
Abstract
Circular RNAs (circRNAs) have been reported to be related to the initiation and progression of chronic obstructive pulmonary disease (COPD) by affecting the function of human bronchial epithelial cells (HBECs). Here, we aimed to investigate the function and mechanism of circ_0006872 in regulating COPD process using cigarette smoke extract (CSE)-induced 16HBEC in vitro. The results showed that circ_0006872 was increased in smokers without or with COPD, especially in smokers with COPD. Also, its expression was dose-dependently up-regulated by CSE exposure in 16HBECs. Functionally, circ_0006872 knockdown dramatically attenuated CSE-evoked proliferation arrest, apoptosis, inflammatory response and oxidative stress in 16HBECs. Mechanistically, circ_0006872/miR-485-3p/cyclin-dependent kinase inhibitor 1B (CDKN1B) formed a competitive endogenous RNA (ceRNA) network. CDKN1B was increased and miR-485-3p was decreased in COPD patients and CSE-induced 16HBECs. MiR-485-3p overexpression or CDKN1B knockdown protected 16HBEC against CSE-induced 16HBEC injury mentioned above. Moreover, rescue experiments showed that circ_0006872 regulated CSE-induced 16HBEC injury via miR-485-3p/CDKN1B axis. Circ_0006872 silencing protected against CSE-induced bronchial epithelial cell injury via miR-485-3p/CDKN1B axis, suggesting the potential application of circ_0006872 in preventing cigarette smoke-induced COPD.
Similar content being viewed by others


Introduction
Chronic obstructive pulmonary disease (COPD) is a common chronic inflammatory respiratory disease mainly caused by cigarette smoking (CS) [1, 2]. Smoking-induced COPD is partly related to the inflammation, which can also give rise to the elevation of reactive oxygen species resulting in oxidative stress activation [3, 4]. Besides, it has been proposed that increased apoptosis mechanisms are positively implicated in COPD pathogenesis [5]. Bronchial epithelial cells are the first barrier to protect airways against harmful substances, the integrity of which is responsible in response to CS [6]. Therefore, in-depth investigations on the molecular mechanism underlying bronchial epithelial cell disruption is of great importance for preventing COPD progression.
Circular RNAs (circRNAs) are a new kind of non-coding molecules possessing a covalently closed continuous loop, so they are failed to be degraded by RNase R exonuclease and more stable than linear RNA [7, 8]. CircRNAs are widely identified in the eukaryotes, and emerging reports suggest that they are engaged in modulating important biological processes [9, 10]. Recently, some reports proposed that dysregulated expression of circRNAs is tightly related to the initiation and progression of CODP. Zheng et al. showed that silencing of circ-OSBPL2 could suppress human bronchial epithelial cells (HBECs) injury in smoke-related COPD by repressing BRD4 through sponging miR-193a-5p [11]. Down-regulation of circ-HACE1 was found to protect HBECs from cigarette smoke extract (CSE)-induced inflammatory response, oxidative stress and apoptosis by miR-485-3p/TLR4 axis [12]. Zhou’s team showed that circFOXO3 knockdown might exert protective effects against pneumonic inflammation in CS-exposed COPD mice model through repressing IKK-β via miR-214-3p [13]. Circ_0006872 is generated by the back-splicing of ASCC3 gene on chr6: 101,073,067–101,086,697. A previous study manifested that circ_0006872 level was higher in COPD patients, and might contribute to CSE-triggered HBECs damage through miR-145-5p/NF-κB pathway [14]. However, large-scale identifications of the mechanism underlying circ_0006872 in CSE-induced HBEC dysfunction were not yet reported.
Hence, this study used CSE-treated 16HBECs, a human bronchial epithelial cell line, to mimic CS-induced COPD in vitro, then the functions of circ_0006872 on CSE-evoked bronchial epithelial cell injury were investigated. It has been proposed that circRNAs can act as the sponges for microRNA (miRNA/miR) to participate in the process of gene translation, namely competitive endogenous RNA (ceRNA) hypothesis [15, 16]. Furthermore, the underlying miRNA/mRNA axis of circ_0006872 on CSE-induced 16HBEC injury was also explored to state the potential regulatory network of circ_0006872 in COPD.
