
Abstract
Background
The limited regenerative capacity of damaged neurons in adult mammals severely restricts neural repair. Although stem cell transplantation is promising, its clinical application remains challenging. Direct reprogramming, which utilizes cell plasticity to regenerate neurons, is an emerging alternative approach.
Methods
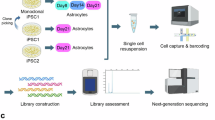
We utilized primary postnatal cortical astrocytes for reprogramming induced neurons (iNs) through the viral-mediated overexpression of the transcription factors Ngn2 and Pax6 (NP). Fluorescence-activated cell sorting (FACS) was used to enrich successfully transfected cells, followed by single-cell RNA sequencing (scRNA-seq) using the 10 × Genomics platform for comprehensive transcriptomic analysis.
Results
The scRNA-seq revealed that NP overexpression led to the differentiation of astrocytes into iNs, with percentages of 36% and 39.3% on days 4 and 7 posttransduction, respectively. CytoTRACE predicted the developmental sequence, identifying astrocytes as the reprogramming starting point. Trajectory analysis depicted the dynamic changes in gene expression during the astrocyte-to-iN transition.
Conclusions
This study elucidates the molecular dynamics underlying astrocyte reprogramming into iNs, revealing key genes and pathways involved in this process. Our research contributes novel insights into the molecular mechanisms of NP-mediated reprogramming, suggesting avenues for optimizing the efficiency of the reprogramming process.
Similar content being viewed by others


Background
In adult mammals, the inability of damaged neurons to regenerate severely limits neural repair. Although stem cell transplantation holds promise, clinical translation remains challenging [1]. Direct lineage reprogramming, which exploits cellular plasticity for neural regeneration, has emerged as an innovative alternative [2,3,4,5,6]. This method converts resident astrocytes into neurons, capitalizing on their similarity to progenitor cells or neural stem cells [7]. Transcription factors such as Ngn2, Ascl1, and Dlx2 can directly differentiate astrocytes from the mouse cerebral cortex into neurons [8, 9].
Previous studies have indicated that reprogramming, driven by factors such as Ascl1, Brn2, and Myt1l, entails epigenetic changes influencing cell fate. For example, Ascl1 initiates rapid chromatin remodeling and nucleosome repositioning, enabling fibroblasts to transform into induced neurons (iNs) within 12 h [10, 11]. Myt1l also plays a crucial role by inhibiting somatic differentiation and promoting neuronal function [12]. Despite these advances, the transcriptional mechanisms underlying direct neuronal fate conversion are not fully understood.
In this study, we utilized single-cell RNA sequencing (scRNA-seq) to investigate the molecular transcriptional mechanisms underlying the transformation of cultured astrocytes into iNs. This research may reveal the underlying mechanisms involved and provide a more in-depth understanding of astrocyte reprogramming.
Materials and methods
Primary culture of postnatal cortical astrocytes
We cultured postnatal astrocytes using a previously described method [13]. After removing the meninges, we dissected and mechanically dissociated cortical tissue from the cerebral cortex of P5‒P7 C57BL/6 J mice. The cells were then centrifuged at 1000 rpm for 2 min, resuspended, and cultured in medium containing DMEM/F12, 10% fetal bovine serum, 5% horse serum, and penicillin/streptomycin. To remove any contaminating oligodendrocyte precursor cells, the culture flasks were vigorously shaken multiple times. The cells were passaged once they reached 90% confluence. The experimental protocol received ethical approval from the Ethics Committee of Guangxi Medical University.
Viral transduction of Ngn2 and Pax6
First, we determined the optimal multiplicity of infection (MOI) value as 20 through gradient infection experiments. Subsequently, we coated a 24-well plate with a 0.1 mg/ml poly-D-lysine (PDL) solution and seeded astrocytes at a density of 1 × 10^4 cells per well. On the day of infection, the cells were divided into a control group and an overexpression group. The control group was infected with a mixture of Ubi-MCS-SV40-EGFP and Ubi-MCS-SV40-mCherry viruses, while the overexpression group was infected with a mixture of Ubi-MCS-SV40-Ngn2-EGFP and Ubi-MCS-SV40-Pax6-mCherry viruses. Sixteen hour postinfection, we replaced the medium with differentiation medium containing 20 ng/mL BDNF and subsequently replaced the medium every four days to maintain cell growth and viral expression. Viruses were directly purchased from Shanghai Genechem Co., Ltd.
