid eye movement (NREM) sleep. Disruptions of the circadian rhythm can profoundly affect health in a wide range of functions, including sleep, alertness, cognition, psychology, motor control and metabolism [3], and have been correlated with several health problems such as neurodegenerative disorders [2].
Neurodegenerative disorders, especially Parkinson's disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD), involve a wide range of clinical symptoms (e.g., motor and non-motor symptoms), many of which exhibit diurnal and nocturnal variations in frequency and intensity. The prevalence of circadian and sleep dysfunction varies greatly across neurodegenerative diseases (Table 1) and plays an important role in differential diagnosis. Meanwhile, circadian and sleep dysfunctions are not only a consequence of neurodegeneration but may also play a causative role. In other words, dysregulated circadian rhythm and sleep could predispose disease onset or exacerbate disease progression, in which circadian dysfunction and neurodegeneration form a detrimental, self-perpetuating transcriptional-translation feedback loop (TTFL) [4]. A deeper understanding of the relationship between circadian rhythms and neurodegeneration is essential for early identification and management of neurodegenerative diseases.
In this review, we summarize the existing literature on the circadian/sleep disruption in neurodegenerative diseases, focusing on the bidirectional links of circadian rhythm and sleep disruptions with neurodegeneration, based on molecular changes, clinical symptom variations, as well as the available treatment options.
Mechanisms underlying circadian rhythm and sleep-wake activity
Circadian rhythms
Key neuroanatomical pathways of the circadian system
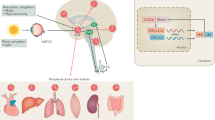
Circadian rhythms govern a wide range of physiological and behavioral processes in organisms, and can be observed at the central and the peripheral. Suprachiasmatic nucleus (SCN), which consists of thousands of neurons that exhibit self-sustaining and synchronous circadian rhythms in their electrical activity, is an important basis for behavioral and physiological rhythms. In humans, the regulation of circadian rhythm begins with the propagation of light information. Light is first detected by intrinsically photoreceptive retinal ganglion cells (ipRGCs) and then delivered to the SCN; the SCN receives and encodes light information, and then synchronizes circadian oscillations and projects signals to other brain regions [5] (Fig. 1).

Key neuroanatomical pathways of the circadian system. (1) SCN indirectly regulates melatonin release from the pineal gland by projecting light signals to PVN. (2) DMH receives light signals from SCN and then projects them to LC and VLPO, which in turn regulates sleep/awake activity. (3) SCN regulates thermoregulation and aggressive tendency by DMH through SPZ or not through SPZ. DMH dorsomedial hypothalamic nucleus; SPZ subparaventricular zone;
The signals encoded by the SCN are mainly projected to the hypothalamus, which acts as a mediator to regulate the specific circadian rhythm. These brain regions include paraventricular nucleus (PVN), dorsomedial hypothalamic nucleus, subparaventricular zone, and medial preoptic nucleus (MPN) [6] (Fig. 1).
Schema of the circadian clock system
Macroscopically, amplitude, phase, and period are key rhythmic parameters driven by the circadian system. Microscopically, circadian rhythm disorders can be limited to alterations in period due to defects in the core molecular mechanism, i.e., the TTFL [7]. TTFL is mainly regulated through heterodimeric partnership between the brain and muscle aryl hydrocarbon receptor nuclear translocator (ARNT)-like 1 (BMAL1) and the circadian locomotor output cycles kaput (CLOCK) (Fig. 2). Period (PER) and cryptochrome (CRY) are involved in this pathway. In addition, the TTFL is complemented by a second loop, in which the REV-ERBα/β repressor and ROR (retinoic acid receptor-related orphan receptor) α/β activator proteins co-maintain the periodic expression of BMAL1 [8, 9] (Fig. 2). The cycle of TTFL is about 24 h [10, 11].

Schema of the circadian clock system—the transcription-translation feedback loops. The circadian clock consists of a network of TTFL that generates endogenous circadian rhythm. TTFL includes two loops: (1) The first loop of TTFL begins with BMAL1:CLOCK complex translocating into the nucleus, activating transcription of target genes containing E-box cis-regulatory enhancer sequences in their promoter regions, such as PER and CRY. The CRY and PER are then transferred to the nucleus and interact with CLOCK:BMAL1 complex to inhibit their own transcription. The decrease of PER and CRY protein levels reduces the suppression of BMAL1:CLOCK activity, which allows for the establishment of a new oscillatory cycle. (2) In the second loop, the REV-ERBα/β repressor and the RORα/β activator proteins co-maintain the periodic expression of BMAL1
Several studies have reported the occurrence of circadian disruption in PD or AD animal models, including changes in circadian rhythm, sleep pattern or clock genes in model animals including mice, rats, Drosophila and zebrafish. The current reports on dysrhythmia in PD and AD models, including neurotoxin-induced and transgenic animal models, are summarized in Tables 2 and 3.
Sleep-wake activity
Sleep-wake activity is regulated by internally driven rhythm of the circadian clock. Disruption of the sleep-wake cycle has been found in a variety of PD and AD models with multiple assessment methods, including running wheels, infrared beams, piezoelectric systems, and electrophysiological measures such as EEG and EMG. Sleep-wake disturbances in PD and AD seem to be associated with genetic mutations. For instance, Drosophila with pink1 (phosphatase and tensin homolog-induced putative kinase 1) and parkin mutants show fragmentation of sleep [12]. Heterozygous (D409V/WT) GBA1 (glucocerebrosidase 1) mutant mice show increased NREM sleep and reduced REM sleep durations [13]. Additionally, α-synuclein BAC transgenic mice exhibit REM sleep without atonia (RSWA), which is a key feature of REM sleep behavior disorder (RBD). Regarding the sleep alterations in animal models of PD and AD, Fifel, Medeiros and their colleagues have comprehensively discussed this topic, together with the strengths and limitations [14, 15].
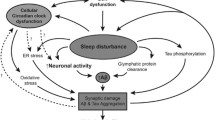
The sleep-wake activity may be related to proteostasis in the brains of patients with neurodegenerative diseases. A few studies have shown that the sleep-wake activity regulates the clearance of misfolded proteins in the glymphatic system of the brain [7]. Interrupted sleep has also been shown to increase the level of Aβ protein in the cerebrospinal fluid [16, 17]. One possible explanation is that Aβ or α-synuclein induces sleep disorders, which in turn hinders their clearance by the lymphatic system, and ultimately accelerates the pathological progression of the disease.
PD
Recent attempts to redefine PD have viewed it as a complex combination of motor and non-motor disorders, with a natural history that includes a prodromal phase dominated by a range of non-motor symptoms (e.g., sleep, psychiatric disorders, autonomic dysfunction, cognitive impairment and sensory deficits) [18]. As with parkinsonian motor abnormalities, non-motor symptoms may also exhibit diurnal fluctuations that change throughout the course of the disease. A dysfunctional circadian system is therefore expected to exacerbate the clinical symptoms of PD patients. It is reasonable to link the symptom fluctuations in PD patients with dysregulation of circadian rhythms affected by chronobiology [19].
Diurnal oscillations are present in characteristic motor and non-motor symptoms of PD. Circadian biomarkers such as melatonin and clock gene, and the neurological processes underlying circadian rhythm, are altered by not only the neurodegeneration of PD, but also dopaminergic treatments used to mitigate parkinsonian symptoms [20]. In general, these circadian dysfunctions in PD can be classified into three categories: behavioral, physiologic, and molecular alterations (Fig. 3).

Circadian dysfunctions in neurodegeneration. Circadian dysfunctions in neurodegeneration can be classified into three categories: behavioral, physiologic, and molecular alterations. There are bi-directional relationships of circadian rhythm and sleep disorder with neurodegeneration
Behavioral alterations in PD
The rest-activity rhythm is often characterized using nonparametric analysis of actigraphy data. The circadian rhythm disruption in PD is characterized by a reduction in the amplitude of the circadian rhythm, resulting in an overall flattening of the rhythm, but with no significant shift in circadian phases [4, 21]. One recent study quantified the actigraphy data, and found that the activity rhythms are associated with disease severity and fluctuations of symptom intensity [22].
Non-motor symptoms in PD are associated to a certain extent with an impaired circadian rhythm. Increasing lines of evidence have shown that circadian dysfunction plays a role in cognitive impairment associated with PD, either by directly affecting cognition or indirectly by exerting effects on sleep and alertness [23]. Several other functions (emotional disorders and gastrointestinal dysfunctions) that are relevant for optimal behavior are known to be altered in PD. However, their link with the circadian clock remains poorly understood [4].
Sleep disorders are the most common non-motor symptoms and comprise the entire spectrum of sleep disorders, mainly presenting with disorders of regulation of sleep and wakefulness (such as insomnia and daytime sleepiness), parasomnias (mainly REM sleep behavior disorders, but also, albeit more rarely, sleepwalking and overlap parasomnia), sleep-related movement disorders (restless leg syndrome, RLS) and sleep‑disordered breathing (SDB) [24]. Insomnia is a frequent symptom in PD and most PD patients often complain about sleep fragmentation and early awakening. The prevalence of insomnia in PD increases over time and requires periodic assessments. PD patients with depressive symptoms, motor fluctuations and the use of higher doses of dopamine agonists tend to suffer more severe insomnia [25]. Interestingly, insomnia is often one of the characteristics of PD where motor symptoms are improved upon awakening from sleep and prior to drug intake. However, the underlying mechanism of this phenomenon (“sleep benefit”) is unclear, and there is no direct evidence for its correlation with the circadian type predominance [26]. Excessive daytime sleepiness (EDS) is a major health hazard in PD, affecting 21%–76% of PD patients with an incidence of 6% per year [27]. In our previous study employing 586 PD patients, male sex, disease duration and depression were found to be main risk factors for EDS in PD patients, while depression was a predictive factor for poor night-time sleep quality in all PD patients, whether they were male or female, and had early- or late-onset PD [28]. Circadian dysfunction may underlie the excessive sleepiness in PD. Compared with PD patients without EDS, patients with EDS have significantly lower amplitude of the melatonin rhythm and 24-h area-under-the-curve for circulating melatonin level [29]. RBD is a parasomnia, characterized by dream-enacting behaviors (DEB) and nightmares linked to RSWA. Compared with PD without RBD, PD with RBD exhibits clinical heterogeneity of motor and non-motor symptoms. More specifically, both DEB and RSWA are associated with the severity of PD. DEB symptom might fluctuate or disappear over time whereas RSWA may continue to develop as PD progresses [30]. RLS has one essential diagnostic characteristic, the presence of circadian variation of symptoms. The circadian clock-controlled gene, Tef, is also associated with sleep disturbances in PD, including RLS symptoms [31]. SDB is not more frequent in PD than in the general population. We previously explored the clinical characteristics of PD with comorbid obstructive sleep apnea (OSA), and found that age and male gender are risk factors for OSA in PD [32]. OSA may exacerbate neurodegenerative processes in PD. It is hypothesized that the OSA-related intermittent hypoxemia leads to oxidative stress, neuroinflammation, cerebrovascular effects, disruption of the glymphatic function by sleep fragmentation, and changes in the integrity of the blood–brain barrier [33].
Nocturia is a condition that is caused by the dampening or reversal of the daily pattern of urine excretion, which is associated with poor quality of life, falls, and institutionalization in PD. The prevalence of nocturia ranges between 76% and 86% according to a previous questionnaire survey [34]. Recent advances in circadian biology and sleep science have raised the importance of considering nocturia as a form of circadian dysfunction, with a focus on the influence of circadian genes on the bladder as studies have demonstrated circadian gene cycling in all levels of bladder tissue, and abnormal nocturnal urine production [35].
Physiologic alterations in PD
The physiological facets of circadian dysfunctions in PD include dysfunction of the autonomic nervous system and dysrhythmias of neuroendocrine secretion.
Rhythmic abnormalities in the autonomic nerve function in PD are well recognized, including reversal or even a full arrhythmia of blood pressure and heart rate variability (HRV), and impairment of the core body temperature (CBT) rhythm [20]. Nocturnal hypertension is nearly ubiquitous in PD, and most PD patients exhibit either a blunted nocturnal fall of blood pressure or higher blood pressure during the night than during the day (known as reverse dipping), leading to the difference between daytime and night-time BP closed [36]. HRV, an index for the autonomic, especially parasympathetic functions, is lower all day but higher at night in individuals with PD than in healthy controls [37]. Research has shown that impaired HRV may be related with the disease severity, motor symptom duration and dopaminergic dose in PD [38]. PD patients also show disruptions of circadian thermoregulation, with significant reductions in the mesor (the mean value around which the core temperature rhythm oscillates) of the CBT rhythm and dampened CBT rhythms, both of which are strongly correlated with REM sleep [39].
Melatonin synthesis and corticosteroid secretion are directly or indirectly regulated by SCN, and can be used as markers of the central clockwork to reflect the endogenous rhythmicity [40]. Two well-known studies have examined rhythms of melatonin in PD. One study showed a significantly diminished amplitude and amount of melatonin secretion in PD patients (n = 20) receiving stable dopaminergic therapy compared with controls (n = 15). Among PD patients, those with EDS exhibit the most prominent impairment in circadian melatonin secretion [29]. The other study also suggested reduced circulating melatonin levels in patients newly diagnosed with PD (n = 30), compared with the matched controls (n = 15) [41]. Neither of these studies found any difference in the timing of melatonin onset or offset [29, 41]. Different from these observations, some other studies [42] found an earlier peak of nocturnal melatonin level in PD patients receiving levodopa than in the control group. However, secretion and diurnal rhythmicity of some other circadian-modulated hormones are unaffected in PD, including growth hormone, thyroid stimulating hormone, prolactin, as well as certain fat tissue-associated hormones [43, 44].
Circadian disruptions in AD and other neurodegenerative disorders
Individuals with AD often experience more severe circadian disruptions than the healthy elderly, which in turn exacerbate neurodegeneration in AD [45]. In the following, we will discuss in detail the behavioral and physiological circadian alterations in AD.
Behavioral alterations in AD
Sleep disturbances
The prevalence of sleep disturbances in AD is roughly 14%–69% [46, 47]. Sleep-wake disorders, including insomnia and EDS, are the most common form of sleep disturbances in AD. Additionally, SDB and RLS are also frequently observed in AD patients. As for parasomnia, RBD is rarely reported in AD when compared to PD and dementia with Lewy bodies (DLB), while evidence for the presence of NREM parasomnia in AD is lacking.
A multicenter study reported that sleep-wake disorders occur in over 50% of AD patients [48] and are primarily manifested as increased sleep latency, nocturnal awakenings, excessive daytime naps, difficulty in maintaining sleep, and early awakening [47, 49]. Sleep-wake disorders could precede the development of classic AD symptoms and may progress throughout the course of the disease [50]. Long-term disruption of sleep could result in exacerbation of neuropsychiatric or behavioral symptoms around the timing of sunset in 2.5%–66% of AD patients [51,52,53], which is called sundowning, or sunset phenomenon. Studies suggest that the phase delay of the body temperature and the hormone secretion patterns may contribute to this unique phenomenon [54].