Materials and methods
Subjects
Blood samples (5 mL) were collected from non-smokers without COPD (n = 35), and smokers without (n = 35) or with COPD (n = 35). All subjects were recruited at Tongde hospital of Zhejiang Province. The diagnosis of COPD patients was based on the guidelines of the Global Initiative for Chronic Obstructive Lung Disease (GOLD) [17]. The exclusion criteria included interstitial lung diseases, asthma, neuromuscular disease, and/or heart failure.
Cigarette smoke extract (CSE) preparation
The mainstream smoke from two cigarettes (Jinsheng Tobacco Corporate Ltd., Nanchang, China) containing 0.1 g nicotine was bubbled through 20 mL of cell growth medium. After removing insoluble particles by filtration with a 0.22 μm filter membrane (Merck Millipore, Shanghai, China), the resultant CSE solution was collected and regarded as 100% CSE solution, and then was diluted with serum-free medium into different concentrations for the use in experiments within 1 h.
Cell culture and treatment
16HBECs were obtained from Procell (Wuhan, China) and grown in RPMI1640 medium (Gibco, Shanghai, China) plus 1% penicillin/streptomycin (Gibco) and 10% FBS (Gibco) at 37 ℃ with 5% CO2. The prepared 16HBECs at 70–80% confluence were exposed to different concentrations of CSE for 24 h for experimental purpose.
Cell transfection
Small interfering RNA (siRNA) targeting circ_0006872 or CDKN1B (si-circ_0006872 or si-CDKN1B) and the nontarget siRNA (si-NC), miR-485-3p mimic (miR-485-3p), inhibitor (anti-miR-485-3p) and negative control oligos (miR-NC or anti-miR-NC), pcDNA3.1 CDKN1B overexpression plasmids (CDKN1B) and scrambled pcDNA3.1 plasmids (pcDNA) was constructed by Sangon (Shanghai, China). After 48 h of transfection, 16HBECs were incubated with 2% CSE for 24 h to conduct subsequent functional experiments.
Reverse transcription, RNase R digestion and qRT-PCR
Total RNA was extracted by the use of TRIzol reagent (Takara, Dalian, China). Approximately 3 µg of total RNAs were mixed with RNase R (3 U/µg) or Mock for 30 min incubation at 37 ℃. The PrimeScript RT Reagent Kit (Takara) was applied for the generation of first-strand cDNA, then qRT-PCR analysis with the TB Green Premix Ex Taq II (Takara) was carried out. U6 or β-actin was utilized as an internal control. The sequences of primers were listed in Table 1.
Cell counting kit-8 (CCK-8) assay
Single 16HBEC suspensions (1 × 105 cells/mL) were cultivated in a 96-well plate (0.1 mL per well) overnight, then per well was added with 10 µL CCK-8 solution for 2 h incubation. At last, OD values at 450 nm were read using a microplate reader (Bio-Rad, Hercules, CA, USA) to represent cell viability ability.
EdU assay
16HBECs were seeded into a 24-well plate all night and incubated with 50 µM EdU labeling solution (RiboBio) in growth medium for 3 h. Then cells were stained with Click-It reaction mixture for 30 min, followed by DAPI staining. Finally, a fluorescence microscope (Leica, Wetzlar, Germany) was utilized to observe and test EdU positive cells.
Flow cytometry
16HBECs were harvested and resuspended in buffer solution (500 µL) to the density of 1 × 106 cells/mL. Then cells (1 × 105) were then stained with Annexin V-FITC and propidium iodide (KenGen Biotech, Nanjing, China) for 20 min under darkness. Cell apoptosis was detected by flow cytometer within 1 h.