Fluorescence-activated cell sorting (FACS)
FACS was utilized to selectively enrich and collect cells that had been successfully transfected. The cells were trypsinized using 0.25% trypsin, followed by centrifugation. After centrifugation, the cells were resuspended in PBS (pH 7.4) at a concentration of 1 × 10^6 cells/mL, supplemented with 0.5% fetal bovine serum, and then placed on ice for further processing. We specifically targeted eGFP + or mCherry + cells, as indicated by the virus labeling, ensuring the accurate selection of the transfected cells, characterized by fluorescence. Using a FACS instrument, we enriched and collected the green (eGFP +) or red (mCherry +) fluorescent cells based on their fluorescence intensity, thereby ensuring high purity of the selected cell populations for subsequent downstream applications.
Single-cell suspension preparation
After sampling, tissue fragments were promptly transferred to a centrifuge tube and incubated with trypsin at 37 °C for 10 min. The tube was then centrifuged at 1000 rpm for 2 min. The cells were resuspended in resuspension buffer consisting of 2.5 ml fetal bovine serum (FBS) and 47.5 ml phosphate-buffered saline (PBS). The cell suspension was passed through a flow cytometry cell strainer into a flow cytometry tube for cell sorting.
10 × Genomics library construction and sequencing
Cellular suspensions were processed using a 10X Genomics GemCode single-cell instrument to generate gel bead-in-EMlusions (GEMs). Libraries were constructed and sequenced from cDNA using Chromium Next GEM Single Cell 3’ Reagent Kits v3.1. Within each GEM, primers carrying an Illumina® R1 sequence, a 16 nt 10 × Barcode, a 10 nt UMI, and a poly-dT primer sequence were released and mixed with the cell lysate and Master Mix to reverse transcribe barcoded, full-length cDNAs from polyadenylated mRNA. The post-GEM reaction mixture was purified by silane magnetic beads, which removed residual reagents and primers. The full-length, barcoded cDNAs were subjected to PCR amplification, yielding sufficient material for library construction.
The R1 primer sequence was added to the molecules during GEM incubation. P5, P7, a sample index, and the R2 primer sequence were incorporated during library construction through processes such as end repair, A-tailing, adaptor ligation, and PCR. The final libraries contained the P5 and P7 primers utilized for Illumina bridge amplification. The P5 and P7 primers were contained in the resulting libraries and were compatible with Illumina sequencing. The standard Illumina paired-end constructs, which begin and end with P5 and P7, comprise a single cell 3’ library.
In Read 1, the single cell 3′ 16 bp 10 × barcode and 10 bp UMI were encoded, while Read 2 was utilized to sequence the cDNA fragment. The sample index sequences were integrated as the i7 index read. Both Read 1 and Read 2, serving as standard Illumina® sequencing primer sites, were used for paired-end sequencing.
scRNA-seq analysis
The Seurat package [14] was utilized to convert 10 × scRNA-seq data into a Seurat object, and the NormalizeData function was employed to normalize the expression matrix. A set of highly variable genes, totaling two thousand, was identified by using the FindVariableFeatures function. To reduce the dimensionality of the data set, principal component analysis (PCA) was performed using the RunPCA method. Further dimensionality reduction and cluster identification were achieved through the uniform manifold approximation and projection (UMAP) algorithm [15]. Significant marker genes for different clusters were identified using the FindAllMarkers function. For the annotation of cell types, marker genes from the literature were used in conjunction with the PanglaoDB database for annotation [16].
Trajectory analysis
We revealed the differentiation trajectories and subtype evolution in the process of cell reprogramming by combining four methods. Gene expression changes were analyzed by the Monocle 2 algorithm using DDRTree technology and pseudotime series [17]. The VECTOR algorithm segments the UMAP into pixels, calculated PC value QP scores, identified developmental origins, and computed pseudotime scores [18]. Monocle 3 was used to sort cells based on pseudotime [19]. The dynamic process of cell state transition was demonstrated through pseudotemporal heatmaps. CytoTRACE scores of astrocytes and iNs were computed using the R package CytoTRACE [20], with a range from 0 to 1. Higher scores indicated less differentiation.