The occurrence of RLS in AD patients is around 4%–6% [55, 56]. In the context of PD, RLS is more frequently observed with a prevalence of 14% [57]. RLS is rarely reported in patients with multiple system atroph (MSA), progressive supranuclear palsy and HD [58,59,60]. The relatively high prevalence of RLS in PD may be attributed to the common dysfunction of the dopaminergic system as some researchers proposed [61]. However, controversies remain on this issue [61]. In fact, diagnosis of RLS in AD can be quite challenging due to the inability of patients to accurately report. Additionally, RLS and sundowning may have common symptoms (i.e., agitation) and timing (i.e., late afternoon), making it more difficult to achieve a reliable diagnosis. Therefore, Richard et al. have further proposed a new diagnostic method, which combines a novel behavior observation test with clinical measurements and comorbidities, and yields a relatively high accuracy [62].
SDB has been reported in 15%–54% of AD patients [56, 63, 64]. Emerging evidence suggests that SDB serves as an independent risk factor for the development of AD [65, 66]. Indeed, there seems to be a bidirectional relationship between SDB and neurodegeneration. A recent meta-analysis pooling several randomized controlled trials indicated that continuous positive airway pressure treatment for SDB in AD patients can ameliorate cognitive performance, mood, EDS, slow-wave sleep (SWS) and apnea–hypopnea index (AHI) [67]. Although studies with a larger sample size are warranted, these studies shed light on the possibility of reversing cognitive decline in AD patients with comorbid SDB.
Accumulating evidence has suggested that RBD should be considered as the prodromal stage of α-synucleinopathies [68]. However, apart from a report of a rare case of drug-induced RBD in AD [69], two cross-sectional studies reported that the prevalence of RBD is approximately 10% in AD [70,71,72] and several longitudinal studies revealed the development of AD in RBD patients [73,74,75,76]. Notably, the diagnosis of AD in these longitudinal studies was not totally confirmed by autopsy. In addition, DLB and AD could be difficult to differentiate due to some overlapping clinical manifestations when postmortem analysis is lacking. Nonetheless, even autopsy findings are obtained, there remains a possibility of misdiagnoses when different neuropathological techniques are applied. For instance, a 72-year-old male patient diagnosed as AD was later defined as ‘Lewy body variant of AD’ when a new staining method was used [77,78,79]. Therefore, whether the development of AD in RBD patients indicates a mixed subtype of dementia or it is merely a technically false diagnosis needs further observations.
As for NREM parasomnia, although AD patients experience a reduction of SWS and spindle activity [80], there is limited evidence for any definitive NREM parasomnia in AD patients.
Rest-activity rhythm changes in AD
The rest-activity rhythm is one of the most commonly studied indicators of circadian disruption in AD. Recent studies showed that AD patients exhibit increased fragmentation of rest-activity rhythm, increased night-time awakening, and decreased daytime activity. However, there are mixed findings regarding alterations of the circadian amplitude or phases [81, 82].
Physiological alterations in AD
Physiological facets of circadian dysfunctions in AD mainly include dysfunctions of the autonomic nervous system and dysrhythmias of hormone secretion.
Dysregulations of the autonomic function in AD primarily manifest as orthostatic hypotension [83], non-dipping or reverse dipping in mainly systolic blood pressure [84], reduction in HRV index [85] and a phase delay in CBT [86]. Intriguingly, Kim et al. reported that HRV might be a potentially useful tool for early differentiation between AD and DLB [87].
Additionally, AD patients exhibit reductions of melatonin levels and a phase delay in melatonin secretion [88, 89]. Findings in HD are largely inconsistent with those in AD [90, 91]. As for cortisol, elevated cortisol levels and a phase-advanced cortisol-secretion pattern are observed in AD patients [92, 93], which bear some resemblance to those in late-stage HD [94]. In addition to higher cortisol levels, early-stage HD also displays an increased amplitude in the cortisol secretion rhythm [90].
Pathogenic mechanisms linking circadian rhythms, sleep and AD
The mechanisms linking circadian rhythms, sleep and AD have not come to a definitive conclusion. Mounting evidence has suggested a bidirectional relationship between them. Previous studies indicated that circadian dysfunctions worsen neurodegeneration in AD through cholinergic disturbances and melatonin loss. Autopsy studies have revealed loss of neurons in the SCN of AD patients, particularly neurons expressing vasopressin, melatonin receptor type 1 and vasoactive intestinal peptide (VIP) [95, 96]. In addition, the decreased melatonin levels [97] are associated with the rest-activity rhythm disorder [95, 98]. Moreover, AD patients with circadian dysfunctions show loss of ipRGCs [82, 99], which was reported to be associated with Aβ deposition in one study [82]. Additionally, AD patients or mouse models with cholinergic disturbances demonstrate impaired circadian pattern [100, 101]. Melatonin suppresses Aβ generation and amyloid fibril formation. There is evidence showing a reduction of melatonin level in the prodromal and progressive stages of AD [102]. On the contrary, circadian disruptions promote neurodegeneration in AD. Sleep deprivation or disruption significantly increases neuroinflammation and subsequent Aβ production in the cerebrospinal fluid (CSF) of AD patients [103], increases Aβ and phosphorylated tau (ptau) levels in transgenic AD mouse models [104, 105], and decreases glymphatic flow and Aβ clearance in human CSF and in animal models [104, 106], which may lead to further progression of neurodegeneration in AD. Notably, chronic sleep disruption may increase the dissemination of tau protein in neural networks [107]. Musiek et al. have reported severe astrogliosis, oxidative injury and synaptic degeneration in Bmal1-deleted mice, indicating that circadian dysregulation of the neuronal redox homeostasis may also contribute to neurodegeneration in AD [1].
Other neurodegenerative disorders
Circadian disruption and sleep disorders are also observed in other neurodegenerative disorders, including MSA, DLB, frontotemporal dementia (FTD), and HD.
Although no α-synuclein deposition is found in either the SCN or the pineal gland in MSA cases, the circadian dysfunction may be secondary to degeneration of other systems, such as the autonomic networks [108]. The circadian rhythm is regulated by VIP-expressing neurons, which are more involved in autonomic control and depleted in the SCN of patients with MSA [95]. MSA has a wider impairment of circadian regulation of endocrine and autonomic functions, such as plasma cortisol concentration [109], blood pressure, gastric myoelectrical activity [110], and nocturnal polyuria.
Patients with DLB present with decreased amplitude of CBT during night and more severe daytime sleepiness than controls, as well as more frequent RBD than AD and healthy controls [111]. Notably, inclusion of RBD has been proven to improve the diagnostic accuracy of DLB [112].
Although neurodegenerative disorders exhibit fragmentation of sleep, patients with FTD [113] show phase-advanced activity rhythm while HD patients [114] are with phase delay.
Bi-directional relationship of circadian rhythm dysregulation and sleep disorder with neurodegeneration
Although initially considered to occur consequently after disease onset, the impairment of sleep and circadian rhythm is now recognized to predate clinical diagnosis or occur in the early stage of neurodegeneration [4, 115, 116], posing individuals at risk of the incidence or progression of neurodegenerative diseases. Therefore, a potential bi-directional relationship can be inferred between sleep disorder/circadian rhythm dysregulation and neurodegeneration.
Sleep disorders accelerate neurodegeneration
RBD
RBD is well recognized as a strong prodromal predictor of α‑synucleinopathies, especially PD. Schenck et al. noted for the first time that 38% of individuals with initial diagnosis of isolated RBD (iRBD) finally developed a parkinsonian disorder within 3.7 years [117]. Now, it is recognized that most iRBD individuals will end up being diagnosed as α‑synucleinopathies, including PD, PD dementia, DLB and MSA. The estimated overall conversion rate is 6.3% per year and 73.5% within 12 years in a large-scale multicenter study [118]. For those with longstanding iRBD without overt conversion, crucial prodromal PD markers such as olfactory loss, constipation and mild parkinsonism are commonly observed in these populations [119]. This suggests that iRBD is consistently influenced by an underlying neurodegenerative process, which is also supported by findings of widespread Lewy body pathology and α-synuclein in iRBD individuals [120]. By pooling studies on biomarkers of iRBD–PD association, a recent meta-analysis showed that motor dysfunction, constipation, orthostatic hypotension, hyposmia, mild cognitive impairment, and abnormal color vision in iRBD are significantly associated with subsequent PD risk [121]. Other biomarkers on imaging, polysomnography (PSG) or EEG can also be used to monitor the neurodegenerative process in iRBD [122]. Considering the long interval between onset of iRBD and overt α‑synucleinopathy, close monitoring of these biomarkers may provide opportunity for use of disease-modifying treatments in the prodromal stage of neurodegeneration. Approximately 3–11% of RBD patients were recorded in previous studies to develop incident clinical AD [73, 74]. However, there is still limited evidence from longitudinal or prospective studies on the conversion of RBD to mild cognitive impairment (MCI) or AD. Compared to individuals without probable RBD (pRBD), those with pRBD have a 2.2-fold increased risk of MCI [123]. In another population-based study, individuals with pRBD who eventually developed MCI and subsequent dementia were actually highly consistent with a diagnosis of DLB [124]. By further evaluating previous neuropathological studies with 16-year follow-up [117], researcher found that the clinically identified AD patients developing from RBD exhibited a ‘mixed pathology’ but not ‘pure AD’, that is, they had histopathological features of both AD and DLB. In sum, the clinically observed RBD–AD association may be due to the presence of dementia in α‑synucleinopathies.
Insomnia
Chronic insufficient sleep plays a vital role in the pathological process of AD pathology. Both animal and human studies showed that sleep deprivation causes increased Aβ formation and deposition [103, 104] and contributes to neurodegenerative process that affects neuroinflammation and synaptic homoeostasis [125]. Besides, objective short sleep duration and circadian rhythm disruption may exert an add-on effect on the risk of AD. Xu et al. [126] systematically reviewed studies on the association of sleep with all-cause cognitive decline or dementia, and found that insomnia significantly contributes to an increased risk of incident AD but not vascular dementia with a pooled relative risk of ~ 1.5. In a subsequent longitudinal analysis of U.S. adults aged over 65 years, individuals with an increase in the severity of insomnia over time have 41%–58% higher risk of memory decline or dementia [127], suggesting the importance of early sleep health for AD prevention. In contrast, studies on the impact of insomnia on the susceptibility to PD are limited. In a large registry-based case–control study, significantly higher incidence of insomnia (RR = 1.38, 95% CI 1.11–1.70) was observed 2 years before diagnosis of PD [128]. A recent study differentiating insomnia subtypes found that sleep-onset insomnia, in comparison to maintenance insomnia, is associated with more motor, cognitive, and autonomic symptoms [129]. To be noted, insomnia is not persistent throughout the disease course [25] and their subtypes [130] may change in PD. In addition, previous studies revealed that the observed insomnia–PD association may disappear with longer follow-up duration [128, 131]. Also, some studies showed that present insomnia could not predict conversion to neurodegeneration in iRBD individuals [132, 133]. Therefore, it is likely that insomnia is more likely to be a prodromal symptom instead of an etiology of PD.
OSA
OSA-related intermittent hypoxia, neuroinflammation and sleep fragmentation have been proven to accelerate neurodegeneration by disturbing Aβ clearance and aggravating neurofibrillary tangles of tau in AD [134], or by damaging the nigrostriatal dopaminergic system and promoting aggregation of α-synuclein in PD [135]. Mounting studies have confirmed the emerging role of SDB, either self-reported sleep apnea or PSG-proven OSA, in the incidence or early progression of neurodegenerative conditions [66, 135, 136]. For example, Yaffe et al. [136] found that women with OSA having AHI > 15 have a higher risk of developing MCI or dementia, with an odds ratio of 1.85. This was further confirmed in a recent prospective cohort with large sample size, which showed that only severe OSA individuals with AHI > 30 have 66%–135% higher risk of developing all-cause dementia or AD [137]. More evidence from the AD biomarker perspective suggests that cognitively intact OSA individuals have higher Aβ burden indicated by blood, CSF and imaging biomarkers compared to individuals free of OSA [138, 139]. Moreover, adherence to positive airway pressure therapy may lower the odds of incident diagnosis of AD or MCI and slow cognitive impairment or its progression to AD [140, 141]. Consistently, another longitudinal analysis revealed increased risk of PD in individuals with sleep apnea [142, 143]. Subsequent meta-analysis confirmed the association of OSA with incident diagnosis of PD by pooling 12 eligible studies and revealed similar risk between males and females [144]. These findings are further supported by Sun et al. showing that levels of plasma total and phosphorylated α-synuclein are significantly elevated in OSA individuals and are inversely correlated with oxyhaemoglobin saturation [145], indicating that OSA-related hypoxia is involved in the pathogenesis of PD pathology. Taking AD and PD together, however, a recent mendelian randomization study failed to reveal a causal association between genetically-predicted OSA and risk of AD or PD [146]. An explanation is that the OSA–neurodegeneration association may be not unidirectional but bi-directional, and that confounding factors such as comorbidities may also play a role [146].
EDS
EDS is another emerging predictor of neurodegeneration. Many studies have identified the temporal association between EDS and risk of dementia or AD, which is independent of comorbid chronic diseases or conditions [147,148,149] but can be partly confounded by lack of daily physical activity or social engagement [149]. In revealing the sleepiness–AD pathologic association, an imaging study by Carvalho et al. reported that EDS patients with normal cognition show more prominent grey matter thinning in age-susceptible regions which is usually observed in AD pathology [150]. In 283 dementia-free participants aged 70 years and over receiving Pittsburgh compound-B positron emission tomography, the same team found that the baseline EDS is longitudinally associated with increased Aβ accumulation in the cingulate gyrus and precuneus regions, indicating that the elderly persons with EDS are more susceptible to AD pathologic alterations [151]. This is supported by similar sleep-related animal studies showing abnormal Aβ generation or Aβ deposition due to impaired glymphatic clearance [104] and dysregulated cortical slow-wave activity [152]. EDS also serves as a robust prodromal marker of PD. Two studies have indicated that daytime sleepiness leads to a 2–3-fold increased risk of incident PD among the general population [153, 154]. However, results were inconsistent on if EDS predicts conversion to neurodegeneration in iRBD individuals [74, 132, 133]. The inconsistency may be due to the differences in sample size as well as ethnic and clinical backgrounds of the participants. In a neuropathological study, Abbott et al. combined EDS assessments and α-synuclein staining in postmortem brain samples from 211 men to identify the relationship between Lewy body pathology (LP) and EDS [155]. They found that the prevalence of EDS became significantly increased only when Lewy body pathology extensively infiltrated into the neocortex (equivalent to Braak stages 5 and 6). However, absence of Lewy body pathology was noticed in 41% cases of EDS at the time of autopsy. In other words, it was uncertain whether EDS preceded Lewy body pathology in the remaining 59% cases, and whether sleep augmentation of the clearance of tau and α-synuclein underlies the relationship between sleepiness and PD pathology remains unknown.