Western blotting
RIPA lysis buffer (KenGen Biotech) harboring 1% proteinase inhibitor was applied for proteins extraction. Then 40 µg of proteins were separated by 8% SDS-PAGE and electrophoretically transferred onto cellulose nitrate membranes. After 1 h block by 5% skim milk powder at 37 ℃, membranes were incubated with the specific primary antibodies against CDKN1B (ab32034, 1:1000), Bcl-2 (ab692, 1:1000), Bax (ab32503, 1:1000), and GAPDH (ab181602, 1:10000) (Abcam, Cambridge, MA, USA) at 4 ℃overnight, followed by probing with secondary antibody (D110058, 1:4000, Sangon Biotech, Shanghai, China) for 2 h at 37 ℃. Protein bands were quantified using an ECL reagent (Beyotime).
ELISA
The culture supernatant of indicated 16HBECs were collected, and then levels of interleukin (IL)-6 and tumor necrosis factor-α (TNF-α) were measured as per the instructions of the corresponding ELISA kits (Abcam).
Measurement of superoxide dismutase (SOD)
16HBECs were lysed using RIPA lysis buffer (KenGen Biotech) and SOD content was determined using a commercial Superoxide Dismutase (SOD) assay kit. Finally, the OD value at 450 nm was assayed.
Dual-luciferase reporter assay
The fragments of circ_0006872 or CDKN1B 3’UTR comprising the binding sites of miR-485-3p or the mutant version without miR-485-3p binding sites were amplified and cloned into psiCHECK™-2 vector (Promega, Madison, WI, USA) to construct wild-type (WT) or mutated (MUT) luciferase reporter vector, named as WT/MUT-circ_0006872 or WT/MUT-CDKN1B 3ʹUTR. Then 50 ng above recombinant plasmids and 50 nM miR-485-3p mimic or the control (miR-NC) were co-transfected into 16HBECs, and firefly activities were detected following 48 h of transfection.
RNA pull-down assay
Biotin-labeled miR-485-3p probe (bio-miR-485-3p) and nonsense control probe (bio-miR-NC) were synthesized by Geneseed Biotech (Shanghai, China) and then incubated with 16HBECs for 2 h. After lysing, the lysates of 16HBECs were incubated with M-280 Streptavidin magnetic beads for 1 h. Finally, the abundance of circ_0006872 or CDKN1B was analyzed.
Statistical analysis
The data from three repetitions were exhibited as mean ± standard deviation (SD). Student’s t test, Mann-Whitney or Analysis of Variance (ANOVA) followed by Tukey’s post-test was used for the group comparisons. Pearson’s correlation coefficient assay was used for correlation analysis between two variables. *P < 0.05, **P < 0.01, ***P < 0.001 or ****P < 0.0001 suggested statistically significant.
Results
CSE treatment induces 16HBECs imjury
To investigate the effects of CS on bronchial epithelial cells, 16HBECs were exposed to different concentrations of CSE (1%, 2%, or 4%) for 24 h. CSE treatment induced arrest of 16HBEC viability and DNA synthesis activity in a dose-dependent manner (Fig. 1A, B). On the contrary, CSE treatment dose-dependently evoked apoptosis in 16HBECs (Fig. 1C), accompanied with the increase of Bax and decrease of Bcl-2 in 16HBECs (Fig. 1D, E). Besides, with the increasing concentrations of CSE, the release of IL-6 and TNF-α was significantly enhanced (Fig. 1F). Moreover, CSE exposure had obstructive effects on SOD content, an endogenous antioxidant enzyme, since SOD level declined upon the exposure of increasing doses of CSE in (Fig. 1G). Thus, these data suggested that CSE could lead to 16HBECs injury.

