
Abstract
A simultaneous analytical method has been developed for quantification and confirmation of the nematicide fluazaindolizine and its seven metabolites (IN-A5760, IN-F4106, IN-QEK31, IN-QZY47, IN-TMQ01, IN-UNS90 and IN-UJV12) in agricultural products. The compounds were extracted with acetonitrile/water (80/20, v/v) and purified using C18 cartridge, and analysis was conducted by liquid chromatography-tandem mass spectrometry in the electrospray positive and negative ion mode. The method has been validated by verifying the performance characteristics such as selectivity, linearity, limit of detection (LOD), limit of quantification (LOQ), accuracy and precision. To prevent the matrix effects, all analytes were quantified with matrix-matched calibration assessed by the determination coefficient (R2) of the range from 0.9988 to 1.0000. The LOD and LOQ were satisfactory to determine the low residual level in agricultural products. The accuracy and precision of the method were evaluated by recoveries with five replicates at three fortification levels (LOQ, 10 × LOQ and 50 × LOQ). The mean recoveries of fluazaindolizine and seven metabolites in agricultural products were 75.6–110.0% with the CV% of 0.2–9.1%. All optimized results were displayed excellent results assessed by the Ministry of Food and Drug Safety guidelines and the Codex Alimentarius Commission guidelines for pesticide residue analysis. This study could use as basic data for setting of residue definition and maximum residue limits of fluazaindolizine in agricultural products.
Similar content being viewed by others


Introduction
Root-knot nematodes are distributed throughout the world and about 2,000 plant species have exposed to infection by them. They are one of the genera of plant-parasitic nematodes that economically damaging on garden and agricultural crops and their infection results in approximately 5% losses of crop over the world [1]. The larvae of root-knot nematodes invades host plants via the roots as second-stage juveniles (J2) and cause adverse effects on plant growth by feeding on plant nutrients after reaching maturity. Also infected roots develop gray or milky galls that are not consistent in shape and size, and rot as they become brown [2]. The main root-knot nematodes that cause a lot of damage in Korea are known as Meloidogyne incognita, M. hapla and M. arenaria [3]. In order to control the root-knot nematodes, pesticides are treated in the soil before planting, and if moisture in the soil is properly maintained and facility is sealed, secondary insecticidal effects can be obtained by increasing the temperature [4].
Fluazaindolizine (CAS No. 1254304-22-7) is a sulfonamide pesticide being developed in 2015 by DuPont Crop Protection. It is a high effective and selective nematicides, especially to control root-knot nematodes such as M. incogn13ita and M. hapla. Fluazaindolizine could destroy the calcium stability in nematode cytoplasm by binding to the special points of the Lyanodine receptor (RyR), a class of intracellular calcium channel. As a result, it affects the movement of muscle and heart and leads to paralysis and death [5, 6]. Lahm et al. reported that fluazaindolizine has a bad influence at concentrations range of 1–50 mg/L on motility, mobility and infectivity of larvae of M.incognita and M. hapla. This shows that the target of fluazaindolizine is plant-parasitic nematodes specific. Also, it has been indicated to be very compatible with a biological agents, such as helpful fungi, bacteria and other nematodes that inhabit the soil [7, 8]. As a result, fluazaindolizine helps to sustain soil health environment and increases the quality of crops and crop yields.
According to report data provided by duPont, fluazaindolizine is usually metabolized via O-demethylation and hydrolysis into the seven major metabolites including IN-A5760, IN-F4106, IN-QEK31, IN-QZY47, IN-TMQ01, IN-UNS90 and IN-UJV12 in the environment. In addition to this, the metabolic pathway of major metabolites is very complex and generated a large number of conjugated metabolites. Applied pesticides may be transformed into a large number of metabolites under degradation by environmental conditions and conjugation with a multitude of natural compounds in the crop and soil. Parent compounds and metabolites may display with very different physicochemical properties and environmental behaviors such as half-life and toxicity [9,10,11]. The metabolites are a matter of concern, because they may act like an active ingredient of the target and may exhibit higher toxicity than parent compounds [5, 12]. Thus, pesticide metabolites that remaining in the environment, especially agricultural products, should also be strictly managed.