RLS and other sleep disorders
In viewing the association between other sleep disorders and neurodegeneration, prospective cohort studies found that RLS is associated with a 1.5–2.5-fold higher risk of incident PD. However, it remains unclear if the dopaminergic system is a pathologically link between RLS and PD due to the absence of evidence from basic research [156]. When examining the overall effect of sleep-related movement disorders (SRMD) on dementia, Lin et al. [157] found individuals with diagnosis of SRMD had a 3.9-fold higher risk of incident all-cause dementia and the observed association was more prominent in women and in those aged 45 to 64 years. For NREM parasomnia, a recent large-scale cross-sectional study including 25,694 men showed that sleepwalking was associated with 4.8-fold odds of having PD, regardless of the confounding factors [158].
Circadian disruption aggregates process of neurodegeneration
Circadian activity disturbance, initially considered as symptoms of neurodegeneration, is believed to be involved in the occurrence or progression of neurodegenerative process. In other words, circadian disruption may precede clinical symptomology or add to the risk of neurodegenerative diseases.
Only few studies have examined the temporal association between circadian rhythm disruption and PD [159,160,161]. In a prospective study including 2930 men, Len et al. found that those who self-reported napping time of more than 1 h/day had a twofold risk of developing PD, compared to those with no EDS or having napping < 1 h/day [159]. From the same cohort, they also reported that weakened circadian rhythmicity was associated with increased risk of incident PD and such association remained significant even after excluding PD diagnosis in the first 2 years of follow-up [160]. These findings indicated that circadian rhythm disruption could be a prodromal marker for PD. To minimize the effect of reverse causality and potential confounding factors, recent mendelian randomization study using UK Biobank from European ancestry found that morning chronotype had an inverse causal effect while M10 (average activity during the most active 10 consecutive hours of the day) had positive causal effect on the later onset age of PD [161]. They also noticed that better sleep efficiency was causally associated with a decreased AD risk. However, temporal associations between circadian rhythm and PD or other neurodegenerative diseases such as HD and amyotrophic lateral sclerosis have not been thoroughly investigated as in AD and dementia. In addition, behavioral indicators like actigraphy do not necessarily parallel endogenous circadian biomarkers which maintain 24-h oscillations even in the case of sleep disturbance. Therefore, comprehensive evaluations of both behavioral and biological markers of circadian rhythm are needed to provide more convincing and detailed evidence on circadian rhythm disruption in PD-related neurodegeneration.
In a case–control study, more evident circadian misalignment, assessed by PSG and dim light melatonin onset, was noticed in MCI patients before AD diagnosis [162]. A recent study by Musiek et al. including 189 healthy elders showed that those with preclinical AD, as assessed by PET, demonstrated increased rest-activity rhythm fragmentation independent of aging and sex [163]. This finding suggested that circadian disruption is involved early in AD pathogenesis. In line with clinical findings, research on animal models revealed disruption of daily Aβ oscillations in the hippocampal interstitial fluid and acceleration of amyloid plaque accumulation in mice with disturbed circadian rhythm [164]. Inhibition of the circadian repressor REV-ERB is also associated with enhanced transcription of core clock gene BMAL1 and increased Aβ clearance [165]. These observations add to the evidence of the potential role of circadian regulation in AD.
The temporal association between circadian rhythm and AD or dementia has been explored in many epidemiological studies [166,167,168,169,170,171,172]. Habits of dysregulated circadian rhythm, such as long-time night-shift or delayed rising time, are associated with increased risk of dementia among healthy populations [166, 167]. Assessment of behavioral indicators of circadian rhythm disruption via actigraphy can more objectively reveal the association [173]. Based on the same cohort of Study of Osteoporotic Fractures including 1283 women, Tranah et al. [174] and Walsh et al. [168] both found an association of decreased circadian amplitude with higher risk of cognitive decline or dementia during the 5-year follow-up. Besides, they reported worse cognitive decline in association with phase delay, different from the report by Rogers-Soeder et al. [169], which showed phase-advanced acrophase in the men-only cohort. In a prospective cohort study including 1401 healthy older adults, lower amplitude of 24-h activity rhythm and higher intradaily variability for hourly fragmentation of activity rhythm are associated with higher risk of developing AD dementia, while lower interdaily stability of 24-h activity rhythm predicts higher risk of transition from MCI to AD dementia [170]. The observed parameters also worsen as dementia deteriorates, indicating that the relationship between circadian disruption and AD progression is bidirectional. To specify the affected aspects of circadian rhythm, novel or refined analysis has been applied in some studies [171, 172]. A 24-h time-limited difference of circadian rhythm was observed in individuals with preclinical PD in various periods in comparison to healthy controls [172]. By using parametric and nonparametric analysis, reduced overall rhythmicity, lower amplitude and activity level, and later activity timing were revealed to be associated with development of MCI and dementia [175]. However, there are also some studies showing no association of 24-h activity rhythm fragmentation with dementia risk or preclinical AD [172]. Discrepancies in these findings could be due to different sample sizes, study populations or statistical methods.
Progression of neurodegenerative disease promotes circadian disruption and sleep disorders
Although sleep-wake disturbance is commonly found in PD, the rhythmic change throughout disease process has been rarely noticed. Using continuous actigraphy, a study showed that PD patients with higher Hoehn and Yahr stage are significantly more active later in the day [22], indicating an alteration of circadian rhythm in relation to disease severity. Additionally, the coexistent sleep disorders in PD might increase the variability of circadian rhythm. In a study of 15 iRBD patients, 31 PD patients and 6 DLB patients [111], Raupach et al. observed an inverse correlation between the CBT amplitude and RBD severity. Interestingly, the alterations in CBT were absent in PD patients free of RBD. This suggests that certain circadian rhythms might be particularly linked to RBD pathology and a further exploration of prodromal sleep disorders in PD is necessary. When discussing the longitudinal contribution of PD progression in relation to sleep disorders or circadian changes, the use of anti-PD medications is also of great importance [4]. In other words, whether these alterations result from dopaminergic treatment or from PD deterioration itself should be clarified. For example, a previous study found that only PD patients with levodopa-induced motor complications showed decreased ratio of melatonin secretion at night while such change was not observed in untreated patients or treated PD patients without motor complication [42]. Two other sleep disorders in PD, EDS and RLS, may also be consequences of dopaminergic therapy when symptoms aggregate as disease progresses. Drug-naive PD patients showed increased EDS severity from baseline to year 3 while no change was observed in the healthy control group. Meanwhile, the influence of dopaminergic medications on EDS was dose-dependent at years 2 and 3 of study follow-up [176]. The prevalence of RLS in untreated PD patients does not differ significantly from that of the general population, but is significantly increased in those receiving dopaminergic medication [177].
Across the disease continuum, studies using functional and biological indicators of circadian disruption in AD showed severer or irregular changes [88, 162, 178]. The severity of increased fragmentation of rest-activity cycle and overnight activity correlates with AD severity, with disturbances being most prominent in institutionalized patients [179]. The phase delay identified in AD patients is more prominent in those with advanced AD pathology [180]. As for core body temperature, a significant phase advance in circadian thermoregulation was observed in MCI patients compared to the healthy group [178] while phase delay was noticed in AD patients [180]. Similarly, for biological indicators such as melatonin level, MCI patients have a phase advance in melatonin secretion whereas those with mild-to-moderate AD exhibit a phase delay [88, 162]. These observations indicate that the neurodegenerative process can alter circadian rhythmicity and the latter can become more irregular as pathology deteriorates. Another question is that whether it is AD itself, but not aging, that promotes circadian disruption and sleep disorders. Initial view contends that individuals with AD have circadian alterations similar to those seen in healthy older adults, but with higher severity [181]. However, a recent study by Musiek et al. showed that preclinical amyloid pathology is associated with worse circadian fragmentation regardless of age [163]. This suggests that despite the same fragmentated circadian pattern, AD and normal aging drive circadian dysfunction in separate ways. Moreover, ageing alone is associated with a dramatically increased prevalence of preclinical AD to 30%–40% [182]. That is to say, in several epidemiological studies examining circadian dysregulation/sleep disorders preceding AD symptomology [168, 171, 175], the enrolled participants of advanced age might be already in the early stage of AD, which provides an interpretation for the observation that an underlying preclinical AD pathology may in turn lead to circadian and sleep dysfunction. The estimated prevalence of overall sleep disorders in individuals of AD is about 39% [46]. With self-reported assessment and overnight PSG, Hita-Yañez et al. found patients with MCI already showing disturbed sleep at both objective and subjective levels [183]. This work was further supported by a subsequent study showing significant relationship between Aβ deposition and sleep quality in preclinical AD [184]. As these cross-sectional observations were from an early stage of AD, this could be interpreted, from one perspective, that sleep is particularly sensitive to AD pathology. As dementia worsens, concurrent sleep disorders in AD such as EDS can be aggregated through potential mediating factors [184]. Comorbid depression and decreased social engagement resulting from impaired cognition in AD may add extra burden to EDS severity [185]. For SDB, around half of individuals with AD experience OSA and show five times higher odds of having OSA compared to age-matched cognitively intact individuals [184]. However, the longitudinal association between OSA or other specific sleep disorders (e.g., RLS) and AD progression has not been well explored to date [186].
As studies focusing on alterations of circadian rhythm/sleep disorder after diagnosis of neurodegenerative diseases are generally lacking, more studies are needed to longitudinally examine alterations of circadian rhythm and sleep disorders with progression of neurodegenerative diseases.
Therapeutic strategies for circadian dysfunction and sleep disorders
Non-pharmacological approaches
Non-pharmacological approaches are considered as the first-line therapies for the management of circadian dysfunction and sleep disorders in neurodegenerative diseases. Treatment plans must be tailored at individual level. Prior to treatment, a comprehensive set of clinical, neuropsychological, neuroimaging, and electrophysiological assessments should be conducted, and the circadian disruption as well as the causes and subtypes of sleep disorders need to be carefully evaluated. Notably, sleep hygiene is recommended as an important behavioral and environmental practice in every treatment plan to promote better-quality sleep in PD [187] and AD [188] patients. As different medications for different types of sleep disorder may interact with each other, it is necessary to identify and treat coexisting or primary sleep disorders before making medication plans (Fig. 4).

Management of circadian disruption and sleep disorders in patients with neurodegenerative diseases in four steps. About circadian disruption, chronopharmacological methods which combine both pharmacological and non-pharmacological approaches can be considered
Physical activity
Exercise is a circadian modulator. In PD, exercise can improve subjective sleep quality and objective PSG parameters. A retrospective study reported that intense physical and multidisciplinary exercise for 28 days improves the total PDSS score in PD patients [189]. Recently, Amara et al. [190] conducted the first study to demonstrate the impact of high-intensity exercise on objective sleep outcomes in PD patients and found that exercise is more effective than sleep hygiene education in improving PSG parameters such as the total sleep time and sleep efficiency. Specifically, recent research suggested traditional Chinese exercise including Tai Chi [191], Baduanjin [192] and Qigong [193] as useful tools to improve PD sleep.
Similarly, walking for 30 min per day reduces the awake time by 33.1 min per night in AD patients [194]. Although one study has found that exercise could improve the daily cortisol rhythm in AD patients [195], the underlying mechanisms remain to be clarified. Adaptive neuroplasticity, which is beneficial for neuron re-organisation, is a suggested mechanism of the effects of physical activity [196].
When applying exercise in patients with neurodegenerative disorders, factors that may cause potential bias, including intensity, modality and compliance, need to be well controlled. A recent meta-analysis pointed out that moderate-to-maximal intensities rather than mild-to-moderate intensities of exercise have significant effects on subjective sleep quality [197]. Multi-modal exercise therapy at vigorous intensities is recommended [197].
Bright light therapy (BLT)
Light exposure is a powerful modulator of circadian rhythm and plays an important role in enhancing the rest-activity rhythm and thus promoting sleep in healthy individuals. Although the mechanisms of light therapy have not been well defined, retina, a key route of light entry into the brain, has been receiving more and more attention with regard to neurodegenerative diseases. Deposition of α-synuclein in retina has been well found in PD patients [198]. Light therapy could stimulate dopamine release by stimulating cells within the retina, thus improving the abnormal circadian rhythm and the motor symptoms of PD patients [199].
Recent studies found that BLT treatment is significantly associated with improvements of circadian rhythm and thus sleep quality of PD patients [200, 201], especially the subjective sleep quality and actigraphic measures, including sleep fragmentation and daily physical activity. In AD, a large double-blind, randomized, placebo-controlled trial found that light therapy (2500 lx) for 2 months could improve sleep quality and restore diurnal activity rhythms as measured by actigraphy [194].
BLT appears to be a feasible treatment for ameliorating sleep disorders in patients with neurodegenerative diseases. Although most studies recommend BLT as an alternative non-pharmacological method with few adverse effects such as headache [200, 202], the conclusion still needs to be confirmed in larger populations. Meanwhile, the effects of BLT may be confounded by medication, lifestyle, severity of disease, compliance of patients and co-morbidities [202].
Repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS)
rTMS and tDCS are two noninvasive brain stimulation techniques that can improve sleep quality in the healthy elderly.
In PD patients, rTMS therapy improves sleep fragmentation and sleep efficiency and reduces the average duration of nocturnal awakenings based on actigraphic results [203] and sleep scales [204]. Recently, rTMS has been demonstrated to improve daytime sleepiness in PD patients [205]. In AD patients, rTMS for 4 weeks significantly improves Pittsburgh Sleep Quality Index scores [206]. So far, only two studies have assessed the therapeutic efficacy on motor symptoms in HD and results are contradictory [207, 208]. tDCS enhances the slow-wave sleep [209], which is thought to play an important role in clearing Aβ during sleep.
Pharmacological approaches
Melatonin and melatonin receptor agonists
Melatonin plays a role in regulating the circadian rhythm and promoting sleep. Replacement therapy with exogenous melatonin may have positive effects against sleep disturbances and even pathological progression of neurodegeneration. Several clinical studies have demonstrated the positive effects of melatonin on insomnia, RBD and rest-activity disruption in neurodegenerative disorders. However, the results are mixed. Melatonin could significantly improve subjective sleep quality and total sleep time in PD [210]. However, the effect of melatonin on EDS remains unclear. Of note, melatonin is recommended as the first-line therapy because it reduces RBD-related injuries with fewer side effects than clonazepam [211]. In the context of AD, one recent systematic review showed that melatonin shortens the sleep onset latency and increases sleep duration [212]. Several studies also proved that prolonged-release melatonin [213] and melatonin receptor agonists [214] can improve subjective sleep quality in both PD and AD patients. Although plasma concentrations of melatonin are shown to be reduced in HD [91], the efficiency of melatonin in HD patients has not been systematically investigated. Recently, a study demonstrated beneficial effects of melatonin in restoring clock gene expression in Drosophila model of HD, suggesting a promising clinical use in the future [215].
However, most melatonin-related studies have similar limitations. (1) The melatonin dose varied widely across studies (mostly 3–5 mg). (2) Most data were derived from case reports and long-term longitudinal studies are lacking. So far, most studies focused on the hypnotic effect rather than effects on the circadian rhythm. More studies evaluating alterations of the biomarkers of circadian rhythm during melatonin treatment are needed. (3) The circadian rhythm of melatonin secretion profile may be different among individuals, and can be influenced by other factors such as food, physical exercise and light. Solutions to these problems can increase the melatonin efficiency in personalized treatments.