CSE treatment induces 16HBECs injury. A–G 16HBECs were exposed to different concentrations of CSE (1%, 2%, or 4%) for 24 h. Analyses of (A, B) 16HBEC proliferation, and C apoptosis. D, E Levels of Bax and Bcl-2 in 16HBECs. F Detection of IL-6 and TNF-α contents in 16HBECs. G Measurement of SOD content in 16HBECs using a commercial kit. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Knockdown of circ_0006872 attenuates CSE-induced 16HBEC injury
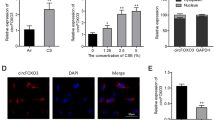
As shown in Fig. 2A, the level of circ_0006872 was higher in smokers with or without COPD than those in non-smokers, especially in COPD group. In addition, circ_0006872 expression was dose-dependently elevated by CSE treatment in 16HBECs (Fig. 2B). Additionally, the experiment showed that RNase R could rapidly degrade linear GAPDH rather than circ_0006872 in 16HBECs, indicating the round structure of circ_0006872 (Fig. 2C). Thereafter, the impacts of circ_0006872 on CSE-induced bronchial epithelial injury were elucidated. Following the transfection of circ_0006872 siRNA in 16HBECs, cells were exposed to 2% CSE for 24 h, then we observed that si-circ_0006872 introduction reduced CSE-induced elevation of circ_0006872 in 16HBECs (Fig. 2D). Functionally, it was proved that circ_0006872 silencing reversed CSE-induced proliferation arrest (Fig. 2E, F) and apoptosis (Fig. 2G, H) in 16HBECs. Furthermore, both the up-regulations of IL-6 and TNF-α mediated by CSE were attenuated by circ_0006872 down-regulation in 16HBECs (Fig. 2I). Besides that, circ_0006872 deletion rescued CSE-evoked decrease of SOD content in 16HBECs (Fig. 2J).

Knockdown of circ_0006872 attenuates CSE-induced 16HBEC injury. A The expression level of circ_0006872 was detected using qRT-PCR in non-smokers (n = 35), smokers (n = 35), and smokers with COPD (n = 35). B Levels of circ_0006872 in 16HBECs exposed to different concentrations of CSE. C Stability analysis by RNase R treatment. D–J 16HBECs were transfected with si-circ_0006872 or si-NC and then exposed with 2% CSE for 24 h. D Transfection efficiency. E, F 16HBEC proliferation analysis. G 16HBEC apoptosis detection. H Levels of Bax and Bcl-2 in 16HBECs. I Levels of IL-6 and TNF-α in 16HBECs. J Measurement of SOD content in 16HBECs using a commercial kit. **P < 0.01, ***P < 0.001, ****P < 0.0001
Circ_0006872 acts as a sponge for miR-485-3p
Circinteractome database predicted that circ_0006872 possesses the binding site of miR-485-3p (Fig. 3A). The miR-485-3p expression was significantly elevated after miR-485-3p mimic introduction in 16HBECs (Fig. 3B). Then we found that miR-485-3p mimics could overtly reduce the luciferase activity of WT group but not the mutant one in 16HBECs (Fig. 3C). Moreover, circ_0006872 was found to be significantly enriched in bio-miR-485-3p group in 16HBECs (Fig. 3D). Thereafter, it was proved that miR-485-3p expression was decreased in smokers, especially in smokers with COPD (Fig. 3E), which was negatively correlated with circ_0006872 expression in COPD patients (Fig. 3F). Besides that, we also showed that CSE treatment dose-dependently decreased miR-485-3p in 16HBECs (Fig. 3G).

Circ_0006872 acts as a sponge for miR-485-3p. A The binding site of miR-485-3p on circ_0006872 predicted by circinteractome database was listed. B The transfection efficiency detection. C, D The interaction between miR-485-3p and circ_0006872 was confirmed using dual-luciferase reporter assay and RNA pull-down assay. E The expression level of miR-485-3p was detected using qRT-PCR in non-smokers (n = 35), smokers (n = 35), and smokers with COPD (n = 35). F The correlation between miR-485-3p and circ_0006872 expression level in smokers with COPD (n = 35) was analyzed by Pearson’s correlation coefficient assay (r=-0.8966, P < 0.001). G Levels of miR-485-3p in16HBECs exposed to different concentrations of CSE. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Knockdown of circ_0006872 attenuates CSE-induced 16HBEC injury via miR-485-3p
To investigate whether circ_0006872 exerted its functions by miR-485-3p, 16HBECs were co-transfected with si-circ_0006872 and/or anti-miR-485-3p, followed by 2% CSE exposure for 24 h. As expected, miR-485-3p inhibitor reduced circ_0006872 knockdown-induced elevation of miR-485-3p in 16HBECs under CSE treatment (Fig. 4A). Functionally, miR-485-3p lack attenuated circ_0006872 down-regulation-evoked promotion of cell proliferation (Fig. 4B–D), arrest of cell apoptosis (Fig. 4E, F), reduction of IL-6 and TNF-α levels (Fig. 4G), as well as elevation of SOD content (Fig. 4H) in 16HBECs exposed with 2% CSE.