High-dimensional weighted gene coexpression network analysis (hdWGCNA)
To explore the molecular functions of iNs, we utilized hdWGCNA to detect genes within coexpression modules that exhibited high levels of aggregation [21]. We applied the TestSoftPowers function to determine an optimal soft-power threshold (β) that facilitated the formation of a scale-free topological network. Subsequently, within each coexpression module, we pinpointed hub genes utilizing eigengene-based connectivity measures to establish their central roles within the network.
Functional enrichment analysis
We utilized the ClusterProfiler R package to perform Gene Ontology (GO) analysis, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis.
Statistical analysis
Statistical analyses were conducted using R (version 4.3.1). Gene expression comparisons between the two groups were assessed using the Wilcoxon test. Differences were considered significant when p < 0.05.
Results
Single-cell atlas
To explore the molecular mechanisms of astrocyte reprogramming, we performed single-cell sequencing analysis on cultured astrocytes. During the early stages of reprogramming, specifically on days 4 and 7 after lentivirus transduction, we isolated astrocytes from the cortex of neonatal mice using FACS and performed high-throughput scRNA-seq. The purpose of this experiment was to evaluate the proportion of iNs in the following groups: the control group on day 4 (D4-C), the control group on day 7 (D7-C), the Ngn2 and Pax6 (NP) overexpression group on day 4 (D4-OE), and the NP overexpression group on day 7 (D7-OE). The results showed the absence of iNs in the D4-C and D7-C groups. The incidence of iNs was 36% and 39.3% in the D4-OE and D7-OE groups, respectively (Fig. 1).

Classification of cell types among the four groups: A UMAP of D4-C, B UMAP of D7-C, C UMAP of D4-OE, and D UMAP of D7-OE. UMAP, uniform manifold approximation and projection
CytoTRACE and expression of marker genes
In this study, we applied the CytoTRACE approach to predict developmental sequences and starting points. In the D4-OE group, the results showed that astrocytes had the highest CytoTRACE scores, indicating a low degree of differentiation and suggesting that they were at the starting point of the cell trajectory (Fig. 2A, B). Additionally, we discovered genes correlated with cell differentiation processes in the D4-OE group, such as H3f3b and Cdkn1a (Fig. 2C). Similarly, in the D7-OE group, astrocytes were at the starting point of the cell trajectory (Fig. 2D, E). Furthermore, we identified genes correlated with cell differentiation processes in the D7-OE group, such as Prdx5 and Gfap (Fig. 2F).

Stemness of astrocytes and iNs was assessed using CytoTRACE to predict the cell differentiation potential of both cell types. A, B tSNE plot and boxplot depicting the distribution of CytoTRACE scores among astrocytes and iNs in the D4-OE group, where dark green indicates lower scores (low stemness) and dark red indicates higher scores (high stemness). C Genes correlated with cell differentiation processes in the D4-OE group, as predicted by CytoTRACE. D, E tSNE plot and boxplot showing the distribution of CytoTRACE scores among astrocytes and iNs in the D7-OE group, with dark green indicating lower scores (low stemness) and dark red indicating higher scores (high stemness). F Genes correlated with cell differentiation processes in the D7-OE group as predicted by CytoTRACE. tSNE, t-distributed dtochastic neighbor embedding
In our study, we examined the expression of astrocytic and neuronal markers in the NP group, focusing on the differences between astrocytes and iNs. Compared to astrocytes iNs in the D4-OE group, exhibited a reduced expression of the astrocytic markers S100a10 and Aqp4 (Fig. 3A, B). Concurrently, these iNs displayed elevated expression of the neuronal markers Sox2 and Crym (Fig. 3C, D). Similarly, in the D7-OE group, compared with astrocytes, iNs exhibited decreased expression of the astrocytic markers Gfap, Aldh1l1, and Aqp4 (Fig. 3E–G). In contrast, these iNs exhibited increased expression levels of the neuronal markers Sox2 and Map2 (Fig. 3H, I).