Hypnotics
Hypnotics including benzodiazepines, non-benzodiazepines drugs, sedative antipsychotics, and sedating antidepressants, are widely used to treat insomnia in healthy adults. Recently, several studies have found its use for various sleep disorders in neurodegenerative disorders.
Clonazepam, a long-lasting benzodiazepine, is the first-line pharmacological option for RBD [216]. Shin et al. showed that clonazepam improves pRBD symptoms in patients with PD [217]. A 6-week randomized controlled trial showed that eszoplicone significantly reduces the number of awakenings after sleep onset and improves subjective sleep quality in PD patients [218]. Similar results have been obtained in AD patients [219]. In fact, current evidence for clonazepam is mainly based on case reports and observational studies [220, 221], thus more clinical trials are needed. Sedative antidepressants drugs like trazodone and doxepin are also proven to be effective for nighttime percent sleep in AD [222] and insomnia in PD patients [223].
It is important to note that hypnotics may also produce adverse effects such as memory deterioration and worsening of daytime sleepiness or sleep-related breathing disorders, especially in elderly adults [24]. Neurologists should well assess the risk/benefit profile before prescribing these hypnotics agents for patients with neurodegenerative diseases.
Chronopharmacological principles: combination of pharmacological and non-pharmacological approaches
Chronotherapy is a therapeutic approach that incorporates an individual's circadian rhythm into disease treatment. It is based on the principle of prescribing drugs according to the different characteristics of an individual's circadian rhythm or combining both pharmacological and non-pharmacological approaches. Characterized by maximizing drug effectiveness and minimizing its side effects, chronotherapy was initially used mainly in the treatment of hypertension and in oncology. Recent studies have found that chronotherapy has promising applications in neurodegenerative diseases [194, 224, 225].
A study suggests that combining BLT with melatonin in demented patients may increase sleep efficiency, attenuate agitated behavior and even improve nocturnal restlessness for 3.5 years [224]. Dowling et al. found that 1 h of morning light exposure (2500 lx) for 10 weeks together with 5-mg melatonin in the evening significantly increases the daytime awake time and activity levels and strengthens the rest-activity rhythm of AD patients, compared to the light therapy alone [225]. MuCurry et al. found that combination treatment (walking, light, and sleep education) and each treatment alone have similar effects in improving sleep outcomes in AD patients [194]. Preliminary studies of chronotherapy in neurodegenerative diseases have shown promising findings. Personalized treatment plans are essential for effective implementation of chronotherapy, as the melatonin secretion curve varies from individual to individual. Therefore, the timing of pharmacological agents such as melatonin should be personalized according to the individual’s circadian rhythm. The implementation time and dose are also to be studied in the future [226].
Neuroprotection and novel interventions
Animal studies have shown that rTMS has positive effects on neural regeneration and neuroprotection through inhibiting apoptotic cell death, as well as regulating neurotransmitters and neurotrophic factors [227]. Light therapy decreases oxidative stress markers [228] and removes Aβ via the lymphatic system of the brain [229] in AD mouse models, and reduces the loss of dopaminergic cells and increases tyrosine hydroxylase-positive cells in PD mouse models [230].
Orexin is a neuropeptide that contributes to the regulation of the sleep-wake cycle by increasing the arousal level and maintaining wakefulness. Suvorexant, one of the orexin receptor antagonists, has been approved to treat insomnia in elderly adults. Animal experiments showed that suvorexant can reduce amyloid-β plaques, improve synaptic plasticity, and restore the circadian phosphorylated CREB (cyclic AMP-response element binding protein) expression in the hippocampus of APP/PS1 mice [231].
Furthermore, some small-molecule modulators have been developed to restore the disrupted circadian system. For example, casein kinase 1 δ/ε inhibitor CKI-7 [232] significantly reduces endogenous Aβ peptide production [233], thus playing an important role in neuroprotection. Rev-erbα is a core negative component of the circadian clock and modulates the cellular clock and energy metabolism [234]. Rev-erbα knock-out mice show disrupted diurnal patterns [235]. The agonists (GSK4112) and antagonists (SR8278) of Rev-erbα could correct the abnormal circadian rhythms [232], providing biological evidence for future trials of these small-molecule modulators as a therapeutic for neurodegeneration.
In addition, traditional Chinese herbs or herbal extracts and non-pharmacological interventions are proven beneficial to patients with neurdegenerative disorders, by exerting antioxidant and anti-inflammatory effects [236, 237]. Clinically, several studies showed that acupuncture [238] and Yang-Xue-Qing-Nao granules [239] improve sleep quality in PD patients. Future research should focus on the quality control of traditional Chinese medicine studies and figuring out the pharmacological mechanisms of main active ingredients.
Conclusions and future prospect
Collectively, all behavioral, physiological and molecular aspects of circadian disturbances in neurodegenerative disorders provide substantial evidence that the circadian system is functionally impaired and most likely contributes to the deterioration of health and quality of life in patients inflicted with neurodegenerative diseases. However, although neurodegenerative disorders have overlapping circadian symptoms, the underlying neuro-pathophysiology may not necessarily be the same. Current evidence shows that dysfunction of the central SCN clock starts very early during the prodromal phase of AD, while the SCN itself functions normally till the early symptomatic phases where its dysfunction starts in both PD and HD [5].
Increasing research indicates that sleep dysfunction and circadian arrhythmicity are key aspects to consider when investigating neurodegenerative diseases. Although generally there are sleep and rhythmic disorders in different neurodegenerative diseases, each disease develops a specific phenotype to some extent [240]. For example, there is early impairment of the circadian homeostasis in AD, while in PD and HD, circadian homeostasis deterioration is more prevalent and occurs after diagnosis [240]. MSA and DLB have a higher prevalence of RBD. Our review reinforces the state of the field that bidirectional links of sleep and circadian rhythms with prevalent forms of neurodegeneration are likely.
Future research efforts are needed to center on the following fields. Despite great progress in understanding the basic mechanisms of the circadian clock and the neural circuitry of sleep, the knowledge of how these systems are affected in the brain in aging and neurodegenerative diseases is still rather superficial. It is necessary to fully characterize the putative bidirectional relationship of sleep and circadian circuits with neurodegeneration, in order to inform therapeutic targets. This will allow the field to expand the use of sleep and circadian rhythms as markers for early treatment of prodromal neurodegenerative disease and enable manipulation of the circadian system at the molecular and behavioral levels in longitudinal animal studies.
Systematically studying the circadian system in neurodegenerative diseases is another direction of future research, which calls for strict control of different circadian parameters, design of a set of evaluation tools, and development of personalized multi-component circadian interventions. Large longitudinal clinical studies are also needed to examine changes in circadian rhythms associated with progression of neurodegeneration and the relationship between different circadian markers and subsequent risk of developing neurodegenerative diseases, and to clarify whether circadian interventions for sleep disorders could prevent or delay the onset of neurodegenerative diseases.
Normalizing sleep and circadian disorders has the potential to reduce risk of neurodegeneration and improve quality of life and symptoms in those with neurodegenerative disorders. First, circadian-based interventions are a critical test of the hypothesis that circadian disruption is an integral component of the disease. Therefore, it is critical to establish a collaborative research program between clinical investigators and basic/translational neuroscientists, in order to advance the understanding of circadian regulation in neurodegeneration and effects of complex medication regimens on circadian function in animal models. Second, developing screen and therapeutic strategies in early (or “prodromal”) stage of neurodegenerative disease may facilitate earlier detection, prevention of disease progression and development of more effective therapeutic interventions. Third, optimization of existing therapies, such as light therapy and chronopharmacological principles, or launching novel neural circuit-based therapeutic interventions to restore the circadian activity, might provide beneficial effects against circadian alterations in patients with neurodegenerative disorders.
Availability of data and materials
Not applicable.
Abbreviations
- PD:
-
Parkinson’s disease
- AD:
-
Alzheimer’s disease
- HD:
-
Huntington’s disease
- SCN:
-
Suprachiasmatic nucleus
- ipRGC:
-
Intrinsically photoreceptive retinal ganglion cell
- PVN:
-
Paraventricular nucleus
- MPN:
-
Medial preoptic nucleus
- TTFL:
-
Transcription-translation feedback loops
- BMAL1:
-
Brain and muscle Arnt-like protein 1
- CLOCK:
-
Circadian locomotor output cycles kaput
- CRY:
-
Cryptochrome
- EEG:
-
Electroencephalography
- EMG:
-
Electromyography
- REM:
-
Rapid eye movement
- NREM:
-
Non-REM
- RSWA:
-
REM sleep without atonia
- RBD:
-
REM sleep behavior disorder
- RLS:
-
Restless leg syndrome
- SDB:
-
Sleep-disordered breathing
- EDS:
-
Excessive daytime sleepiness
- DEB:
-
Dream-enacting behaviors
- OSA:
-
Obstructive sleep apnea
- AHI:
-
Apnea hypopnea index
- HRV:
-
Heart rate variability
- CBT:
-
Core body temperature
- MSA:
-
Multiple system atrophy
- SWS:
-
Slow wave sleep
- VIP:
-
Vasoactive intestinal peptide
- DLB:
-
Dementia with lewy bodies
- FTD:
-
Frontotemporal dementia
- iRBD:
-
Isolated RBD
- MCI:
-
Mild cognitive impairment
- pRBD:
-
Probable RBD
- PSG:
-
Polysomnography
- SRMD:
-
Sleep-related movement disorders
- BLT:
-
Bright light therapy
- rTMS:
-
Repetitive transcranial magnetic stimulation
- tDCS:
-
Transcranial direct current stimulation
References
Fifel K. Alterations of the circadian system in Parkinson’s disease patients. Mov Disord. 2017;32(5):682–92.
Steele TA, St Louis EK, Videnovic A, Auger RR. Circadian rhythm sleep-wake disorders: a contemporary review of neurobiology, treatment, and dysregulation in neurodegenerative disease. Neurotherapeutics. 2021;18(1):53–74.
Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science. 2016;354(6315):1004–8.
Leng Y, Musiek ES, Hu K, Cappuccio FP, Yaffe K. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol. 2019;18(3):307–18.
Lucas RJ. Mammalian inner retinal photoreception. Curr Biol. 2013;23(3):R125–33.
Harvey J, Plante AE, Meredith AL. Ion channels controlling circadian rhythms in suprachiasmatic nucleus excitability. Physiol Rev. 2020;100(4):1415–54.
Colwell CS. Defining circadian disruption in neurodegenerative disorders. J Clin Invest. 2021. https://doi.org/10.1172/JCI148288.
Lee Y. Roles of circadian clocks in cancer pathogenesis and treatment. Exp Mol Med. 2021;53(10):1529–38.
Arafa K, Emara M. Insights about circadian clock and molecular pathogenesis in gliomas. Front Oncol. 2020;10:199.
Rasmussen ES, Takahashi JS, Green CB. Time to target the circadian clock for drug discovery. Trends Biochem Sci. 2022;47(9):745–58.
Fagiani F, Di Marino D, Romagnoli A, Travelli C, Voltan D, Di Cesare ML, et al. Molecular regulations of circadian rhythm and implications for physiology and diseases. Signal Transduct Target Ther. 2022;7(1):41.
Valadas JS, Esposito G, Vandekerkhove D, Miskiewicz K, Deaulmerie L, Raitano S, et al. ER lipid defects in neuropeptidergic neurons impair sleep patterns in Parkinson’s disease. Neuron. 2018;98(6):1155-69.e6.
Gelegen C, Cash D, Ilic K, Sander M, Kim E, Simmons C, et al. Relevance of sleep and associated structural changes in GBA1 mouse to human rapid eye movement behavior disorder. Sci Rep. 2022;12(1):7973.
Medeiros DC, Lopes Aguiar C, Moraes M, Fisone G. Sleep disorders in rodent models of Parkinson’s disease. Front Pharmacol. 2019;10:1414.
Fifel K, Piggins H, Deboer T. Modeling sleep alterations in Parkinson’s disease: how close are we to valid translational animal models. Sleep Med Rev. 2016;25:95–111.
Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA. Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 2014;71(8):971–7.
Ju YS, Ooms SJ, Sutphen C, Macauley SL, Zangrilli MA, Jerome G, et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain. 2017;140(8):2104–11.
Titova N, Chaudhuri KR. Non-motor Parkinson disease: new concepts and personalised management. Med J Aust. 2018;208(9):404–9.
Videnovic A, Willis GL. Circadian system—a novel diagnostic and therapeutic target in Parkinson’s disease. Mov Disord. 2016;31(3):260–9.
Liu Y, Niu L, Liu X, Cheng C, Le W. Recent progress in non-motor features of Parkinson’s disease with a focus on circadian rhythm dysregulation. Neurosci Bull. 2021;37(7):1010–24.
Obayashi K, Saeki K, Yamagami Y, Kurumatani N, Sugie K, Kataoka H. Circadian activity rhythm in Parkinson’s disease: findings from the PHASE study. Sleep Med. 2021;85:8–14.
Brooks C, Shaafi Kabiri N, Mortazavi F, Auerbach S, Bonato P, Erb MK, et al. Variations in rest-activity rhythm are associated with clinically measured disease severity in Parkinson’s disease. Chronobiol Int. 2020;37(5):699–711.
Wu JQ, Li P, Stavitsky Gilbert K, Hu K, Cronin-Golomb A. Circadian rest-activity rhythms predict cognitive function in early Parkinson’s disease independently of sleep. Mov Disord Clin Pract. 2018;5(6):614–9.
Stefani A, Högl B. Sleep in Parkinson’s disease. Neuropsychopharmacology. 2020;45(1):121–8.
Zhu K, van Hilten JJ, Marinus J. The course of insomnia in Parkinson’s disease. Parkinsonism Relat Disord. 2016;33:51–7.
Rui Z, Qingling C, Xinyue Z, Xin Z, Weihong L. The related factors of sleep benefit in Parkinson’s disease: a systematic review and meta-analysis. PLoS ONE. 2019;14(3): e0212951.
Shen Y, Huang JY, Li J, Liu CF. Excessive daytime sleepiness in Parkinson’s disease: clinical implications and management. Chin Med J. 2018;131(8):974–81.
Liu M, Luo YJ, Gu HY, Wang YM, Liu MH, Li K, et al. Sex and onset-age-related features of excessive daytime sleepiness and night-time sleep in patients with Parkinson’s disease. BMC Neurol. 2021;21(1):165.
Videnovic A, Noble C, Reid KJ, Peng J, Turek FW, Marconi A, et al. Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurol. 2014;71(4):463–9.
Shen Y, Dai YP, Wang Y, Li J, Xiong KP, Mao CJ, et al. Two polysomnographic features of REM sleep behavior disorder: clinical variations insight for Parkinson’s disease. Parkinsonism Relat Disord. 2017;44:66–72.
Hua P, Liu W, Zhao Y, Ding H, Wang L, Xiao H. Tef polymorphism is associated with sleep disturbances in patients with Parkinson’s disease. Sleep Med. 2012;13(3):297–300.
Shen Y, Shen Y, Dong ZF, Pan PL, Shi HC, Liu CF. Obstructive sleep apnea in Parkinson’s disease: a study in 239 Chinese patients. Sleep Med. 2020;67:237–43.