Knockdown of circ_0006872 attenuates CSE-induced 16HBEC injury via miR-485-3p. A–H 16HBECs were co-transfected with si-circ_0006872 and/or anti-miR-485-3p, followed by 2% CSE exposure for 24 h. A Transfection efficiency. B–D 16HBEC proliferation analysis. E 16HBEC apoptosis detection. F Levels of Bax and Bcl-2 in 16HBECs. G Levels of IL-6 and TNF-α in 16HBECs. H Measurement of SOD content in 16HBECs using a commercial kit. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
CDKN1B is targeted by miR-485-3p, and circ_0006872/miR-485-3p/CDKN1B constitutes a feedback loop in 16HBECs
Next, starbase software showed that CDKN1B contains conserved target site of miR-485-3p (Fig. 5A). Then it was showed the luciferase activity of wild-type CDKN1B 3′UTR reporter vector was significantly reduced by miR-485-3p overexpression (Fig. 5B). Besides that, RNA pull-down analysis indicated a specific enrichment of CDKN1B in the biotin-labeled miR-485-3p probe group (Fig. 5C). CDKN1B mRNA was found to be increased in smokers, especially in smokers with COPD (Fig. 5D), and was negatively correlated with miR-485-3p expression in COPD patients (Fig. 5E). Similarly, its protein expression was also elevated in smokers with COPD (Fig. 5F). In addition, CSE dose-dependently elevated CDKN1B expression in 16HBECs (Fig. 5G). Importantly, circ_0006872 down-regulation was accompanied by the decrease of CDKN1B, which was subsequently rescued by miR-485-3p inhibitor (Fig. 5H). In all, circ_0006872 could indirectly regulated CDKN1B through sponging miR-485-3p in 16HBECs.

CDKN1B is a target of miR-485-3p, and circ_0006872/miR-485-3p/CDKN1B constitutes a feedback loop in 16HBECs. A The conserved target site of miR-485-3p on CDKN1B predicted by starbase software was listed. B, C The interaction between miR-485-3p and CDKN1B was confirmed using dual-luciferase reporter assay and RNA pull-down assay. D Levels of CDKN1B mRNA in non-smokers (n = 35), smokers (n = 35), and smokers with COPD (n = 35). E The correlation between miR-485-3p and CDKN1B mRNA expression level in smokers with COPD (n = 35) was assessed by Pearson’s correlation coefficient assay (r=-0.8606, p < 0.001). F CDKN1B protein level in non-smokers (n = 35), smokers (n = 35), and smokers with COPD (n = 35). G CDKN1B expression in 16HBECs exposed to different concentrations of CSE. H The impacts of circ_0006872/miR-485-3p axis on CDKN1B expression in 16HBECs under CSE treatment. **P < 0.01, ***P < 0.001, ****P < 0.0001
CDKN1B silencing reverses CSE-induced 16HBEC injury
Next, we evaluated the functions of CDKN1B on CSE-induced bronchial epithelial injury. CDKN1B siRNAs were established to knock down CDKN1B expression in 16HBECs. As exhibited in Fig. 6A, the introduction of si-CDKN1B in 16HBECs significantly reduced CSE-evoked elevation of CDKN1B expression level. Then transfected 16HBECs were treated with 2% CSE for 24 h. In CCK-8 and EdU assays, CDKN1B knockdown suppressed CSE-induced inhibition of 16HBEC proliferation (Fig. 6B–D). Flow cytometric analysis suggested that the apoptosis of CSE-induced 16HBECs was decreased after CDKN1B silencing (Fig. 6E). And western blotting showed the decrease of Bax protein level and increase of Bcl-2 level in 16HBECs after CDKN1B down-regulation in the presence of CSE (Fig. 6F). Additionally, CDKN1B depletion in ELISA analysis gave rise to IL-6 and TNF-α release enhancement in CSE-induced 16HBECs (Fig. 6G). In final SOD measurement, a remarkable elevation of SOD level was unveiled in response to CDKN1B siRNA in CSE-treated 16HBECs (Fig. 6H). These data suggested that knockdown of CDKN1B protected CSE-induced 16HBEC injury.