For monitoring and risk assessment of pesticides, several foreign regulatory authorities, including the Codex Alimentarius Commission, US Environmental Protection Agency and European Commission, have been established the maximum residue limit (MRL) and residue definition in or on food of plant and animal origin. Inclusion of metabolites in the residue definition of pesticide is considered when they detected at significant proportions of the total radioactive residue (TRR) and residue levels [13]. The MRL and residue definition of fluazaindolizine have not been established in any countries, and when fluazaindolizine is detected on the domestic and imported foods in Korea, the residual amount should be less than 0.01 mg/kg according to the positive list system (PLS). However, if an MRL based on appropriate experimental data is not established and the residual amounts is continuously regulated by PLS, the violation rate of particular crops may increase. Therefore, it is important to develop a simple and accurate method for further establishment of fluazaindolizine.
Chen et al. [5] developed the analytical method for fluazaindolizine and seven metabolites in tomato using LC–MS/MS and evaluated the dissipation behavior and residue distribution under open-field conditions. The mean recoveries of parent and seven metabolites in tomato were 81–117% with the RSDs less than 11%, and the method was suitably for multi-quantitation of fluazaindolizine and seven metabolites residue in tomato. However, an analytical method with satisfactory results in five agricultural products (hulled rice, potato, soybean, mandarin and green pepper) is required in order to become an official method in Korea. In this study, as the residue definition of fluazaindolizine is expected to be established as the sum of parent compound plus its seven metabolites, an analytical method was developed that can analyze all compounds simultaneously in five crops.
Experimental
Standard, reagents and samples
The pesticide standard of fluazaindolizine (99.7%) and its seven metabolites, including IN-A5760 (98.7%), IN- F4106 (99.6%), IN-QEK31 (100.0%), IN-QZY47 (85.2%), IN-TMQ01 (98.1%), IN-UNS90 (92.9%) and IN-UJV12 (97.0%), were provided by Corteva Agriscience, agriculture division of DowDuPont™ (Wilmington, Delaware, United States). HPLC-grade acetonitrile and methanol were purchased from Merck (Darmstadt, Germany) and hydrochloric acid was supplied by Fisher Chemical (Quebec, Canada). C18 SPE cartridges (6 cc, 500 mg) was purchased from Waters (Dublin, Ireland) and the PVDF syringe filter (0.2 μm ×13 mm) was obtained from Teknokroma (Barcelona, Spain). All agricultural samples used to develop the method were purchased, which were not treated with pesticides. A five samples of mandarin, potato, soybean, green pepper and hulled rice were purchased from the local supermarket (E-mart, homeplus) in the city of Cheongju. The hulled rice and soybean samples were crushed with a blender and then filtered through a standard sieve of 420 μm, and potato, mandarin and green pepper samples were chopped and homogenized. The processed samples were placed in a plastic container and stored at − 50 °C freezer until used in experiments.
Preparation of stock solutions and standard solutions
A stock solutions of fluazaindolizine, IN-A5760 and IN-F4106 were prepared in acetonitrile at a concentration of 1000 mg/L, respectively. On the other hand, stock solutions of IN-QEK31, IN-QZY47, IN-TMQ01, IN-UNS90 and IN-UJV12 were individually dissolved in methanol/water (80/20, v/v) solution at a concentration of 1,000 mg/L. A standard solutions of fluazaindolizine for the recovery test were diluted of the stock solution with the same solvent to concentrations of 12.5, 2.5 and 0.25 mg/L. Calibration solutions were prepared by serially diluting the 2.5 mg/L with acetonitrile to yield solutions of concentrations 0.02, 0.025, 0.05, 0.1, 0.25 and 0.5 mg/L. The matrix-matched solutions were prepared by dilution 100 μL of the calibration solutions with 900 μL of the blank samples in order to include more than 90% of the matrix. The mixed standard solutions of seven metabolites (mixture A) were prepared by mixing stock solutions with methanol/water (80/20, v/v) solution at a concentrations of 62.5 mg/L (IN-A5760, IN-F4106, IN-QZY47 and IN-UJV12) and 12.5 mg/L (IN-QEK31, IN-TMQ01 and IN-UNS90). Mixture B was made by diluting five times of mixture A and mixture C was made by diluting 10 times of mixture B. The mixed calibration solutions were gradually diluted the mixture B with methanol/water (80/20, v/v) solution following the concentrations of 0.1, 0.125, 0.25, 0.5, 1.25 and 2.5 mg/L for four metabolites and the concentrations of 0.02, 0.025, 0.05, 0.1, 0.25 and 0.5 mg/L for three metabolites. The matrix-matched solutions of metabolites were prepared in the same way as parent compound. The stock solutions were stored at − 20 °C refrigerator in amber glass vials, and the standard solutions were freshly prepared and diluted before each analysis.