Lajoie AC, Lafontaine AL, Kaminska M. The spectrum of sleep disorders in Parkinson disease: a review. Chest. 2021;159(2):818–27.
Batla A, Phé V, De Min L, Panicker JN. Nocturia in Parkinson’s disease: why does it occur and how to manage. Mov Disord Clin Pract. 2016;3(5):443–51.
Kim JW, Moon YT, Kim KD. Nocturia: the circadian voiding disorder. Investig Clin Urol. 2016;57(3):165–73.
Stuebner E, Vichayanrat E, Low DA, Mathias CJ, Isenmann S, Haensch CA. Twenty-four hour non-invasive ambulatory blood pressure and heart rate monitoring in Parkinson’s disease. Front Neurol. 2013;4:49.
Salsone M, Vescio B, Fratto A, Sturniolo M, Arabia G, Gambardella A, et al. Cardiac sympathetic index identifies patients with Parkinson’s disease and REM behavior disorder. Parkinsonism Relat Disord. 2016;26:62–6.
Arnao V, Cinturino A, Mastrilli S, Buttà C, Maida C, Tuttolomondo A, et al. Impaired circadian heart rate variability in Parkinson’s disease: a time-domain analysis in ambulatory setting. BMC Neurol. 2020;20(1):152.
Zhong G, Bolitho S, Grunstein R, Naismith SL, Lewis SJ. The relationship between thermoregulation and REM sleep behaviour disorder in Parkinson’s disease. PLoS ONE. 2013;8(8): e72661.
Li S, Wang Y, Wang F, Hu LF, Liu CF. A new perspective for Parkinson’s disease: circadian rhythm. Neurosci Bull. 2017;33(1):62–72.
Breen DP, Vuono R, Nawarathna U, Fisher K, Shneerson JM, Reddy AB, et al. Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 2014;71(5):589–95.
Bordet R, Devos D, Brique S, Touitou Y, Guieu JD, Libersa C, et al. Study of circadian melatonin secretion pattern at different stages of Parkinson’s disease. Clin Neuropharmacol. 2003;26(2):65–72.
Aziz NA, Pijl H, Frölich M, Roelfsema F, Roos RA. Leptin, adiponectin, and resistin secretion and diurnal rhythmicity are unaltered in Parkinson’s disease. Mov Disord. 2011;26(4):760–1.
Aziz NA, Pijl H, Frölich M, Roelfsema F, Roos RA. Diurnal secretion profiles of growth hormone, thyrotrophin and prolactin in Parkinson’s disease. J Neuroendocrinol. 2011;23(6):519–24.
Nassan M, Videnovic A. Circadian rhythms in neurodegenerative disorders. Nat Rev Neurol. 2022;18(1):7–24.
Zhao QF, Tan L, Wang HF, Jiang T, Tan MS, Tan L, et al. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: systematic review and meta-analysis. J Affect Disord. 2016;190:264–71.
Bubu OM, Brannick M, Mortimer J, Umasabor-Bubu O, Sebastião YV, Wen Y, et al. Sleep, Cognitive impairment, and Alzheimer’s disease: a systematic review and meta-analysis. Sleep. 2017;40(1):zsw032.
Roland JP, Bliwise DL. Impact of pharmacotherapy on insomnia in patients with Alzheimer’s disease. Drugs Aging. 2021;38(11):951–66.
Musiek ES, Xiong DD, Holtzman DM. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp Mol Med. 2015;47(3): e148.
Abbott SM, Videnovic A. Chronic sleep disturbance and neural injury: links to neurodegenerative disease. Nat Sci Sleep. 2016;8:55–61.
Evans LK. Sundown syndrome in institutionalized elderly. J Am Geriatr Soc. 1987;35(2):101–8.
Martin J, Marler M, Shochat T, Ancoli-Israel S. Circadian rhythms of agitation in institutionalized patients with Alzheimer’s disease. Chronobiol Int. 2000;17(3):405–18.
Gnanasekaran G. “Sundowning” as a biological phenomenon: current understandings and future directions: an update. Aging Clin Exp Res. 2016;28(3):383–92.
Canevelli M, Valletta M, Trebbastoni A, Sarli G, D’Antonio F, Tariciotti L, et al. Sundowning in dementia: clinical relevance, pathophysiological determinants, and therapeutic approaches. Front Med. 2016;3:73.
Talarico G, Canevelli M, Tosto G, Vanacore N, Letteri F, Prastaro M, et al. Restless legs syndrome in a group of patients with Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2013;28(2):165–70.
Guarnieri B, Adorni F, Musicco M, Appollonio I, Bonanni E, Caffarra P, et al. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement Geriatr Cogn Disord. 2012;33(1):50–8.
Yang X, Liu B, Shen H, Li S, Zhao Q, An R, et al. Prevalence of restless legs syndrome in Parkinson’s disease: a systematic review and meta-analysis of observational studies. Sleep Med. 2018;43:40–6.
Bhalsing K, Suresh K, Muthane UB, Pal PK. Prevalence and profile of restless legs syndrome in Parkinson’s disease and other neurodegenerative disorders: a case-control study. Parkinsonism Relat Disord. 2013;19(4):426–30.
Savva E, Schnorf H, Burkhard PR. Restless legs syndrome: an early manifestation of Huntington’s disease. Acta Neurol Scand. 2009;119(4):274–6.
Evers S, Stögbauer F. Genetic association of Huntington’s disease and restless legs syndrome? A family report Mov Disord. 2003;18(2):225–7.
Peeraully T, Tan EK. Linking restless legs syndrome with Parkinson’s disease: clinical, imaging and genetic evidence. Transl Neurodegener. 2012;1(1):6.
Richards KC, Bost JE, Rogers VE, Hutchison LC, Beck CK, Bliwise DL, et al. Diagnostic accuracy of behavioral, activity, ferritin, and clinical indicators of restless legs syndrome. Sleep. 2015;38(3):371–80.
Bombois S, Derambure P, Pasquier F, Monaca C. Sleep disorders in aging and dementia. J Nutr Health Aging. 2010;14(3):212–7.
Emamian F, Khazaie H, Tahmasian M, Leschziner GD, Morrell MJ, Hsiung GY, et al. The association between obstructive sleep apnea and Alzheimer’s disease: a meta-analysis perspective. Front Aging Neurosci. 2016;8:78.
Lee JE, Yang SW, Ju YJ, Ki SK, Chun KH. Sleep-disordered breathing and Alzheimer’s disease: a nationwide cohort study. Psychiatry Res. 2019;273:624–30.
Liguori C, Maestri M, Spanetta M, Placidi F, Bonanni E, Mercuri NB, et al. Sleep-disordered breathing and the risk of Alzheimer’s disease. Sleep Med Rev. 2021;55: 101375.
Bubu OM, Andrade AG, Umasabor-Bubu OQ, Hogan MM, Turner AD, de Leon MJ, et al. Obstructive sleep apnea, cognition and Alzheimer’s disease: a systematic review integrating three decades of multidisciplinary research. Sleep Med Rev. 2020;50: 101250.
de Natale ER, Wilson H, Politis M. Predictors of RBD progression and conversion to synucleinopathies. Curr Neurol Neurosci Rep. 2022;22(2):93–104.
Yeh SB, Yeh PY, Schenck CH. Rivastigmine-induced REM sleep behavior disorder (RBD) in a 88-year-old man with Alzheimer’s disease. J Clin Sleep Med. 2010;6(2):192–5.
Gagnon JF, Petit D, Fantini ML, Rompré S, Gauthier S, Panisset M, et al. REM sleep behavior disorder and REM sleep without atonia in probable Alzheimer disease. Sleep. 2006;29(10):1321–5.
Wang P, Wing YK, Xing J, Liu Y, Zhou B, Zhang Z, et al. Rapid eye movement sleep behavior disorder in patients with probable Alzheimer’s disease. Aging Clin Exp Res. 2016;28(5):951–7.
Boeve BF, Silber MH, Parisi JE, Dickson DW, Ferman TJ, Benarroch EE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61(1):40–5.
Youn S, Kim T, Yoon IY, Jeong J, Kim HY, Han JW, et al. Progression of cognitive impairments in idiopathic REM sleep behaviour disorder. J Neurol Neurosurg Psychiatry. 2016;87(8):890–6.
Zhou J, Zhang J, Lam SP, Chan JW, Mok V, Chan A, et al. Excessive daytime sleepiness predicts neurodegeneration in idiopathic REM sleep behavior disorder. Sleep. 2017;40(5):zsx041.
Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. 2009;72(15):1296–300.
Schenck CH, Montplaisir JY, Frauscher B, Hogl B, Gagnon JF, Postuma R, et al. Rapid eye movement sleep behavior disorder: devising controlled active treatment studies for symptomatic and neuroprotective therapy–a consensus statement from the international rapid eye movement sleep behavior disorder study group. Sleep Med. 2013;14(8):795–806.
Schenck CH, Mahowald MW, Anderson ML, Silber MH, Boeve BF, Parisi JE. Lewy body variant of Alzheimer’s disease (AD) identified by postmortem ubiquitin staining in a previously reported case of AD associated with REM sleep behavior disorder. Biol Psychiatry. 1997;42(6):527–8.
Hansen LA, Samuel W. Criteria for Alzheimer’s disease and the nosology of dementia with Lewy bodies. Neurology. 1997;48(1):126–32.
Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology. 1996;46(2):388–93.
Lu W, Göder R. Does abnormal non-rapid eye movement sleep impair declarative memory consolidation?: Disturbed thalamic functions in sleep and memory processing. Sleep Med Rev. 2012;16(4):389–94.
Weissová K, Bartoš A, Sládek M, Nováková M, Sumová A. Moderate changes in the circadian system of Alzheimer’s disease patients detected in their home environment. PLoS One. 2016;11(1): e0146200.
La Morgia C, Ross-Cisneros FN, Koronyo Y, Hannibal J, Gallassi R, Cantalupo G, et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann Neurol. 2016;79(1):90–109.
Jensen-Dahm C, Waldemar G, Staehelin Jensen T, Malmqvist L, Moeller MM, Andersen BB, et al. Autonomic dysfunction in patients with mild to moderate Alzheimer’s disease. J Alzheimers Dis. 2015;47(3):681–9.
Chen HF, Chang-Quan H, You C, Wang ZR, Hui W, Liu QX, et al. The circadian rhythm of arterial blood pressure in Alzheimer disease (AD) patients without hypertension. Blood Press. 2013;22(2):101–5.
Ramos Bernardes Da Silva Filho S, Oliveira Barbosa JH, Rondinoni C, Dos Santos AC, Garrido Salmon CE, da Costa Lima NK, et al. Neuro-degeneration profile of Alzheimer’s patients: a brain morphometry study. Neuroimage Clin. 2017;15:15–24.
Harper DG, Volicer L, Stopa EG, McKee AC, Nitta M, Satlin A. Disturbance of endogenous circadian rhythm in aging and Alzheimer disease. Am J Geriatr Psychiatry. 2005;13(5):359–68.
Kim MS, Yoon JH, Hong JM. Early differentiation of dementia with Lewy bodies and Alzheimer’s disease: heart rate variability at mild cognitive impairment stage. Clin Neurophysiol. 2018;129(8):1570–8.
Manni R, Cremascoli R, Perretti C, De Icco R, Picascia M, Ghezzi C, et al. Evening melatonin timing secretion in real life conditions in patients with Alzheimer disease of mild to moderate severity. Sleep Med. 2019;63:122–6.
Nous A, Engelborghs S, Smolders I. Melatonin levels in the Alzheimer’s disease continuum: a systematic review. Alzheimers Res Ther. 2021;13(1):52.
Aziz NA, Pijl H, Frölich M, van der Graaf AW, Roelfsema F, Roos RA. Increased hypothalamic-pituitary-adrenal axis activity in Huntington’s disease. J Clin Endocrinol Metab. 2009;94(4):1223–8.
Kalliolia E, Silajdžić E, Nambron R, Hill NR, Doshi A, Frost C, et al. Plasma melatonin is reduced in Huntington’s disease. Mov Disord. 2014;29(12):1511–5.
Saelzler UG, Verhaeghen P, Panizzon MS, Moffat SD. Intact circadian rhythm despite cortisol hypersecretion in Alzheimer’s disease: a meta-analysis. Psychoneuroendocrinology. 2021;132: 105367.
Hartmann A, Veldhuis JD, Deuschle M, Standhardt H, Heuser I. Twenty-four hour cortisol release profiles in patients with Alzheimer’s and Parkinson’s disease compared to normal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol Aging. 1997;18(3):285–9.
Heuser IJ, Chase TN, Mouradian MM. The limbic-hypothalamic-pituitary-adrenal axis in Huntington’s disease. Biol Psychiatry. 1991;30(9):943–52.
Wang JL, Lim AS, Chiang WY, Hsieh WH, Lo MT, Schneider JA, et al. Suprachiasmatic neuron numbers and rest-activity circadian rhythms in older humans. Ann Neurol. 2015;78(2):317–22.
Slats D, Claassen JA, Verbeek MM, Overeem S. Reciprocal interactions between sleep, circadian rhythms and Alzheimer’s disease: focus on the role of hypocretin and melatonin. Ageing Res Rev. 2013;12(1):188–200.
Mishima K, Tozawa T, Satoh K, Matsumoto Y, Hishikawa Y, Okawa M. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol Psychiatry. 1999;45(4):417–21.
Mattis J, Sehgal A. Circadian rhythms, sleep, and disorders of aging. Trends Endocrinol Metab. 2016;27(4):192–203.
Feng R, Li L, Yu H, Liu M, Zhao W. Melanopsin retinal ganglion cell loss and circadian dysfunction in Alzheimer’s disease (Review). Mol Med Rep. 2016;13(4):3397–400.
Davis B, Sadik K. Circadian cholinergic rhythms: implications for cholinesterase inhibitor therapy. Dement Geriatr Cogn Disord. 2006;21(2):120–9.
Morley JE, Farr SA, Kumar VB, Armbrecht HJ. The SAMP8 mouse: a model to develop therapeutic interventions for Alzheimer’s disease. Curr Pharm Des. 2012;18(8):1123–30.
Urrestarazu E, Iriarte J. Clinical management of sleep disturbances in Alzheimer’s disease: current and emerging strategies. Nat Sci Sleep. 2016;8:21–33.
Lucey BP, Hicks TJ, McLeland JS, Toedebusch CD, Boyd J, Elbert DL, et al. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann Neurol. 2018;83(1):197–204.
Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–7.
Niu L, Zhang F, Xu X, Yang Y, Li S, Liu H, et al. Chronic sleep deprivation altered the expression of circadian clock genes and aggravated Alzheimer’s disease neuropathology. Brain Pathol. 2022;32(3): e13028.
Benveniste H, Lee H, Volkow ND. The glymphatic pathway: waste removal from the CNS via cerebrospinal fluid transport. Neuroscientist. 2017;23(5):454–65.
Wang C, Holtzman DM. Bidirectional relationship between sleep and Alzheimer’s disease: role of amyloid, tau, and other factors. Neuropsychopharmacology. 2020;45(1):104–20.
De Pablo-Fernández E, Courtney R, Warner TT, Holton JL. A histologic study of the circadian system in Parkinson Disease, multiple system atrophy, and progressive supranuclear palsy. JAMA Neurol. 2018;75(8):1008–12.