CDKN1B silencing reverses CSE-induced 16HBEC injury. A–H 16HBECs were transfected with si-CDKN1B or si-NC, followed by treatment with 2% CSE for 24 h. (A) Transfection efficiency. B–D 16HBEC proliferation analysis. E 16HBEC apoptosis detection. F Levels of Bax and Bcl-2 in 16HBECs. G Levels of IL-6 and TNF-α in 16HBECs. H Measurement of SOD content in 16HBECs using a commercial kit. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Mir-485-3p protects 16HBEC from CSE-induced injury via CDKN1B
In order to investigate the action of miR-485-3p/CDKN1B axis on CSE-induced bronchial epithelial injury, we conducted rescue experiments by transfecting miR-485-3p mimics and/or CDKN1B overexpression plasmids into 16HBECs. After treating with 2% CSE for 24 h, the transfection efficiency was validated by CDKN1B expression in 16HBECs (Fig. 7A). Functionally, 16HBEC proliferative ability was potentiated upon miR-485-3p overexpression in the presence of CSE, while CDKN1B overexpression could reverse this condition (Fig. 7B–D). Furthermore, miR-485-3p mimic reversed CSE evoked apoptosis in 16HBECs, and this phenomenon was counteracted via CDKN1B up-regulation (Fig. 7E, F). Both the increases of IL-6 and TNF-α mediated by CSE in 16HBECs were reduced by miR-485-3p mimic, and subsequently promoted in response to CDKN1B plasmids (Fig. 7G). In addition, the SOD content was increased with miR-485-3p mimic introduction in CSE-induced 16HBECs, which was abolished by CDKN1B overexpression (Fig. 7H). Collectively, miR-485-3p suppressed CSE-induced16HBEC injury via CDKN1B.

miR-485-3p protects 16HBEC from CSE-induced injury via CDKN1B. A–H 16HBECs were co-transfected with miR-485-3p mimics and/or CDKN1B overexpression and then treated with 2% CSE for 24 h. A Transfection efficiency. B–D 16HBEC proliferation analysis. E 16HBEC apoptosis detection. F Levels of Bax and Bcl-2 in 16HBECs. G Levels of IL-6 and TNF-α in 16HBECs. H Measurement of SOD content in 16HBECs using a commercial kit. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Discussion
As the fourth leading cause of death through the world, over 40% of COPD deaths are attributed to smoking [18, 19]. Long-term exposure to CS is the leading cause of COPD through inducing the activation of epithelial cells and macrophages to cause apoptosis and inflammation and the subsequent release of mediators in response to oxidative stress [20]. Studies have exhibited that circRNA dysregulation is related to CSE-induced HBECs damage [11, 12, 21]. CircRNAs hold great promise as potential biomarkers in COPD since their highly stable structure and ubiquitous abundance in eukaryotes [22]. In our study, we found an increased circ_0006872 in smokers without or with COPD, especially in smokers with COPD. Furthermore, its expression was also up-regulated by CSE exposure in 16HBECs in a dose-dependent manner. Furthermore, deletion of circ_0006872 abolished CSE-triggered proliferation arrest, inflammation, apoptosis, and oxidative stress in 16HBECs. Thus, we speculated that knockdown of circ_0006872 might exert protective effects against CS-evoked bronchial epithelial cell injury in the process of COPD.