Instrumental conditions
Fluazaindolizine and seven metabolites were analyzed by LC (Acquity UPLC, Milford, MA, USA) equipped with tandem mass spectrometer (Xevo TQ-S, Milford, MA, USA) and were separated on a Candenza CD C18 HT (2.0 mm i.d. ×150 mm, 3.0 μm particle size) analytical column. The mobile phase was used using a gradient system that started at 10% of 0.1% formic acid in acetonitrile (A) and 90% of 0.1% formic acid in water (B). The linear mobile phase kept at 10% of A (0–2 min), increased to 70% of A (2–5 min), increased to 90% of A (5–7 min), increased to 100% of A (7–8 min), held at 100% of A (8–9 min), and reduced back to the initial conditions (9–10 min), where it was held (10–12 min). The flow rate and injection volume were 0.2 mL/min and 5 μL, respectively, and column temperature was maintained at 40 °C. An electrospray ionization (ESI) source was operated in positive ion mode for IN-QEK31 and negative ion mode for fluazaindolizine and six metabolites. Capillary voltage was 1 kV, and source and desolvation temperature were controlled at 150 °C and 500 °C, respectively. Cone gas and desolvation gas flow were 150 L/hr and 1000 L/hr, respectively. Multiple reaction monitoring (MRM) mode was selected for simultaneous analysis.
Sample preparation
To analyze fluazaindolizine in five agricultural samples, the processed sample (5 g) was weighed into 50 mL centrifuge tube. In the case of dry samples, a certain amount of water was added to the samples and waited for it to be wet sufficiently. 10 mL of acetonitrile/water (80/20, v/v) was added into the tube, and then shaken for 10 min. After first extraction, the tube was centrifuged for 10 min at 4 °C, 4000 G and the supernatant was decanted into a new 50 mL centrifuge tube. A second extraction was conducted in the same procedure as above. After then, the extracts were combined and adjusted total volume to 25 mL with acetonitrile/water (80/20, v/v), followed by centrifugation for 10 min at 4 °C, 4000 G. The 5 mL of the supernatant was transferred to a 15 mL centrifuge tube and it was evaporated to a volume less than 1 mL under a nitrogen stream. The residue was redissolved with water to a volume of 3 mL. C18 cartridges were conditioned with 3 mL of methanol and 3 mL of water, and were then loaded with 3 mL of redissolved residue. The cartridges were washed with 3 mL of methanol/water (30/70, v/v), and were eluted with 4 mL of methanol/water (70/30, v/v) into a 15 mL centrifuge tube. The eluent was filtered through a PVDF syringe filter prior to LC–MS/MS analysis.
To quantify seven metabolites in the crop samples, 5 g of the homogenized sample was extracted following the extraction procedure of fluazaindolizine. After taking 5 mL of the supernatant which was adjusted volume, the samples were concentrated to a volume less than 1 mL and reconstituted with 2 M hydrochloric acid to a volume of 3 mL. The tubes were capped loosely and were placed in a heating block set to 80 °C for 16 h. After hydrolysis step, the tubes were placed in a room temperature and were allowed them to cool. C18 cartridges were conditioned following the above step, and hydrolyzed samples were loaded onto the cartridge. The compounds were washed with 1 mL of methanol/water (40/60, v/v), and were eluted with 4 mL of methanol/water (50/50, v/v) into a 15 mL centrifuge tube. The eluent was filtered via PVDF syringe filter and analyzed using LC–MS/MS.