Ozawa T, Soma Y, Yoshimura N, Fukuhara N, Tanaka M, Tsuji S. Reduced morning cortisol secretion in patients with multiple system atrophy. Clin Auton Res. 2001;11(4):271–2.
Suzuki A, Asahina M, Ishikawa C, Asahina KM, Honma K, Fukutake T, et al. Impaired circadian rhythm of gastric myoelectrical activity in patients with multiple system atrophy. Clin Auton Res. 2005;15(6):368–72.
Raupach AK, Ehgoetz Martens KA, Memarian N, Zhong G, Matar E, Halliday GM, et al. Assessing the role of nocturnal core body temperature dysregulation as a biomarker of neurodegeneration. J Sleep Res. 2020;29(5): e12939.
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88–100.
Harper DG, Stopa EG, McKee AC, Satlin A, Harlan PC, Goldstein R, et al. Differential circadian rhythm disturbances in men with Alzheimer disease and frontotemporal degeneration. Arch Gen Psychiatry. 2001;58(4):353–60.
Aziz NA, Anguelova GV, Marinus J, Lammers GJ, Roos RA. Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease. Parkinsonism Relat Disord. 2010;16(5):345–50.
Fifel K, Videnovic A. Circadian and sleep dysfunctions in neurodegenerative disorders-an update. Front Neurosci. 2020;14: 627330.
Owen JE, Veasey SC. Impact of sleep disturbances on neurodegeneration: insight from studies in animal models. Neurobiol Dis. 2020;139: 104820.
Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med. 2013;14(8):744–8.
Postuma RB, Iranzo A, Hu M, Högl B, Boeve BF, Manni R, et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain. 2019;142(3):744–59.
Iranzo A, Stefani A, Serradell M, Martí MJ, Lomeña F, Mahlknecht P, et al. Characterization of patients with longstanding idiopathic REM sleep behavior disorder. Neurology. 2017;89(3):242–8.
Iranzo A, Fairfoul G, Ayudhaya A, Serradell M, Gelpi E, Vilaseca I, et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol. 2021;20(3):203–12.
Wang C, Chen F, Li Y, Liu J. Possible predictors of phenoconversion in isolated REM sleep behaviour disorder: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2022;93(4):395–403.
Miglis MG, Adler CH, Antelmi E, Arnaldi D, Baldelli L, Boeve BF, et al. Biomarkers of conversion to α-synucleinopathy in isolated rapid-eye-movement sleep behaviour disorder. Lancet Neurol. 2021;20(8):671–84.
Boot BP, Boeve BF, Roberts RO, Ferman TJ, Geda YE, Pankratz VS, et al. Probable rapid eye movement sleep behavior disorder increases risk for mild cognitive impairment and Parkinson disease: a population-based study. Ann Neurol. 2012;71(1):49–56.
Jung Y, Boot BP, Mielke MM, Ferman TJ, Geda YE, McDade E, et al. Phenoconversion from probable rapid eye movement sleep behavior disorder to mild cognitive impairment to dementia in a population-based sample. Alzheimers Dement. 2017;8:127–30.
Pak VM, Onen SH, Bliwise DL, Kutner NG, Russell KL, Onen F. Sleep Disturbances in MCI and AD: neuroinflammation as a possible mediating pathway. Front Aging Neurosci. 2020;12:69.
Xu W, Tan CC, Zou JJ, Cao XP, Tan L. Sleep problems and risk of all-cause cognitive decline or dementia: an updated systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2020;91(3):236–44.
Beydoun HA, Beydoun MA, Weiss J, Hossain S, Huang S, Alemu BT, et al. Insomnia as a predictor of diagnosed memory problems: 2006–2016 health and retirement study. Sleep Med. 2021;80:158–66.
Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 2015;14(1):57–64.
Yao CW, Pelletier A, Fereshtehnejad SM, Cross N, Dang-Vu T, Postuma RB. Insomnia symptom subtypes and manifestations of prodromal neurodegeneration: a population-based study in the Canadian longitudinal study on aging. J Clin Sleep Med. 2022;18(2):345–59.
Tholfsen LK, Larsen JP, Schulz J, Tysnes OB, Gjerstad MD. Changes in insomnia subtypes in early Parkinson disease. Neurology. 2017;88(4):352–8.
Lysen TS, Darweesh S, Ikram MK, Luik AI, Ikram MA. Sleep and risk of parkinsonism and Parkinson’s disease: a population-based study. Brain. 2019;142(7):2013–22.
Iranzo A, Serradell M, Santamaria J. Excessive daytime sleepiness does not predict neurodegeneration in idiopathic REM sleep behavior disorder. Sleep. 2017;40(12):zsx161.
Postuma RB, Gagnon JF, Pelletier A, Montplaisir JY. Insomnia and somnolence in idiopathic RBD: a prospective cohort study. NPJ Parkinsons Dis. 2017;3:9.
Rosenzweig I, Glasser M, Polsek D, Leschziner GD, Williams SC, Morrell MJ. Sleep apnoea and the brain: a complex relationship. Lancet Respir Med. 2015;3(5):404–14.
Bohnen NI, Hu M. Sleep disturbance as potential risk and progression factor for Parkinson’s disease. J Parkinsons Dis. 2019;9(3):603–14.
Yaffe K, Laffan AM, Harrison SL, Redline S, Spira AP, Ensrud KE, et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA. 2011;306(6):613–9.
Lutsey PL, Misialek JR, Mosley TH, Gottesman RF, Punjabi NM, Shahar E, et al. Sleep characteristics and risk of dementia and Alzheimer’s disease: the atherosclerosis risk in communities study. Alzheimers Dement. 2018;14(2):157–66.
Kang J, Tian Z, Wei J, Mu Z, Liang J, Li M. Association between obstructive sleep apnea and Alzheimer’s disease-related blood and cerebrospinal fluid biomarkers: a meta-analysis. J Clin Neurosci. 2022;102:87–94.
Ylä-Herttuala S, Hakulinen M, Poutiainen P, Laitinen TM, Koivisto AM, Remes AM, et al. Severe obstructive sleep apnea and increased cortical amyloid-β deposition. J Alzheimers Dis. 2021;79(1):153–61.
Shieu MM, Zaheed A, Shannon C, Chervin RD, Conceicao A, Paulson HL, et al. Positive airway pressure and cognitive disorders in adults with obstructive sleep apnea: a systematic review of the literature. Neurology. 2022;99(4):e334–46.
Dunietz GL, Chervin RD, Burke JF, Conceicao AS, Braley TJ. Obstructive sleep apnea treatment and dementia risk in older adults. Sleep. 2021;44(9):zsab076.
Chou PS, Lai CL, Chou YH, Chang WP. Sleep apnea and the subsequent risk of Parkinson’s disease: a 3-year nationwide population-based study. Neuropsychiatr Dis Treat. 2017;13:959–65.
Sheu JJ, Lee HC, Lin HC, Kao LT, Chung SD. A 5-year follow-up study on the relationship between obstructive sleep apnea and Parkinson disease. J Clin Sleep Med. 2015;11(12):1403–8.
Sun AP, Liu N, Zhang YS, Zhao HY, Liu XL. The relationship between obstructive sleep apnea and Parkinson’s disease: a systematic review and meta-analysis. Neurol Sci. 2020;41(5):1153–62.
Sun HL, Sun BL, Chen DW, Chen Y, Li WW, Xu MY, et al. Plasma α-synuclein levels are increased in patients with obstructive sleep apnea syndrome. Ann Clin Transl Neurol. 2019;6(4):788–94.
Li J, Zhao L, Ding X, Cui X, Qi L, Chen Y. Obstructive sleep apnea and the risk of Alzheimer’s disease and Parkinson disease: a Mendelian randomization study OSA, Alzheimer’s disease and Parkinson disease. Sleep Med. 2022;97:55–63.
Tsapanou A, Gu Y, O’Shea D, Eich T, Tang MX, Schupf N, et al. Daytime somnolence as an early sign of cognitive decline in a community-based study of older people. Int J Geriatr Psychiatry. 2016;31(3):247–55.
Sung PS, Yeh CC, Wang LC, Hung PH, Muo CH, Sung FC, et al. Increased risk of dementia in patients with non-apnea sleep disorder. Curr Alzheimer Res. 2017;14(3):309–16.
Smagula SF, Jia Y, Chang CH, Cohen A, Ganguli M. Trajectories of daytime sleepiness and their associations with dementia incidence. J Sleep Res. 2020;29(6): e12952.
Carvalho DZ, St Louis EK, Boeve BF, Mielke MM, Przybelski SA, Knopman DS, et al. Excessive daytime sleepiness and fatigue may indicate accelerated brain aging in cognitively normal late middle-aged and older adults. Sleep Med. 2017;32:236–43.
Carvalho DZ, St Louis EK, Knopman DS, Boeve BF, Lowe VJ, Roberts RO, et al. Association of excessive daytime sleepiness with longitudinal β-amyloid accumulation in elderly persons without dementia. JAMA Neurol. 2018;75(6):672–80.
Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, et al. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 2011;14(6):750–6.
Abbott RD, Ross GW, White LR, Tanner CM, Masaki KH, Nelson JS, et al. Excessive daytime sleepiness and subsequent development of Parkinson disease. Neurology. 2005;65(9):1442–6.
Gao J, Huang X, Park Y, Hollenbeck A, Blair A, Schatzkin A, et al. Daytime napping, nighttime sleeping, and Parkinson disease. Am J Epidemiol. 2011;173(9):1032–8.
Abbott RD, Ross GW, Duda JE, Shin C, Uyehara-Lock JH, Masaki KH, et al. Excessive daytime sleepiness and topographic expansion of Lewy pathology. Neurology. 2019;93(15):e1425–32.
Alonso-Navarro H, García-Martín E, Agúndez J, Jiménez-Jiménez FJ. Association between restless legs syndrome and other movement disorders. Neurology. 2019;92(20):948–64.
Lin CC, Chou CH, Fan YM, Yin JH, Chung CH, Chien WC, et al. Increased risk of dementia among sleep-related movement disorders: a population-based longitudinal study in Taiwan. Medicine. 2015;94(51): e2331.
Zhang X, Molsberry SA, Pavlova M, Schwarzschild MA, Ascherio A, Gao X. Association of sleepwalking and REM sleep behavior disorder with Parkinson disease in men. JAMA Netw Open. 2021;4(4): e215713.
Leng Y, Goldman SM, Cawthon PM, Stone KL, Ancoli-Israel S, Yaffe K. Excessive daytime sleepiness, objective napping and 11-year risk of Parkinson’s disease in older men. Int J Epidemiol. 2018;47(5):1679–86.
Leng Y, Blackwell T, Cawthon PM, Ancoli-Israel S, Stone KL, Yaffe K. Association of circadian abnormalities in older adults with an increased risk of developing Parkinson disease. JAMA Neurol. 2020;77(10):1270–8.
Cullell N, Cárcel-Márquez J, Gallego-Fábrega C, Muiño E, Llucià-Carol L, Lledós M, et al. Sleep/wake cycle alterations as a cause of neurodegenerative diseases: a Mendelian randomization study. Neurobiol Aging. 2021;106:320.e1-320.e12.
Naismith SL, Hickie IB, Terpening Z, Rajaratnam SM, Hodges JR, Bolitho S, et al. Circadian misalignment and sleep disruption in mild cognitive impairment. J Alzheimers Dis. 2014;38(4):857–66.
Musiek ES, Bhimasani M, Zangrilli MA, Morris JC, Holtzman DM, Ju YS. Circadian rest-activity pattern changes in aging and preclinical Alzheimer disease. JAMA Neurol. 2018;75(5):582–90.
Kress GJ, Liao F, Dimitry J, Cedeno MR, FitzGerald GA, Holtzman DM, et al. Regulation of amyloid-β dynamics and pathology by the circadian clock. J Exp Med. 2018;215(4):1059–68.
Lee J, Kim DE, Griffin P, Sheehan PW, Kim DH, Musiek ES, et al. Inhibition of REV-ERBs stimulates microglial amyloid-beta clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer’s disease. Aging Cell. 2020;19(2): e13078.
Bokenberger K, Sjölander A, Dahl Aslan AK, Karlsson IK, Åkerstedt T, Pedersen NL. Shift work and risk of incident dementia: a study of two population-based cohorts. Eur J Epidemiol. 2018;33(10):977–87.
Bokenberger K, Ström P, Dahl Aslan AK, Johansson AL, Gatz M, Pedersen NL, et al. Association between sleep characteristics and incident dementia accounting for baseline cognitive status: a prospective population-based study. J Gerontol A Biol Sci Med Sci. 2017;72(1):134–9.
Walsh CM, Blackwell T, Tranah GJ, Stone KL, Ancoli-Israel S, Redline S, et al. Weaker circadian activity rhythms are associated with poorer executive function in older women. Sleep. 2014;37(12):2009–16.
Rogers-Soeder TS, Blackwell T, Yaffe K, Ancoli-Israel S, Redline S, Cauley JA, et al. Rest-activity rhythms and cognitive decline in older men: the osteoporotic fractures in men sleep study. J Am Geriatr Soc. 2018;66(11):2136–43.
Li P, Gao L, Gaba A, Yu L, Cui L, Fan W, et al. Circadian disturbances in Alzheimer’s disease progression: a prospective observational cohort study of community-based older adults. Lancet Healthy Longev. 2020;1(3):e96-96e105.
Posner AB, Tranah GJ, Blackwell T, Yaffe K, Ancoli-Israel S, Redline S, et al. Predicting incident dementia and mild cognitive impairment in older women with nonparametric analysis of circadian activity rhythms in the study of osteoporotic fractures. Sleep. 2021;44(10):zsab19.
Spira AP, Zipunnikov V, Raman R, Choi J, Di J, Bai J, et al. Brain amyloid burden, sleep, and 24-hour rest/activity rhythms: screening findings from the anti-amyloid treatment in asymptomatic Alzheimer’s and longitudinal evaluation of amyloid risk and neurodegeneration studies. Sleep Adv. 2021;2(1):zpab015.
Smith MT, McCrae CS, Cheung J, Martin JL, Harrod CG, Heald JL, et al. Use of actigraphy for the evaluation of sleep disorders and circadian rhythm sleep-wake disorders: an American academy of sleep medicine clinical practice guideline. J Clin Sleep Med. 2018;14(7):1231–7.
Tranah GJ, Blackwell T, Stone KL, Ancoli-Israel S, Paudel ML, Ensrud KE, et al. Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann Neurol. 2011;70(5):722–32.
Xiao Q, Shadyab AH, Rapp SR, Stone KL, Yaffe K, Sampson JN, et al. Rest-activity rhythms and cognitive impairment and dementia in older women: results from the women’s health initiative. J Am Geriatr Soc. 2022;70(10):2925–37.
Amara AW, Chahine LM, Caspell-Garcia C, Long JD, Coffey C, Högl B, et al. Longitudinal assessment of excessive daytime sleepiness in early Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2017;88(8):653–62.
Trenkwalder C, Allen R, Högl B, Paulus W, Winkelmann J. Restless legs syndrome associated with major diseases: a systematic review and new concept. Neurology. 2016;86(14):1336–43.