According to the ceRNA hypothesis [15, 16], circRNAs are able to act as miRNA sponges to extricate the degradation of the downstream mRNA mediated by miRNAs. Thus, the underlying miRNA/mRNA axis of circ_0006872 in 16HBECs was explored. This work confirmed the circ_0006872/miR-485-3p/CDKN1B feedback loop in 16HBECs. Previous studies have showed the implication of miRNAs in multiple processes of COPD, modulating pathways related to apoptosis, inflammation and stress response [23,24,25]. miR-485-3p had been unveiled to be down-regulated in patients with COPD [26]. Besides that, miR-485-3p could attenuate CSE-triggered 16HBEC dysfunction via circ-HACE/miR-485-3p/TLR4 axis [12]. Consistent with previous findings, we also confirmed the protective action of miR-485-3p on CSE-induced 16HBECs. Moreover, miR-485-3p could abolish the functions of circ_0006872 knockdown on 16HBEC injury caused by CSE. CDKN1B belongs to the Kip/Cip family of CDK inhibitors, and suppresses the function of multiple cyclin-CDK complexes, thus showing anti-proliferative activity by impairing cell cycle progression [27]. In COPD, Yang et al. showed that CDKN1B was decreased in COPD patients, and could abate the action of miR-221-3p on the inhibition of CSE-evoked apoptotic and inflammatory damages in 16HBECs [28]. In our study, the increased expression of CDKN1B in smokers without or with COPD, and CSE-treated 16HBECs was observed, moreover, we proved that CDKN1B down-regulation could prevent CSE-induced16HBEC injury. In addition, miR-485-3p exerted its protective functions by targeting CDKN1B.
In all, this work demonstrated that circ_0006872 silencing could protect against CSE-induced inflammatory, apoptotic, and oxidative injury via miR-485-3p/CDKN1B axis, which may provide a novel insight into COPD prevention in subjects with smoking addiction.
Availability of data and materials
The analyzed data sets generated during the present study are available from the corresponding author on reasonable request.
References
Rabe KF, Watz H (2017) Chronic obstructive pulmonary disease. Lancet 389:1931–1940
Labaki WW, Rosenberg SR (2020) Chronic obstructive pulmonary disease. Ann Intern Med. https://doi.org/10.7326/AITC202008040
Zuo L, Prather ER, Stetskiv M, Garrison DE, Meade JR, Peace TI, Zhou T (2019) Inflammaging and oxidative stress in human diseases: from molecular mechanisms to novel treatments. International J Mol Sci 20:4472
Wiegman CH, Li F, Ryffel B, Togbe D, Chung KF (2020) Oxidative stress in ozone-induced chronic lung inflammation and emphysema: a facet of chronic obstructive pulmonary disease. Front Immunol 11:1957
Tan WSD, Shen HM, Wong WSF (2019) Dysregulated autophagy in COPD: a pathogenic process to be deciphered. Pharmacol Res 144:1–7
Gohy ST, Hupin C, Pilette C, Ladjemi MZ (2016) Chronic inflammatory airway diseases: the central role of the epithelium revisited. Clin Exp Allergy 46:529–542
Chen LL, Yang L (2015) Regulation of circRNA biogenesis. RNA Biol 12:381–388
Geng Y, Jiang J, Wu C (2018) Function and clinical significance of circRNAs in solid tumors. J Hematol Oncol 11:98
Han B, Chao J, Yao H (2018) Circular RNA and its mechanisms in disease: from the bench to the clinic. Pharmacol Ther 187:31–44
Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J (2019) The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20:675–691
Zheng C, Zhang Y, Zhao Y, Duan Y, Mu Q, Wang X (2021) Circ-OSBPL2 contributes to smoke-related chronic obstructive pulmonary disease by targeting miR-193a-5p/BRD4 axis. Int J Chronic Obstr Pulm Dis 16:919–931
Zhou F, Cao C, Chai H, Hong J, Zhu M (2021) Circ-HACE1 aggravates cigarette smoke Extract-Induced Injury in Human bronchial epithelial cells via regulating toll-like receptor 4 by sponging miR-485-3p. Int J Chronic Obstr Pulm Dis 16:1535–1547
Zhou L, Wu B, Yang J, Wang B, Pan J, Xu D, Du C (2021) Knockdown of circFOXO3 ameliorates cigarette smoke-induced lung injury in mice. Respir Res 22:294