Method validation
Method validation was conducted by verifying the performance characteristics of analytical method such as selectivity, linearity, limit of detection (LOD), limit of quantification (LOQ), accuracy and precision. Validation results were assessed by the MFDS guidelines on the standard procedures for preparing an analysis method [14] and Codex Alimentarius Commission guidelines for pesticide residue analysis [15]. Selectivity was assessed by comparing the chromatograms of standard solutions, the blank sample and samples spiked with target compounds. Linearity was evaluated by calculating the determination coefficient (R2) of a matrix-matched calibration curve over six concentration levels. The LOD is defined as the smallest concentration of analyte that can be clearly distinguished from zero and LOQ is the lowest concentration of analyte that can be quantitatively detected. The LOD and LOQ were calculated as the signal-to-noise ratios (S/N) at three and ten times, respectively. The accuracy test was performed by fortifying at three spiking levels (LOQ, 10 ×LOQ and 50 ×LOQ) onto blank samples and divided into two experiments because fluazaindolizine degrades into IN-F4106 and IN-QEK31 during hydrolysis. The accuracy was assessed in terms of recovery by calculating the average of five replicates and the precision was estimated from the coefficient of variation (CV%) of within-laboratory recovery analyses.
The matrix effects (ME%) are caused by interaction of the target compound and co-eluting components of matrix. It can be obtained by calculating the average values of the peak area between the compound spiked onto blank sample and compound dissolved in pure solvent, and assessed in terms of ion suppression (loss in response) or ion enhancement (increase in response) [16, 17]. ME% of fluazaindolizine and seven metabolites was calculated as follows.
Results and discussion
LC–MS/MS conditions
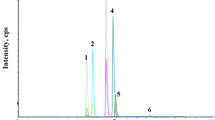
Table 1 indicated summary of properties of fluazaindolizine and its seven metabolites and instrumental parameters of the optimized MS/MS analysis. Each standard solutions were directly injected into the mass detector at a constant rate (10 μL/min) in order to optimize the precursor ion and product ions and obtain the optimal cone voltage and collision energy, which maximize the intensity. Two or three MS/MS transitions were selected for the quantification and confirmation of the analytes. The transition with the highest intensity was used for quantification, and the second and third highest intensity were utilized for confirmation. IN-UNS90 and IN-JV12 showed the highest intensity at m/z 206, which is precursor ion of IN-A5760, and IN-QZY47 and IN-TMQ01 showed the highest intensity at m/z 220, which is precursor ion of IN-F4106 (Fig. 1). When the compounds are analyzed with the optimized transition, IN-A5760 co-eluted with IN-UNS90 and IN-UJV12, and IN-F4106 co-eluted with IN-QZY47 and IN-TMQ01. This phenomenon may lead to ion-suppression and cross-talk effects. The cross-talk effect mainly occurs in MRM mode and appears when analyzing compounds with the same product ion or structural similarities [17]. And that effect is more pronounced when it is influenced by the matrix interference compared to the analysis of the pure standard solutions. To solve this phenomenon, an isotopically labeled internal standards are sometimes used to distinguish which compound the product ion is derived from. However, internal standards are not always available and also there is the problem of cost to get it particularly in the case of metabolites [18, 19]. Therefore, the compounds are classified by different retention time under gradient conditions in this study.

Daughter scan spectrum of a standard solution of A IN-A5760, B IN-UNS90, C IN-UJV12, D IN-F4106, E IN-QZY47 and F IN-TMQ01
Optimization of sample extraction and acid hydrolysis
Fluazaindolizine is degraded into metabolites IN-F4106 and IN-QEK31 due to the breakdown of amide bond (-CONH-) in the molecular structure during hydrolysis. Therefore, the recovery experiments of the parent compound and seven metabolites were carried out by fortifying onto different sample, respectively. In addition, since information on the physicochemical properties of metabolites is insufficient, the recovery tests of the metabolites were conducted first and the parent compound was reviewed using a method that satisfies all the criteria. Chen, X. et al. [5] used methanol/water (70/30, v/v) to extract fluazaindolizine and seven metabolites residue in tomato, tomato plant, and soil. In order to compare the extraction efficiency according to the mixture ratio of methanol and water and the number of extractions (one time of 20 mL or two times of 10 mL), metabolites were spiked in mandarin sample. As a result, the recoveries were slightly higher in the two times extractions and there was no significant difference depending on the ratio of the mixture. The extraction solvent was selected with methanol which was easy to concentrate before the purification process, and applied to soybean sample. The recoveries of all metabolites have tended to decrease, and recoveries of IN-A5760, IN-QEK31, and IN-TMQ01 were less than 70% which did not meet the criteria for pesticide residue analysis. Because soybean contains a large amount of fats and proteins that act as emulsifiers, separation between water and organic solvent is not clear and the amount of extracted solution decreases [20]. Next, the extraction efficiency of acetonitrile, a polar solvent such as methanol, was examined in order to lower the extraction of non-polar interfering substances. As a result of two times extractions of soybean sample moistened with 5 mL of water, IN-UJV12 showed that the recoveries increased as the ratio of water (Table 2). However, when the ratio of acetonitrile is 70%, the recovery of IN-A5760 was decreased sharply. Finally, the extraction solvent was selected with acetonitrile/water (80/20, v/v) and the soybean sample was determined to be moistened with 10 mL of water. As a result of examining the recovery of parent compound through the optimized extraction process, satisfactory results were obtained with an average recovery of 101.7% in mandarin and 86.4% in soybean sample.