Differential expression of astrocytic and neuronal markers in iNs and astrocytes across the D4-OE and D7-OE groups. A, B Expression of the astrocytic markers S100a10 and Aqp4 in the D4-OE group. C–D Expression of the neuronal markers Sox2 and Crym in the D4-OE group. E–G Expression of the astrocytic markers Gfap, Aldh1l1, and Aqp4 in the D7-OE group. H, I Expression of neuronal Sox2 and Map2 in the D7-OE group. ****p < 0.0001
Trajectory analysis of marker genes
To deepen our understanding of astrocyte differentiation mechanisms, we performed a dynamic gene expression analysis utilizing Monocle 2. This analysis revealed temporal shifts in gene expression as astrocytes transitioned to iNs. Specifically, in the D4-OE group, during the process of differentiation, we observed the upregulation of Ascl1, Cux1, Map2, and Tubb3 in the early stages (Fig. 4A–D). Similarly, in the D7-OE group, Cux1 and Map2 exhibited increased expression levels during the transformation process of astrocytes compared to those at the beginning of the transformation (Fig. 4E, F).

Relative expression profiles of marker genes in the differentiation pseudotime trajectory. A–D D4-OE. A, Expression analysis of Ascl1; B Expression analysis of Cux1, C Expression analysis of Map2; D Expression analysis of Tubb3. E, F D7-OE. E Expression analysis of Cux1; F Expression analysis of Map2
Pseudotemporal heatmap and functional enrichment analysis
We utilized pseudotemporal heatmaps to analyze the temporal dynamics of gene expression throughout the astrocyte-to-iNs transition. We detected concurrent activation of genes such as Prdx1 and Slc1a3 in the D4-OE stage. Functional enrichment analysis indicated that these activated genes were predominantly involved in pathways such as glial cell differentiation, neuron apoptotic process, response to oxidative stress and neurotransmitter uptake (Fig. 5A).

Heatmap illustrating gene expression patterns over pseudotime and functional enrichment analysis of genes throughout the astrocyte-to-iN conversion process. A D4-OE; B D7-OE
Furthermore, in the D7-OE stage, we detected an upregulation of genes, including Fth1, Tmsb4x, Hpca, Cadm3, and Gm42418, as astrocytes converted into iNs. Functional enrichment analysis revealed enrichment of genes involved in forebrain development, telencephalon development, hindbrain development and centrosome duplication (Fig. 5B).
High-dimensional weighted gene coexpression network analysis (hdWGCNA) and functional enrichment analysis
We constructed a weighted coexpression network to detect gene functional modules, as many genes exert their effects through close interactions and exhibit similar expression patterns. We used hdWGCNA to identify the major molecular features of iNs on D4-OE. With a soft threshold of 4, we constructed a scale-free network of iNs to obtain optimal connectivity and identified 7 gene modules (Fig. 6A, B). We performed GO enrichment analysis on the hub genes and found that their functions were mainly related to glial cell differentiation, regulation of cell growth, and regulation of neurogenesis (Fig. 6C). KEGG pathway enrichment analysis revealed significant enrichment in pathways associated with ferroptosis, fluid shear stress and atherosclerosis, and mineral absorption (Fig. 6D). We also identified the top five hub genes of iNs on D4-OE (Fig. 6E).

hdWGCNA revealed hub genes in the iNs module of the D4-OE group. A Fit indices of the scale-free topology model and the average connectivity were evaluated for different soft-thresholding powers. After careful analysis, the optimal soft threshold was determined to be 4. B iNs were subjected to high dimensional weighted gene coexpression analysis. C GO analysis of the hub genes associated with the iNs module. D KEGG analysis of the hub genes associated with the iNs module. E Visualization of hub genes associated with the iNs module
Furthermore, we identified the major molecular features of iNs on D7-OE. With a soft threshold of 3, we constructed a scale-free network of iNs and identified 7 gene modules (Fig. 7A, B). We performed GO enrichment analysis on the hub genes and found that their functions involved mainly positive regulation of nervous system development, regulation of neurogenesis, positive regulation of neurogenesis, axon guidance, and neuron projection guidance (Fig. 7C). KEGG pathway enrichment analysis revealed that the main enriched pathways were the JAK − STAT signaling pathway and the PI3K–Akt signaling pathway (Fig. 7D). We also identified the top five hub genes of iNs on D7-OE (Fig. 7E).