Xue M, Peng N, Zhu X, Zhang H (2021) Hsa_circ_0006872 promotes cigarette smoke-induced apoptosis, inflammation and oxidative stress in HPMECs and BEAS-2B cells through the miR-145-5p/NF-κB axis. Biochem Biophys Res Commun. 534:553–560
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495:384–388
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell 146:353–358
Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M, Stockley RA, Sin DD (2013) Rodriguez-Roisin, global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 187:347–365
Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O’Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez-Ruiz F, Perico N, Phillips D, Pierce K, Pope CA, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De León FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA (2012) Global andregional mortality from 235 causes of death for 20 age groups in 1990 and 2010:a systematic analysis for the global burden of disease study 2010. Lancet 380:2095–2128
Adeloye D, Chua S, Lee C, Basquill C, Papana A, Theodoratou E, Nair H, Gasevic D, Sridhar D, Campbell H, Chan KY, Sheikh A, Rudan I (2015) Global and regional estimates of COPD prevalence: systematic review and meta-analysis. J global health 5:020415
Salvi SS, Barnes PJ (2009) Chronic obstructive pulmonary disease in non-smokers. Lancet (London England) 374:733–743
Wang Z, Zuo Y, Gao Z (2021) CircANKRD11 Knockdown protects HPMECs from cigarette smoke extract-induced injury by regulating miR-145-5p/BRD4 axis. Int J Chronic Obstr Pulm Dis 16:887–899
Patop IL, Wüst S, Kadener S (2019) Past, present, and future of circRNAs. EMBO J 38:e100836
Cañas JA, Rodrigo-Muñoz JM, Sastre B, Gil-Martinez M, Redondo N (2020) Del Pozo, MicroRNAs as potential regulators of immune response networks in asthma and chronic obstructive pulmonary disease. Front Immunol 11:608666
Tan BWQ, Sim WL, Cheong JK, Kuan WS, Tran T, Lim HF (2020) MicroRNAs in chronic airway diseases: clinical correlation and translational applications. Pharmacol Res 160:105045
Baker JR, Vuppusetty C, Colley T, Papaioannou AI, Fenwick P, Donnelly L, Ito K, Barnes PJ (2016) Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci Rep 6:35871
Musri MM, Coll-Bonfill N, Maron BA, Peinado VI, Wang RS, Altirriba J, Blanco I, Oldham WM, Tura-Ceide O, García-Lucio J, de la Cruz-Thea B, Meister G, Loscalzo J, Barberà JA (2018) MicroRNA dysregulation in pulmonary arteries from chronic obstructive pulmonary disease. Relationships with vascular remodeling. Am J Respir Cell Mol Biol 59:490–499
Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A (1994) p27Kip1, a cyclin-cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev 8:9–22
Yang H, Zhang L, Wang Q (2021) MicroRNA-221-3p alleviates cell apoptosis and inflammatory response by targeting cyclin dependent kinase inhibitor 1B in chronic obstructive pulmonary disease. Bioengineered 12:5705–5715
Acknowledgements
Not applicable.
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
Conceptualization and Methodology: ZL and LZ; Formal analysis and Data curation: JT and CW; Validation and Investigation: JW and ZL; Writing - original draft preparation and Writing - review and editing: JW, ZL and LZ; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The present study was approved by the ethical review committee of Tongde hospital of Zhejiang Province. Written informed consent was obtained from all enrolled patients.
Consent for publication
Patients agree to participate in this work.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Li, Z., Zheng, L. et al. Knockdown of circ_0006872 alleviates CSE-induced human bronchial epithelial cells injury in chronic obstructive pulmonary disease. Appl Biol Chem 66, 14 (2023). https://doi.org/10.1186/s13765-023-00772-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-023-00772-8