Fluazaindolizine is applied to the soil at transplant and is generally metabolized, and the metabolites including seven major metabolites are remained conjugated state. Due to the complexity of metabolic pathway, analyzing the residue in a crop or soil sample is impossible and also it is impractical to synthesize standards for each metabolite. If the residue definition includes the conjugated compound of parent pesticide, the analytical method must conduct appropriate procedure for releasing the conjugated moiety [21]. So the acid hydrolysis step was utilized in this research, the metabolite conjugates was converted back to seven core molecules.
Optimization of solid-phase extraction
Considering that the target components are dissolved in hydrochloric acid before the purification process, the purification efficiency was examined with SPE (Solid-Phase Extraction) procedure using the HLB (Hydrophobic Liphophilic Balanced reversed-phase) cartridge, which is an adsorption cartridge that is stable even in a low pH range and has hydrophilic and wettability characteristics. The cartridge was conditioned with 3 mL of methanol and 3 mL of water, loaded with a mixed standard solution dissolved in 2 M HCl, and eluted according to the ratio of the mixture of methanol and water. At this time, the elution solvent was made to contain 1% formic acid in order to increase the elution strength. Most recoveries were satisfactory, with more than 87%. IN-UJV12 was first eluted from the methanol/water (20/80, v/v) and IN-QEK31 was finally eluted from the methanol/water (80/20, v/v). To reduce loss of seven metabolites and increase the recoveries, washing should be performed with a methanol/water (10/90, v/v) and elution should be performed with a methanol/water (90/10, v/v). The parent compound was also conducted by the above experiment procedure, it was eluted for the first time in a methanol/water (90/10, v/v) and was eluted at more than 70% in 100% methanol. As a result, when the HLB cartridge is selected as a purification cartridge, the elution solvent must have a methanol content of more than 90%. So a high methanol ratio has a disadvantage that interfering substances may be eluted together. Accordingly, it was attempted to lower the content of methanol in the elution solvent by examining the C18 cartridge, which is more hydrophobic than the HLB cartridge.
In experiments with a C18 cartridge, IN-UJV12 was first eluted in the first fraction of methanol/water (10/90, v/v) and IN-QEK31 was eluted in the second fraction of methanol/water (40/60, v/v). And all compounds were eluted in methanol with a lower content than in the HLB cartridge (Table 3). Therefore, it was established that metabolites were washed with 1 mL of methanol/water (40/60, v/v) and then eluted with 4 mL of methanol/water (50/50, v/v). The parent compound was tend to show a different elution characteristics per each sample that was first eluted with methanol/water (40/60, v/v) in soybean and first eluted with methanol/water (60/40, v/v) in mandarin. Therefore, it was established that the parent compound was washed with 3 mL of methanol/water (30/70, v/v) and then eluted with 4 mL of methanol/water (70/30, v/v).