Ortiz-Tudela E, Martinez-Nicolas A, Díaz-Mardomingo C, García-Herranz S, Pereda-Pérez I, Valencia A, et al. The characterization of biological rhythms in mild cognitive impairment. Biomed Res Int. 2014;2014: 524971.
van Someren EJ, Hagebeuk EE, Lijzenga C, Scheltens P, de Rooij SE, Jonker C, et al. Circadian rest-activity rhythm disturbances in Alzheimer’s disease. Biol Psychiatry. 1996;40(4):259–70.
Harper DG, Stopa EG, McKee AC, Satlin A, Fish D, Volicer L. Dementia severity and Lewy bodies affect circadian rhythms in Alzheimer disease. Neurobiol Aging. 2004;25(6):771–81.
Videnovic A, Lazar AS, Barker RA, Overeem S. ’The clocks that time us’–circadian rhythms in neurodegenerative disorders. Nat Rev Neurol. 2014;10(12):683–93.
Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313(19):1924–38.
Hita-Yañez E, Atienza M, Cantero JL. Polysomnographic and subjective sleep markers of mild cognitive impairment. Sleep. 2013;36(9):1327–34.
Ju YE, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70(5):587–93.
Penninkilampi R, Casey AN, Singh MF, Brodaty H. The Association between social engagement, loneliness, and risk of dementia: a systematic review and meta-analysis. J Alzheimers Dis. 2018;66(4):1619–33.
Brzecka A, Leszek J, Ashraf GM, Ejma M, Ávila-Rodriguez MF, Yarla NS, et al. Sleep disorders associated with Alzheimer’s disease: a perspective. Front Neurosci. 2018;12:330.
Leroi I, Baker P, Kehoe P, Daniel E, Byrne EJ. A pilot randomized controlled trial of sleep therapy in Parkinson’s disease: effect on patients and caregivers. Int J Geriatr Psychiatry. 2010;25(10):1073–9.
McCurry SM, LaFazia DM, Pike KC, Logsdon RG, Teri L. Development and evaluation of a sleep education program for older adults with dementia living in adult family homes. Am J Geriatr Psychiatry. 2012;20(6):494–504.
Frazzitta G, Maestri R, Ferrazzoli D, Riboldazzi G, Bera R, Fontanesi C, et al. Multidisciplinary intensive rehabilitation treatment improves sleep quality in Parkinson’s disease. J Clin Mov Disord. 2015;2:11.
Amara AW, Wood KH, Joop A, Memon RA, Pilkington J, Tuggle SC, et al. Randomized, controlled trial of exercise on objective and subjective sleep in Parkinson’s disease. Mov Disord. 2020;35(6):947–58.
Yang JH, Wang YQ, Ye SQ, Cheng YG, Chen Y, Feng XZ. The effects of group-based versus individual-based tai chi training on nonmotor symptoms in patients with mild to moderate Parkinson’s disease: a randomized controlled pilot trial. Parkinsons Dis. 2017;2017:8562867.
Xiao CM, Zhuang YC. Effect of health Baduanjin Qigong for mild to moderate Parkinson’s disease. Geriatr Gerontol Int. 2016;16(8):911–9.
Wassom DJ, Lyons KE, Pahwa R, Liu W. Qigong exercise may improve sleep quality and gait performance in Parkinson’s disease: a pilot study. Int J Neurosci. 2015;125(8):578–84.
McCurry SM, Pike KC, Vitiello MV, Logsdon RG, Larson EB, Teri L. Increasing walking and bright light exposure to improve sleep in community-dwelling persons with Alzheimer’s disease: results of a randomized, controlled trial. J Am Geriatr Soc. 2011;59(8):1393–402.
Venturelli M, Sollima A, Cè E, Limonta E, Bisconti AV, Brasioli A, et al. Effectiveness of exercise- and cognitive-based treatments on salivary cortisol levels and sundowning syndrome symptoms in patients with Alzheimer’s disease. J Alzheimers Dis. 2016;53(4):1631–40.
Hillman CH, Erickson KI, Kramer AF. Be smart, exercise your heart: exercise effects on brain and cognition. Nat Rev Neurosci. 2008;9(1):58–65.
Cristini J, Weiss M, De Las HB, Medina-Rincón A, Dagher A, Postuma RB, et al. The effects of exercise on sleep quality in persons with Parkinson’s disease: a systematic review with meta-analysis. Sleep Med Rev. 2021;55: 101384.
Ortuño-Lizarán I, Beach TG, Serrano GE, Walker DG, Adler CH, Cuenca N. Phosphorylated α-synuclein in the retina is a biomarker of Parkinson’s disease pathology severity. Mov Disord. 2018;33(8):1315–24.
Li Z, Tian T. Light therapy promoting dopamine release by stimulating retina in Parkinson disease. JAMA Neurol. 2017;74(10):1267–8.
Lin F, Su Y, Weng Y, Lin X, Weng H, Cai G, et al. The effects of bright light therapy on depression and sleep disturbances in patients with Parkinson’s disease: a systematic review and meta-analysis of randomized controlled trials. Sleep Med. 2021;83:280–9.
Videnovic A, Klerman EB, Wang W, Marconi A, Kuhta T, Zee PC. Timed light therapy for sleep and daytime sleepiness associated with Parkinson disease: a randomized clinical trial. JAMA Neurol. 2017;74(4):411–8.
Shen Y, Gong S, Liu Y, Li J, Xiong K, Mao C, et al. Therapeutic potential of bright light therapy for the non-motor symptoms in Parkinson’s disease. Chin Med J. 2021;135(2):243–4.
van Dijk KD, Møst EI, Van Someren EJ, Berendse HW, van der Werf YD. Beneficial effect of transcranial magnetic stimulation on sleep in Parkinson’s disease. Mov Disord. 2009;24(6):878–84.
Zhuang S, Wang FY, Gu X, Wu JJ, Mao CJ, Gui H, et al. Low-frequency repetitive transcranial magnetic stimulation over right dorsolateral prefrontal cortex in Parkinson’s disease. Parkinsons Dis. 2020;2020:7295414.
Zhang X, Zhuang S, Wu J, Wang L, Mao C, Chen J, et al. Effects of repetitive transcranial magnetic stimulation over right dorsolateral prefrontal cortex on excessive daytime sleepiness in patients with Parkinson’s disease. Sleep Med. 2022;100:133–8.
Zhou X, Wang Y, Lv S, Li Y, Jia S, Niu X, et al. Transcranial magnetic stimulation for sleep disorders in Alzheimer’s disease: a double-blind, randomized, and sham-controlled pilot study. Neurosci Lett. 2022;766: 136337.
Brusa L, Versace V, Koch G, Bernardi G, Iani C, Stanzione P, et al. Improvement of choreic movements by 1 Hz repetitive transcranial magnetic stimulation in Huntington’s disease patients. Ann Neurol. 2005;58(4):655–6.
Shukla A, Jayarajan RN, Muralidharan K, Jain S. Repetitive transcranial magnetic stimulation not beneficial in severe choreiform movements of Huntington disease. J ECT. 2013;29(2):e16–7.
Romanella SM, Roe D, Paciorek R, Cappon D, Ruffini G, Menardi A, et al. Sleep, noninvasive brain stimulation, and the aging brain: challenges and opportunities. Ageing Res Rev. 2020;61: 101067.
Ma H, Yan J, Sun W, Jiang M, Zhang Y. Melatonin treatment for sleep disorders in Parkinson’s disease: a meta-analysis and systematic review. Front Aging Neurosci. 2022;14: 784314.
Taximaimaiti R, Luo X, Wang XP. Pharmacological and non-pharmacological treatments of sleep disorders in Parkinson’s disease. Curr Neuropharmacol. 2021;19(12):2233–49.
Blackman J, Swirski M, Clynes J, Harding S, Leng Y, Coulthard E. Pharmacological and non-pharmacological interventions to enhance sleep in mild cognitive impairment and mild Alzheimer’s disease: a systematic review. J Sleep Res. 2021;30(4): e13229.
Ahn JH, Kim M, Park S, Jang W, Park J, Oh E, et al. Prolonged-release melatonin in Parkinson’s disease patients with a poor sleep quality: a randomized trial. Parkinsonism Relat Disord. 2020;75:50–4.
De Berardis D, Fornaro M, Serroni N, Olivieri L, Marini S, Moschetta FS, et al. Agomelatine treatment of major depressive disorder in Parkinson’s disease: a case series. J Neuropsychiatry Clin Neurosci. 2013;25(4):343–5.
Khyati, Malik I, Agrawal N, Kumar V. Melatonin and curcumin reestablish disturbed circadian gene expressions and restore locomotion ability and eclosion behavior in Drosophila model of Huntington’s disease. Chronobiol Int. 2021;38(1):61–78.
Gilat M, Marshall NS, Testelmans D, Buyse B, Lewis S. A critical review of the pharmacological treatment of REM sleep behavior disorder in adults: time for more and larger randomized placebo-controlled trials. J Neurol. 2022;269(1):125–48.
Shin C, Park H, Lee WW, Kim HJ, Kim HJ, Jeon B. Clonazepam for probable REM sleep behavior disorder in Parkinson’s disease: a randomized placebo-controlled trial. J Neurol Sci. 2019;401:81–6.
Menza M, Dobkin RD, Marin H, Gara M, Bienfait K, Dicke A, et al. Treatment of insomnia in Parkinson’s disease: a controlled trial of eszopiclone and placebo. Mov Disord. 2010;25(11):1708–14.
Huo S, Cheng L, Li S, Xu F. Effects of eszopiclone on sleep quality and cognitive function in elderly patients with Alzheimer’s disease and sleep disorder: a randomized controlled trial. Brain Behav. 2022;12(2): e2488.
Aurora RN, Zak RS, Maganti RK, Auerbach SH, Casey KR, Chowdhuri S, et al. Best practice guide for the treatment of REM sleep behavior disorder (RBD). J Clin Sleep Med. 2010;6(1):85–95.
Li SX, Lam SP, Zhang J, Yu MW, Chan JW, Liu Y, et al. A prospective, naturalistic follow-up study of treatment outcomes with clonazepam in rapid eye movement sleep behavior disorder. Sleep Med. 2016;21:114–20.
Camargos EF, Louzada LL, Quintas JL, Naves JO, Louzada FM, Nóbrega OT. Trazodone improves sleep parameters in Alzheimer disease patients: a randomized, double-blind, and placebo-controlled study. Am J Geriatr Psychiatry. 2014;22(12):1565–74.
Rios Romenets S, Creti L, Fichten C, Bailes S, Libman E, Pelletier A, et al. Doxepin and cognitive behavioural therapy for insomnia in patients with Parkinson’s disease—a randomized study. Parkinsonism Relat Disord. 2013;19(7):670–5.
Riemersma-van Der Lek RF, Swaab DF, Twisk J, Hol EM, Hoogendijk WJ, Van Someren EJ. Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: a randomized controlled trial. JAMA. 2008;299(22):2642–55.
Dowling GA, Burr RL, Van Someren EJ, Hubbard EM, Luxenberg JS, Mastick J, et al. Melatonin and bright-light treatment for rest-activity disruption in institutionalized patients with Alzheimer’s disease. J Am Geriatr Soc. 2008;56(2):239–46.
Fifel K, Videnovic A. Chronotherapies for Parkinson’s disease. Prog Neurobiol. 2019;174:16–27.
Uzair M, Abualait T, Arshad M, Yoo WK, Mir A, Bunyan RF, et al. Transcranial magnetic stimulation in animal models of neurodegeneration. Neural Regen Res. 2022;17(2):251–65.
Purushothuman S, Johnstone DM, Nandasena C, Mitrofanis J, Stone J. Photobiomodulation with near infrared light mitigates Alzheimer’s disease-related pathology in cerebral cortex—evidence from two transgenic mouse models. Alzheimers Res Ther. 2014;6(1):2.
Zinchenko E, Navolokin N, Shirokov A, Khlebtsov B, Dubrovsky A, Saranceva E, et al. Pilot study of transcranial photobiomodulation of lymphatic clearance of beta-amyloid from the mouse brain: breakthrough strategies for non-pharmacologic therapy of Alzheimer’s disease. Biomed Opt Express. 2019;10(8):4003–17.
El Massri N, Lemgruber AP, Rowe IJ, Moro C, Torres N, Reinhart F, et al. Photobiomodulation-induced changes in a monkey model of Parkinson’s disease: changes in tyrosine hydroxylase cells and GDNF expression in the striatum. Exp Brain Res. 2017;235(6):1861–74.
Zhou F, Yan XD, Wang C, He YX, Li YY, Zhang J, et al. Suvorexant ameliorates cognitive impairments and pathology in APP/PS1 transgenic mice. Neurobiol Aging. 2020;91:66–75.
Chang YC, Kim JY. Therapeutic implications of circadian clocks in neurodegenerative diseases. J Neurosci Res. 2020;98(6):1095–113.
Flajolet M, He G, Heiman M, Lin A, Nairn AC, Greengard P. Regulation of Alzheimer’s disease amyloid-beta formation by casein kinase I. Proc Natl Acad Sci U S A. 2007;104(10):4159–64.
Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, et al. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318(5857):1786–9.
Schnell A, Chappuis S, Schmutz I, Brai E, Ripperger JA, Schaad O, et al. The nuclear receptor REV-ERBα regulates Fabp7 and modulates adult hippocampal neurogenesis. PLoS ONE. 2014;9(6): e99883.
Han L, Xie YH, Wu R, Chen C, Zhang Y, Wang XP. Traditional Chinese medicine for modern treatment of Parkinson’s disease. Chin J Integr Med. 2017;23(8):635–40.
Zeng BY. Effect and mechanism of Chinese herbal medicine on Parkinson’s disease. Int Rev Neurobiol. 2017;135:57–76.
Cheng FK. The use of acupuncture in patients with Parkinson’s disease. Geriatr Nurs. 2017;38(4):302–14.
Pan W, Kwak S, Li G, Chen Y, Cai D. Therapeutic effect of Yang-Xue-Qing-Nao granules on sleep dysfunction in Parkinson’s disease. Chin Med. 2013;8:14.
Fifel K, Videnovic A. Circadian alterations in patients with neurodegenerative diseases: neuropathological basis of underlying network mechanisms. Neurobiol Dis. 2020;144: 105029.
Ylikoski A, Martikainen K, Partinen M. Parasomnias and isolated sleep symptoms in Parkinson’s disease: a questionnaire study on 661 patients. J Neurol Sci. 2014;346(1–2):204–8.
Chahine LM, Amara AW, Videnovic A. A systematic review of the literature on disorders of sleep and wakefulness in Parkinson’s disease from 2005 to 2015. Sleep Med Rev. 2017;35:33–50.
Maggi G, Vitale C, Cerciello F, Santangelo G. Sleep and wakefulness disturbances in Parkinson’s disease: a meta-analysis on prevalence and clinical aspects of REM sleep behavior disorder, excessive daytime sleepiness and insomnia. Sleep Med Rev. 2023;68: 101759.
Kumar S, Bhatia M, Behari M. Sleep disorders in Parkinson’s disease. Mov Disord. 2002;17(4):775–81.