hdWGCNA revealed hub genes in the iNs module of the D7-OE group. A Fit indices of the scale-free topology model and the average connectivity were evaluated for different soft-thresholding powers. After careful analysis, the optimal soft threshold was determined to be 3. B iNs were subjected to high dimensional weighted gene coexpression analysis. C GO analysis of the hub genes associated with the iNs module. D KEGG analysis of the hub genes associated with the iNs module. E Visualization of hub genes associated with the iNs module
Discussion
In this study, compared with those in control astrocytes, the proportions of iNs generated from NP-mediated astrocytes in the D4-OE and D7-OE groups were 36% and 39.3%, respectively. Dynamic gene expression during the conversion process was described, and it was found that the expression of certain neuronal markers (Map2, Cux1) gradually increased during the conversion. Through pseudotemporal heatmaps, we observed that during astrocyte-to-iN conversion, a set of genes were simultaneously activated in D4-OE cells, participated in glial cell differentiation and neurotransmitter uptake. On the other hand, in D7-OE, another set of genes was simultaneously activated, involving pathways such as forebrain development, telencephalon development, hindbrain development and centrosome duplication. By constructing a weighted coexpression network, we determined the major molecular features of iNs during D4-OE and D7-OE. On D4-OE, genes related to glial cell differentiation, regulation of cell growth, and regulation of neurogenesis were associated with iNs. Moreover, in D7-OE, genes related to the positive regulation of nervous system development, regulation of neurogenesis, positive regulation of neurogenesis, axon guidance, and neuron projection guidance were associated with iNs. These results revealed the molecular characteristics and regulatory mechanisms underlying the astrocyte-to-iN conversion.
Nng2 is essential for the glutamatergic neurotransmitter phenotype in the embryonic neocortex and effectively converts astrocytes into glutamatergic neurons, underscoring its role in excitatory neurogenesis. Additionally, Ngn2 induces both excitatory and inhibitory neurons in the spinal cord [22]. Through the CRISPRa system, the activation of Ngn2 and Isl1 can convert astrocytes into functional motor neurons [23]. Furthermore, when Ngn2 combines with Nurr1, it can induce the formation of dopaminergic neurons [24]. As a key regulator of neurogenesis, Pax6 serves as an intrinsic determinant of the neurogenic potential of glial cells [13]. Pax6 transduction in proliferative glial cells induces the expression of early neuronal markers [25].
This study employed the CytoTRACE method, a novel approach to delineate the developmental trajectory and pinpoint the initiation points of astrocytes as they undergo reprogramming into iNs. Our findings in the D4-OE and D7-OE groups revealed that astrocytes with elevated CytoTRACE scores are less differentiated, supporting the premise that these cells are poised at the onset of their cellular developmental pathway. Genes associated with cell differentiation processes, such as H3f3b, Cdkn1a, and Prdx5, were identified. H3f3b, the gene encoding histone H3.3, is crucial for establishing the identity of mitotic neurons after key developmental stages [26]. CDKN1A/p21, a cyclin-dependent kinase inhibitor, plays a pivotal role in the suppression of ferroptosis through its induction by p53 [27]. This is particularly relevant in the context of neuronal reprogramming, where the generation of reactive oxygen species (ROS) has been reported to impede the reprogramming process [28].
Additionally, PRDX5, an antioxidant protein, is associated with aberrant ROS [29]. Given that oxidative stress-induced ferroptosis can impose limitations on neuronal reprogramming [30], the potential of PRDX5 as a key mediator in safeguarding cells against ROS, particularly those originating from mitochondrial sources, is highlighted [31]. These insights collectively enhance our understanding of the molecular dynamics during the reprogramming of astrocytes into iNs and offer avenues for developing targeted strategies to optimize this process.