Method validation
By comparing the chromatograms of standard in pure solvent, blank sample and recovery samples, the analysis results has high selectivity that did not contain any interfering peaks at the retention times of the target analytes (Fig. 2). The matrix effects values which are more than 20% indicated signal enhancement, while values which are less than − 20% means signal suppression. Values between from − 20–20% are considered that matrix effects could be negligible [22]. Otherwise, it need to be addressed in calibration according to SANTE/11813/2017 [23]. Table 4 shows the matrix effects, regression equation, and linearity of fluazaindolizine and seven metabolites in five agricultural samples. IN-A5760, IN-F4106, and IN-QZY47 showed ion suppression in all crops, and IN-UNS90 showed ion enhancement. Therefore, matrix-matched calibrations were adopted for accurate quantification in this study. The linearity of the matrix-matched calibrations at 0.002 to 0.05 mg/L for parent compound and three metabolites (IN-QEK31, IN-TMQ01 and IN-UNS90) and 0.01 to 0.25 mg/L for four metabolites (IN-A5760, IN-F4106, IN-QZY47 and IN-UJV12) were excellent, as assessed by the determination coefficient (R2) of the range from 0.9988 to 1.0000 in all agricultural samples. The LOD and LOQ of fluazaindolizine, IN-QEK31, IN-TMQ01 and IN-UNS90 was estimated to be 0.00075 mg/kg and 0.0025 mg/kg, respectively. The LOD and LOQ of IN-A5760, IN-F4106, IN-QZY47 and IN-UJV12 were 0.0042 mg/kg and 0.0125 mg/kg, respectively. The accuracy and precision of the present method could be evaluated by recoveries with five replicates at three fortification levels. The mean recoveries of fluazaindolizine and seven metabolites in agricultural products were 75.6–110.0% with the CV% of 0.2–9.1% (Table 5). All validated results demonstrated that the method was reliable for simultaneous analysis of fluazaindolizine and its metabolites residue in agricultural samples.

Representative MRM LC–MS/MS chromatograms of A fluazaindolizine, B IN-A5760, C IN-F4106, D IN-QEK31, E IN-QZY47, F IN-TMQ01, G IN-UNS90 and H IN-UJV12 at a blank sample, b matrix-matched standard, and c recovery sample spiked at 10 ×LOQ in hulled rice
Conclusions
An effective and accurate analytical method for simultaneous quantification of fluazaindolizine and its seven metabolites in agricultural products was established and validated. The metabolites were obtained from convert conjugated compounds into core molecules through acid hydrolysis. C18 SPE procedure combined with LC–MS/MS ESI mode technique was simple and convenient to analyze fluazaindolizine, IN-A5760, IN-F4106, IN-QEK31, IN-QZY47, IN-TMQ01, IN-UNS90 and IN-UJV12. The validation results were satisfied according to the guidelines formally provided by the pesticide residue regulatory authorities. The method could apply for monitoring of fluazaindolizine and major metabolites residue in domestic and imported agricultural samples, and utilize as scientific references on establishment of residue definition and maximum residue limits of fluazaindolizine.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Wikipedia, Root-knot nematode. https://en.wikipedia.org/wiki/Root-knot_nematode
Alford DV (2012) Chapter 4—Miscellaneous pests. In: Pests of ornamental trees, shrubs and flowers, 2nd edn. CRC Press, New York. https://books.google.co.kr/books?hl=ko&lr=&id=fWNNjWFwKwgC&oi=fnd&pg=PP1&ots=MyDQrchaKc&sig=x91MTsGdbuB7ldBrOrmNJjzco-o#v=onepage&q&f=false
Park JY (2013) Biological control of the root-knot nematode, Meloidogyne hapla with an antagonistic bacterium a thesis for the degree of master of science. Seoul National University. https://s-space.snu.ac.kr/handle/10371/125795
Doopedia, northern root-knot nematode. https://www.doopedia.co.kr/doopedia/master/master.do?_method=view&MAS_IDX=101013000792890
Chen X, Li X, Pang K, Fan X, Ma Y, Hu J (2018) Dissipation behavior and residue distribution of fluazaindolizine and its seven metabolites in tomato ecosystem based on SAX SPE procedure using HPLC-QqQ-MS/MS technique. J Hazard Mater 342:698–704
Thoden T, Pardavella IV, Tzortzakakis EA (2019) In vitro sensitivity of different populations of Meloidogyne javanica and M incognita to the nematicides Salibro™ and Vydate®. Nematology 21(8):889–893
Lahm GP, Desaeger J, Smith BK, Pahutski TF, Rivera MA, Meloro T, Kucharczyk R, Lett RM, Daly A, Smith BT, Cordova D, Thoden T, Wiles JA (2017) The discovery of fluazaindolizine: a new product for the control of plant parasitic nematodes. Bioorg Med Chem Lett 27:1572–1575