Terzaghi M, Minafra B, Zangaglia R, Picascia M, Pozzi N, Cremascoli R, et al. NREM sleep arousal-related disorders reflect cognitive impairment in Parkinson’s disease. Sleep Med. 2020;75:491–6.
Valko PO, Hauser S, Sommerauer M, Werth E, Baumann CR. Observations on sleep-disordered breathing in idiopathic Parkinson’s disease. PLoS ONE. 2014;9(6): e100828.
Stefanova N, Bücke P, Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol. 2009;8(12):1172–8.
Moreno-López C, Santamaría J, Salamero M, Del Sorbo F, Albanese A, Pellecchia MT, et al. Excessive daytime sleepiness in multiple system atrophy (SLEEMSA study). Arch Neurol. 2011;68(2):223–30.
Palma JA, Fernandez-Cordon C, Coon EA, Low PA, Miglis MG, Jaradeh S, et al. Prevalence of REM sleep behavior disorder in multiple system atrophy: a multicenter study and meta-analysis. Clin Auton Res. 2015;25(1):69–75.
Gaig C, Iranzo A. Sleep-disordered breathing in neurodegenerative diseases. Curr Neurol Neurosci Rep. 2012;12(2):205–17.
Cochen De Cock V. Sleep abnormalities in multiple system atrophy. Curr Treat Options Neurol. 2018;20(6):16.
Elder GJ, Lazar AS, Alfonso-Miller P, Taylor JP. Sleep disturbances in Lewy body dementia: a systematic review. Int J Geriatr Psychiatry. 2022. https://doi.org/10.1002/gps.5814.
Manni R, Terzaghi M. Sleep-disordered breathing in dementia with Lewy bodies. Curr Neurol Neurosci Rep. 2015;15(3):7.
McCarter SJ, St Louis EK, Boeve BF. Sleep disturbances in frontotemporal dementia. Curr Neurol Neurosci Rep. 2016;16(9):85.
Trotti LM, Bliwise DL, Keating GL, Rye DB, Hu WT. Cerebrospinal fluid hypocretin and nightmares in dementia syndromes. Dement Geriatr Cogn Dis Extra. 2021;11(1):19–25.
Roche S, Jacquesson JM, Destée A, Defebvre L, Derambure P, Monaca C. Sleep and vigilance in corticobasal degeneration: a descriptive study. Neurophysiol Clin. 2007;37(4):261–4.
Hurtado González CA, Piedrahita C, Vivas Álzate D, García Borrero JJ, Marmolejo Escobar CS, Ospina Otalvaro S, et al. Neuropsychiatric aspects in a patient diagnosed with corticobasal degeneration: clinical case of low incidence and prevalence in Colombia. Case Rep Neurol. 2020;12(3):387–401.
Baumann-Vogel H, Hor H, Poryazova R, Valko P, Werth E, Baumann CR. REM sleep behavior in Parkinson disease: frequent, particularly with higher age. PLoS ONE. 2020;15(12): e0243454.
Abbott SM, Videnovic A. Sleep disorders in atypical Parkinsonism. Mov Disord Clin Pract. 2014;1(2):89–96.
Arena JE, Weigand SD, Whitwell JL, Hassan A, Eggers SD, Höglinger GU, et al. Progressive supranuclear palsy: progression and survival. J Neurol. 2016;263(2):380–9.
Arnulf I, Merino-Andreu M, Bloch F, Konofal E, Vidailhet M, Cochen V, et al. REM sleep behavior disorder and REM sleep without atonia in patients with progressive supranuclear palsy. Sleep. 2005;28(3):349–54.
Sixel-Döring F, Schweitzer M, Mollenhauer B, Trenkwalder C. Polysomnographic findings, video-based sleep analysis and sleep perception in progressive supranuclear palsy. Sleep Med. 2009;10(4):407–15.
Moccia M, Picillo M, Erro R, Allocca R, Barone P, Vitale C. Diagnosis and treatment of restless legs syndrome in progressive supranuclear palsy. J Neurol Sci. 2015;350(1–2):103–4.
Boeve A, Ferman TJ, Aakre J, St Louis E, Silber M, Machulda M, et al. Excessive daytime sleepiness in major dementia syndromes. Am J Alzheimers Dis Other Demen. 2019;34(4):261–4.
Matsuzaki A, Shiroki K, Kimura G. Induction of cellular DNA synthesis by adenovirus type 12 in a set of temperature-sensitive mutants of rat 3Y1 fibroblasts blocked in G1 phase. Virology. 1987;160(1):227–35.
Furby H, Siadimas A, Rutten-Jacobs L, Rodrigues FB, Wild EJ. Natural history and burden of Huntington’s disease in the UK: a population-based cohort study. Eur J Neurol. 2022;29(8):2249–57.
Aldaz T, Nigro P, Sánchez-Gómez A, Painous C, Planellas L, Santacruz P, et al. Non-motor symptoms in Huntington’s disease: a comparative study with Parkinson’s disease. J Neurol. 2019;266(6):1340–50.
Videnovic A, Leurgans S, Fan W, Jaglin J, Shannon KM. Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism Relat Disord. 2009;15(6):471–4.
Jha M, Kamble N, Lenka A, Yadav R, Purushottam M, Jain S, et al. Sleep disturbances in patients with Huntington’s disease: a questionnaire-based study. Annals Mov Disord. 2019;1:9–14.
Arnulf I, Nielsen J, Lohmann E, Schiefer J, Wild E, Jennum P, et al. Rapid eye movement sleep disturbances in Huntington disease. Arch Neurol. 2008;65(4):482–8.
Piano C, Losurdo A, Della Marca G, Solito M, Calandra-Buonaura G, Provini F, et al. Polysomnographic findings and clinical correlates in Huntington disease: a cross-sectional cohort study. Sleep. 2015;38(9):1489–95.
Fifel K, Cooper HM. Loss of dopamine disrupts circadian rhythms in a mouse model of Parkinson’s disease. Neurobiol Dis. 2014;71:359–69.
Pfeffer M, Zimmermann Z, Gispert S, Auburger G, Korf HW, von Gall C. Impaired photic entrainment of spontaneous locomotor activity in mice overexpressing human mutant α-synuclein. Int J Mol Sci. 2018;19(6):1651.
Kudo T, Loh DH, Truong D, Wu Y, Colwell CS. Circadian dysfunction in a mouse model of Parkinson’s disease. Exp Neurol. 2011;232(1):66–75.
Hayashi A, Matsunaga N, Okazaki H, Kakimoto K, Kimura Y, Azuma H, et al. A disruption mechanism of the molecular clock in a MPTP mouse model of Parkinson’s disease. Neuromolecular Med. 2013;15(2):238–51.
Masini D, Lopes-Aguiar C, Bonito-Oliva A, Papadia D, Andersson R, Fisahn A, et al. The histamine H3 receptor antagonist thioperamide rescues circadian rhythm and memory function in experimental parkinsonism. Transl Psychiatry. 2017;7(4): e1088.
Wang Y, Lv D, Liu W, Li S, Chen J, Shen Y, et al. Disruption of the circadian clock alters antioxidative defense via the SIRT1-BMAL1 pathway in 6-OHDA-induced models of Parkinson’s disease. Oxid Med Cell Longev. 2018;2018:4854732.
Li H, Song S, Wang Y, Huang C, Zhang F, Liu J, et al. Low-grade inflammation aggravates rotenone neurotoxicity and disrupts circadian clock gene expression in rats. Neurotox Res. 2019;35(2):421–31.
Gravotta L, Gavrila AM, Hood S, Amir S. Global depletion of dopamine using intracerebroventricular 6-hydroxydopamine injection disrupts normal circadian wheel-running patterns and PERIOD2 expression in the rat forebrain. J Mol Neurosci. 2011;45(2):162–71.
Isobe Y, Nishino H. Circadian rhythm of drinking and running-wheel activity in rats with 6-hydroxydopamine lesions of the ventral tegmental area. Brain Res. 2001;899(1–2):187–92.
Ben V, Bruguerolle B. Effects of bilateral striatal 6-OHDA lesions on circadian rhythms in the rat: a radiotelemetric study. Life Sci. 2000;67(13):1549–58.
Yang S, Wan Y, Wu N, Song L, Liu Z, Zhao J, et al. L-3,4-dihydroxyphenylalanine recovers circadian rhythm disturbances in the rat models of Parkinson’s disease by regulating the D1R-ERK1/2-mTOR pathway. Front Aging Neurosci. 2021;13: 719885.
Fifel K, Vezoli J, Dzahini K, Claustrat B, Leviel V, Kennedy H, et al. Alteration of daily and circadian rhythms following dopamine depletion in MPTP treated non-human primates. PLoS ONE. 2014;9(1): e86240.
Julienne H, Buhl E, Leslie DS, Hodge J. Drosophila PINK1 and parkin loss-of-function mutants display a range of non-motor Parkinson’s disease phenotypes. Neurobiol Dis. 2017;104:15–23.
Gajula Balija MB, Griesinger C, Herzig A, Zweckstetter M, Jäckle H. Pre-fibrillar α-synuclein mutants cause Parkinson’s disease-like non-motor symptoms in Drosophila. PLoS ONE. 2011;6(9): e24701.
Silva R, Pinho BR, Santos MM, Oliveira J. Disruptions of circadian rhythms, sleep, and stress responses in zebrafish: New infrared-based activity monitoring assays for toxicity assessment. Chemosphere. 2022;305: 135449.
Sterniczuk R, Dyck RH, Laferla FM, Antle MC. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 1. Circadian changes Brain Res. 2010;1348:139–48.
Ambrée O, Touma C, Görtz N, Keyvani K, Paulus W, Palme R, et al. Activity changes and marked stereotypic behavior precede Abeta pathology in TgCRND8 Alzheimer mice. Neurobiol Aging. 2006;27(7):955–64.
Han SM, Jang YJ, Kim EY, Park SA. The change in circadian rhythms in P301S transgenic mice is linked to variability in Hsp70-related tau disaggregation. Exp Neurobiol. 2022;31(3):196–207.
Huang J, Peng X, Fan R, Dong K, Shi X, Zhang S, et al. Disruption of circadian clocks promotes progression of Alzheimer’s disease in diabetic mice. Mol Neurobiol. 2021;58(9):4404–12.
Wang L, Zhao J, Wang CT, Hou XH, Ning N, Sun C, et al. D-Ser2-oxyntomodulin ameliorated Aβ31-35-induced circadian rhythm disorder in mice. CNS Neurosci Ther. 2020;26(3):343–54.
Li X, Guan J, Sun T, Yang J, Yu H, Yao J, et al. Circadian learning and memory changes in Aβ1-42 induced Alzheimer’s mice. Metab Brain Dis. 2020;35(3):463–71.
Filon MJ, Wallace E, Wright S, Douglas DJ, Steinberg LI, Verkuilen CL, et al. Sleep and diurnal rest-activity rhythm disturbances in a mouse model of Alzheimer’s disease. Sleep. 2020;43(11):087.
Furtado A, Astaburuaga R, Costa A, Duarte AC, Gonçalves I, Cipolla-Neto J, et al. The rhythmicity of clock genes is disrupted in the choroid plexus of the APP/PS1 mouse model of Alzheimer’s disease. J Alzheimers Dis. 2020;77(2):795–806.
Tate B, Aboody-Guterman KS, Morris AM, Walcott EC, Majocha RE, Marotta CA. Disruption of circadian regulation by brain grafts that overexpress Alzheimer beta/A4 amyloid. Proc Natl Acad Sci USA. 1992;89(15):7090–4.
Duncan MJ, Smith JT, Franklin KM, Beckett TL, Murphy MP, St Clair DK, et al. Effects of aging and genotype on circadian rhythms, sleep, and clock gene expression in APPxPS1 knock-in mice, a model for Alzheimer’s disease. Exp Neurol. 2012;236(2):249–58.
Graybeal JJ, Bozzelli PL, Graybeal LL, Groeber CM, McKnight PE, Cox DN, et al. Human ApoE ε4 alters circadian rhythm activity, IL-1β, and GFAP in CRND8 mice. J Alzheimers Dis. 2015;43(3):823–34.
Song H, Moon M, Choe HK, Han DH, Jang C, Kim A, et al. Aβ-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol Neurodegener. 2015;10:13.
Wang X, Wang L, Xu Y, Yu Q, Li L, Guo Y. Intranasal administration of exendin-4 antagonizes Aβ31-35-induced disruption of circadian rhythm and impairment of learning and memory. Aging Clin Exp Res. 2016;28(6):1259–66.
Wang X, Wang L, Yu Q, Xu Y, Zhang L, Zhao X, et al. Alterations in the expression of Per1 and Per2 induced by Aβ31-35 in the suprachiasmatic nucleus, hippocampus, and heart of C57BL/6 mouse. Brain Res. 2016;1642:51–8.
Coronas-Samano G, Baker KL, Tan WJ, Ivanova AV, Verhagen JV. Fus1 KO mouse as a model of oxidative stress-mediated sporadic Alzheimer’s disease: circadian disruption and long-term spatial and olfactory memory impairments. Front Aging Neurosci. 2016;8:268.
Oyegbami O, Collins HM, Pardon MC, Ebling F, Heery DM, Moran PM. Abnormal clock gene expression and locomotor activity rhythms in two month-old female APPSwe/PS1dE9 mice. Curr Alzheimer Res. 2017;14(8):850–60.
Wu M, Zhou F, Cao X, Yang J, Bai Y, Yan X, et al. Abnormal circadian locomotor rhythms and Per gene expression in six-month-old triple transgenic mice model of Alzheimer’s disease. Neurosci Lett. 2018;676:13–8.
Wang L, Zhang R, Hou X, Wang C, Guo S, Ning N, et al. DA-JC1 improves learning and memory by antagonizing Aβ31-35-induced circadian rhythm disorder. Mol Brain. 2019;12(1):14.
Kent BA, Michalik M, Marchant EG, Yau KW, Feldman HH, Mistlberger RE, et al. Delayed daily activity and reduced NREM slow-wave power in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neurobiol Aging. 2019;78:74–86.
Gerstner JR, Lenz O, Vanderheyden WM, Chan MT, Pfeiffenberger C, Pack AI. Amyloid-β induces sleep fragmentation that is rescued by fatty acid binding proteins in Drosophila. J Neurosci Res. 2017;95(8):1548–64.
Arnes M, Alaniz ME, Karam CS, Cho JD, Lopez G, Javitch JA, et al. Role of tau protein in remodeling of circadian neuronal circuits and sleep. Front Aging Neurosci. 2019;11:320.
Acknowledgements
Not applicable.
Funding
This review was funded by the National Natural Science Foundation of China (82071420), the Discipline Construction Program of the Second Affiliated Hospital of Soochow University (XKTJ-XK202001), and Suzhou Medical and Health Technology Innovation Project (SKJY2021090).
Author information
Authors and Affiliations
Contributions
All authors participated in drafting the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors claim that there are no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shen, Y., Lv, Qk., Xie, Wy. et al. Circadian disruption and sleep disorders in neurodegeneration. Transl Neurodegener 12, 8 (2023). https://doi.org/10.1186/s40035-023-00340-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-023-00340-6