The reprogramming of astrocytes into iNs is a complex and intricate process that involves a delicate interplay of transcription factors and cellular markers, which are pivotal in determining cell fate. Following the overexpression of NP in astrocytes, we observed a rapid downregulation of astrocytic genes in both the D4-OE and D7-OE groups. The rapid transcriptomic changes observed after overexpressing neural transcription factors are consistent with previous reports on NeuroD1, Ascl1, or Ngn2 [10, 32,33,34]. The observed downregulation of astrocytic markers such as S100a10, Aqp4, and Gfap, coupled with the upregulation of neuronal markers such as Sox2, Map2, and Crym, underscores the phenotypic metamorphosis that astrocytes undergo as they are reprogrammed into iNs. As revealed by our Monocle 2 analysis, the temporal dynamics of gene expression provided deeper insights into the molecular timeline of the reprogramming process from astrocytes to iNs. There was early upregulation of Ascl1, Cux1, Map2, and Tubb3 in the D4-OE group, with increased expression of Map2 and Cux1 in the D7-OE group during the transformation process. Previous studies have shown that the direct reprogramming of fibroblasts into neurons involves two stages: an initiation stage in which Ascl1 induces neuronal fate, and a maturation stage in which reprogrammed fibroblasts permanently acquire a neuronal identity [12]. This may also hold true for astrocytes, as Ascl1 expression increases and then decreases during the process of astrocyte-to-iN differentiation. In the initial stage, Ascl1 expression increases, and Ascl1 functions as a core driver of reprogramming [10], with its overexpression capable of inducing rapid neuronal differentiation [35]. Map2, which is predominantly expressed in neurons, is integral to axonal and dendritic growth, synaptic plasticity, and overall neuronal development [36]. Cux1 promotes the integration of layer II–III neurons in the cortical network in a highly specific manner [37]. The orchestrated modulation of astrocytic and neuronal markers during the reprogramming of astrocytes into iNs highlights the complexity of cell fate determination. Understanding the molecular choreography that drives this process is vital for harnessing the therapeutic potential of cellular reprogramming in neurological applications.
The pseudotemporal heatmaps allowed us to dissect the temporal dynamics of this transition, revealing significant insights into the genes and pathways involved. In the D4-OE stage, the activation of genes such as Prdx1 and Slc1a3 is noteworthy. Antioxidant proteins play a crucial role in maintaining body homeostasis. PRDX1 possesses an antioxidant function and is capable of clearing ROS within the body [38]. It interacts with various kinases and enzymes and can prevent cell apoptosis caused by oxidative stress [39]. Its upregulation during the astrocyte-to-iN transition suggests a potential protective mechanism against oxidative stress, which is known to be a critical factor in neurogenesis [40]. Glutamate levels are regulated by SLC1A3, which reduces excitotoxicity and helps to prevent damage to the nervous system caused by excitotoxicity [41]. The functional enrichment analysis revealed that these activated genes are predominantly involved in pathways associated with glial cell differentiation, neuron apoptotic process, response to oxidative stress and neurotransmitter uptake. Moreover, direct neuronal reprogramming to obtain neurons is a process that can induce oxidative stress [42], which necessitates an appropriate response to oxidative stress to maintain cellular homeostasis and ensure the successful differentiation of reprogrammed cells.
In the subsequent D7-OE stage, genes such as Fth1, Tmsb4x, Hpca, Cadm3, and Gm42418 were upregulated. TMSB4X plays a crucial role in the transdifferentiation of human fibroblasts into myogenic cells, enhancing the efficiency of the process [43]. The expression level of HPCA increases during neuronal differentiation, and overexpression of HPCA enhances neuronal differentiation [44]. CADM3 mediates direct contact and interaction between axons and glial cells [47].
The hdWGCNA was used to identify gene modules and hub genes that play a central role in the reprogramming process. GO enrichment analysis of the hub genes in the D7-OE group revealed functions that are essential for the development and maturation of the nervous system, including positive regulation of nervous system development, regulation of neurogenesis, axon guidance, and neuron projection guidance. Neurons undergo processes such as neuronal migration, axonal elongation, axon pruning, dendritic morphogenesis, synaptic maturation, and plasticity to form neural circuits [45]. Moreover, KEGG pathway enrichment analysis revealed pathways that are implicated in neurodegenerative diseases, such as ferroptosis, and other pathways related to the JAK–STAT and PI3K–Akt signaling pathways. The JAK–STAT3 pathway is not only recognized as the core signaling pathway for maintaining pluripotency but also has recently been shown to be essential for the complete reprogramming of mouse somatic cells [46]. The dedifferentiation of astrocytes into an undifferentiated state is induced through the activation of the PI3K/Akt/p21 signaling pathway, thereby promoting their transdifferentiation into neurons [48].
There are limitations to this study. Our research is based on in vitro experiments, and the results need to be validated in vivo to ensure their reliability and reproducibility. The specific mechanisms and processes of transformation still require further investigation. There may be unknown factors and obstacles during the cellular conversion process, which could affect the efficiency of conversion and the stability of cell characteristics. Our study revealed the potential of the NP to reprogram astrocytes into iNs, explored the molecular features and regulatory mechanisms of astrocyte-to-iN conversion.