Wram CL, Zasada IA (2019) Short-term effects of sublethal doses of nematicides on Meloidogyne incognita. Phytopathology 109:1605–1613
Fava L, Orru MA, Crobe A, Barra Caracciolo A, Bottoni P, Funari E (2005) Pesticide metabolites as contaminants of groundwater resources: assessment of the leaching potential of endosulfan sulfate, 2,6-dichlorobenzoic acid, 3,4-dichloroaniline, 2,4-dichlorophenol and 4-chloro-2-methylphenol. Microchem J 79:207–211
Soler C, Pico Y (2007) Recent trends in liquid chromatography-tandem mass spectrometry to determine pesticides and their metabolites in food. Trends Anal Chem 26:103–115
Farlin J, Gallé T, Bayerle M, Pittois D, Braun C, El Khabbaz H, Lallement C, Leopold U, Vanderborght J, Weihermueller L (2013) Using the long-term memory effect of pesticide and metabolite soil residues to estimate field degradation half-life and test leaching predictions. Geoderma 207–208:15–24
Hernandez F, Sancho JV, Ibanez M, Grimalt S (2008) Investigation of pesticide metabolites in food and water by LC-TOF-MS. Trends Anal Chem 27:862–872
EFSA Panel on Plant Protection Products and their Residues (PPR) (2016) Guidance on the establishment of the residue definition for dietary risk assessment. EFSA J. https://doi.org/10.2903/j.efsa.2016.4549
Ministry of Food and Drug Safety (2016) Guidelines on standard procedures for preparing analysis method. https://www.mfds.go.kr/brd/m_210/view.do?seq=12920&srchFr=&srchTo=&srchWord=%EC%8B%9D%ED%92%88%EB%93%B1+%EC%8B%9C%ED%97%98%EB%B2%95+%EB%A7%88%EB%A0%A8&srchTp=0&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&company_cd=&company_nm=&page=1
Codex Alimentarius Commission (2010) Guidelines on good laboratory practice in pesticide residue analysis. CAC/GL 40–1993 (amendment 2010). http://www.fao.org/input/download/standards/378/cxg_040e.pdf
Zhou W, Yang S, Wang PG (2017) Matrix effects and application of matrix effect factor. Bioanalysis 9:1839–1844
Nischwitz V, Pergantisa SA (2006) Optimisation of an HPLC selected reaction monitoring electrospray tandem mass spectrometry method for the detection of 50 arsenic species. J Anal At Spectrom 21:1277–1286
Kwon JW, Cho YJ, Rhee GS (2015) Identification of pitfalls related to the analysis of liquid chromatography-tandem mass spectrometry and liquid chromatography-time of flight mass spectrometry. Korean J Environ Agric 34:230–237
Tokman N, Soler C, Farré ML, Picó Y, Barceló D (2009) Determination of amitraz and its transformation products in pears by ethyl acetate extraction and liquid chromatography–tandem mass spectrometry. J Chromatogr A 1216:3138–3146
Lee SJ, McClements DJ (2010) Fabrication of protein-stabilized nanoemulsions using a combined homogenization and amphiphilic solvent dissolution/evaporation approach. Food Hydrocoll 24:560–569
Steinborn A, Alder L, Spitzke M, Dörk D, Anastassiades M (2017) Development of a QuEChERS-based method for the simultaneous determination of acidic pesticides, their esters, and conjugates following alkaline hydrolysis. J Agric Food Chem 65:1296–1305
García AJN, González RR, Frenich AG (2015) Multi-pesticide residue analysis in nutraceuticals from grape seed extracts by gas chromatography coupled to triple quadrupole mass spectrometry. Food Control 47:369–380
Commission E (2017) SANTE/11813/2017-Guidance document on analytical quality control and method validation procedures for pesticide residues and analysis in food and feed. Off J Eur Union 46:1–42
Acknowledgements
Not applicable.
Funding
This research was financially supported by a Grant (21161MFDS364) from the Ministry of Food and Drug Safety, Republic of Korea, in 2021.
Author information
Authors and Affiliations
Contributions
SJL and SSY conceived and designed the experiments. HSL and SYG reviewed the literature and elucidated the compound structures. SJL performed the LC–MS/MS measurements and wrote the manuscript. SJL, HSL, SYG, HSS, SGK and SSY analyzed the experimental data. HJY and YHJ provided guidance and supervised the study. All authors helped prepare the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, S.J., Lee, H.S., Gu, S.Y. et al. Development of simultaneous analytical method for the determination of fluazaindolizine and its seven metabolites in agricultural products by liquid chromatography tandem mass spectrometry. Appl Biol Chem 65, 74 (2022). https://doi.org/10.1186/s13765-022-00739-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-022-00739-1