Conclusion
This study elucidated the molecular dynamics of astrocyte reprogramming into iNs by identifying key genes and pathways involved in this process. The CytoTRACE method revealed less differentiated astrocytes as the starting point for reprogramming. Overexpression of Ngn2 and Pax6 led to rapid downregulation of astrocytic genes and upregulation of neuronal markers, indicative of phenotypic transformation. This study also highlighted the role of antioxidants in countering oxidative stress during reprogramming, which is essential for maintaining cellular homeostasis and successful neuronal differentiation. Despite in vitro limitations, the findings offer insights into the regulatory mechanisms of astrocyte-to-iN conversion, potentially aiding in developing therapeutic strategies for neurodegenerative diseases.
Availability of data and materials
All the data generated or analyzed during this study are included in the methods section of this article. Other data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Lindvall O. Why is it taking so long to develop clinically competitive stem cell therapies for CNS disorders? Cell Stem Cell. 2012;10(6):660–2.
Heinrich C, Spagnoli FM, Berninger B. In vivo reprogramming for tissue repair. Nat Cell Biol. 2015;17(3):204–11.
Amamoto R, Arlotta P. Development-inspired reprogramming of the mammalian central nervous system. Science. 2014;343(6170):1239882.
Mertens J, et al. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat Rev Neurosci. 2016;17(7):424–37.
Srivastava D, DeWitt N. In vivo cellular reprogramming: the next generation. Cell. 2016;166(6):1386–96.
Chen G, et al. In vivo reprogramming for brain and spinal cord repair. eNeuro. 2015;2(5):ENEURO.0106-15.2015.
Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–84.
Heinrich C, et al. Directing astroglia from the cerebral cortex into subtype specific functional neurons. PLoS Biol. 2010;8(5): e1000373.
Heinrich C, et al. Generation of subtype-specific neurons from postnatal astroglia of the mouse cerebral cortex. Nat Protoc. 2011;6(2):214–28.
Wapinski OL, et al. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell. 2013;155(3):621–35.
Wapinski OL, et al. Rapid chromatin switch in the direct reprogramming of fibroblasts to neurons. Cell Rep. 2017;20(13):3236–47.
Mall M, et al. Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature. 2017;544(7649):245–9.
Heins N, et al. Glial cells generate neurons: the role of the transcription factor Pax6. Nat Neurosci. 2002;5(4):308–15.
Butler A, et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20.
Becht E, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2018;37:38–44. https://doi.org/10.1038/nbt.4314.
Franzén O, Gan LM, Björkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford). 2019;2019:baz046.
Qiu X, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14(10):979–82.
Zhang F, Li X, Tian W. Unsupervised inference of developmental directions for single cells using VECTOR. Cell Rep. 2020;32(8): 108069.
Cao J, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566(7745):496–502.
Gulati GS, et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science. 2020;367(6476):405–11.
Li JD, et al. Triosephosphate isomerase 1 may be a risk predictor in laryngeal squamous cell carcinoma: a multi-centered study integrating bulk RNA, single-cell RNA, and protein immunohistochemistry. Eur J Med Res. 2023;28(1):591.
Liu F, et al. Neurog2 directly converts astrocytes into functional neurons in midbrain and spinal cord. Cell Death Dis. 2021;12(3):225.
Zhou M, et al. Reprogramming astrocytes to motor neurons by activation of endogenous Ngn2 and Isl1. Stem Cell Reports. 2021;16(7):1777–91.
Rodríguez-Traver E, et al. Role of Nurr1 in the generation and differentiation of dopaminergic neurons from stem cells. Neurotox Res. 2016;30(1):14–31.
Buffo A, et al. Expression pattern of the transcription factor Olig2 in response to brain injuries: implications for neuronal repair. Proc Natl Acad Sci USA. 2005;102(50):18183–8.
Funk OH, et al. Postmitotic accumulation of histone variant H3.3 in new cortical neurons establishes neuronal chromatin, transcriptome, and identity. Proc Natl Acad Sci USA. 2022;119(32):e2116956119.
Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162–8.
Esteban MA, et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell. 2010;6(1):71–9.
Qian S, et al. The synergistic effects of PRDX5 and Nrf2 on lung cancer progression and drug resistance under oxidative stress in the zebrafish models. Oncol Res. 2022;30(2):53–64.
Gascón S, et al. Identification and successful negotiation of a metabolic checkpoint in direct neuronal reprogramming. Cell Stem Cell. 2016;18(3):396–409.
Kropotov A, et al. Mitochondrial targeting of human peroxiredoxin V protein and regulation of PRDX5 gene expression by nuclear transcription factors controlling biogenesis of mitochondria. Febs j. 2007;274(22):5804–14.
Ma NX, Puls B, Chen G. Transcriptomic analyses of NeuroD1-mediated astrocyte-to-neuron conversion. Dev Neurobiol. 2022;82(5):375–91.
Masserdotti G, et al. Transcriptional mechanisms of proneural factors and REST in regulating neuronal reprogramming of astrocytes. Cell Stem Cell. 2015;17(1):74–88.
Rao Z, et al. Molecular mechanisms underlying Ascl1-mediated astrocyte-to-neuron conversion. Stem Cell Rep. 2021;16(3):534–47.
Ma Q, et al. Distinct domains within Mash1 and Math1 are required for function in neuronal differentiation versus neuronal cell-type specification. Genes Dev. 2004;131(6):1319–30.
Sánchez C, Díaz-Nido J, Avila J. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol. 2000;61(2):133–68.
Cubelos B, et al. Cux1 and Cux2 selectively target basal and apical dendritic compartments of layer II-III cortical neurons. Dev Neurobiol. 2015;75(2):163–72.
Poynton RA, Hampton MB. Peroxiredoxins as biomarkers of oxidative stress. Biochim Biophys Acta. 2014;1840(2):906–12.
Kim YJ, et al. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 2006;66(14):7136–42.
Yuan TF, et al. Oxidative stress and adult neurogenesis. Stem Cell Rev Rep. 2015;11(5):706–9.
Dahlmanns M, et al. Glial glutamate transporter-mediated plasticity: system x(c)(-)/xCT/SLC7A11 and EAAT1/2 in brain diseases. Front Biosci (Landmark Ed). 2023;28(3):57.
Falco A, Bartolomé-Cabrero R, Gascón S. Bcl-2-assisted reprogramming of mouse astrocytes and human fibroblasts into induced neurons. Methods Mol Biol. 2021;2352:57–71.
Abdel-Raouf KMA, et al. Transdifferentiation of human fibroblasts into skeletal muscle cells: optimization and assembly into engineered tissue constructs through biological ligands. Biology (Basel). 2021;10(6):539.
Kang MJ, Park SY, Han JS. MicroRNA-24-3p regulates neuronal differentiation by controlling hippocalcin expression. Cell Mol Life Sci. 2019;76(22):4569–80.
Agostini M, et al. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016;23(9):1502–14.
Tang Y, Tian XC. JAK-STAT3 and somatic cell reprogramming. Jakstat. 2013;2(4): e24935.
Rebelo AP, et al. A CADM3 variant causes Charcot-Marie-Tooth disease with marked upper limb involvement. Brain. 2021;144(4):1197-1213.
Feng GD, et al. Fibroblast growth factor 4 is required but not sufficient for the astrocyte dedifferentiation. Mol Neurobiol. 2014;50(3):997-1012.
Acknowledgements
None.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82271371 and 82260367), the High-Level Medical Expert Training Program of Guangxi “139” Plan Funding, the Guangxi Medical and Health Appropriate Technology Development and Application Project (S2021107), the Clinical Research “Climbing” Program of the First Affiliated Hospital of Guangxi Medical University (YYZS2021002), the Advanced Innovation Teams and Xinghu Scholars Program of Guangxi Medical University, and the Innovation Project of Guangxi Graduate Education (YCSW2023242).
Author information
Authors and Affiliations
Contributions
LC designed and supervised the study. RXQ analyzed and interpreted the results and drafted the manuscript. YDZ performed the experiments. YY, JFC, LJH, WX, QCQ, XJL, XYL, XYH, and MSX screened the data and drafted the manuscript. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The experimental protocol was approved by the Ethics Committee of Guangxi Medical University.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qin, R., Zhang, Y., Yang, Y. et al. Decoding single-cell molecular mechanisms in astrocyte-to-iN reprogramming via Ngn2- and Pax6-mediated direct lineage switching. Eur J Med Res 29, 390 (2024). https://doi.org/10.1186/s40001-024-01989-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-024-01989-z