
Abstract
To manage the safety of the herbicide metazachlor, analytical methods are required for the determination of metazachlor metabolites in agricultural crops. Herein, a liquid chromatography–tandem mass spectrometry (LC–MS/MS) method was developed for the simultaneous determination of metazachlor metabolites (479M04, 479M08, and 479M16) in various agricultural commodities. After extraction using acetonitrile and adjusting the pH to 3, the samples were purified using a hydrophilic–lipophilic balance cartridge. The matrix-matched calibration curves (0.002–0.2 μg/mL) were linear (r2 > 0.99). For validation, recovery tests were carried out at three fortification levels (limit of quantification (LOQ), 10 LOQ, and 50 LOQ) in various agricultural samples. The recoveries of 479M04, 479M08, and 479M16 were 79.6–113.0, 76.9–97.7, and 79.1–102.1%, respectively, with relative standard deviation values of less than 17.0%. Furthermore, inter-laboratory testing was conducted to validate the method. All the values corresponded to the criteria of both the CODEX (CAC/GL 40-1993, 2003) and Ministry of Food and Drug Safety guidelines. Therefore, the proposed LC–MS/MS method can be used as an analytical method for the determination of metazachlor.
Similar content being viewed by others

Introduction
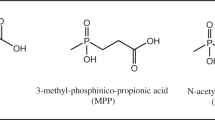
Metazachlor [2-chloro-N-(pyrazol-1-ylmethyl)acet-2′,6′-xylidide] (Fig. 1), a chloroacetamide herbicide developed by BASF, was produced by introducing 2,6-dimethylaniline (2,6-DMA; C8H11N) into the structure of chloroacetamide (C2H4ClNO) [1,2,3]. Metazachlor is mainly used to control annual weeds and broadleaf weeds before or after germination, and is absorbed through the roots or stems. Absorbed metazachlor affects elongase activity during lipid biosynthesis, thereby inhibiting the synthesis of very-long-chain fatty acids, as well as interfering with cell division and tissue differentiation during growth, resulting in the production of deformed seeds [1,2,3,4,5,6]. Metazachlor was completely metabolized in the plant and was not detected at harvest [7]. Furthermore, metazachlor residues were not detected in kohlrabi after pesticide application [6], as it is quickly metabolized in plants [8]. Therefore, to monitor metazachlor in plants, analytical methods are required for determination of its metabolites Pesticide metabolism can be divided into two phases. By these processes, nonpolar pesticides are generally converted into polar pesticides [9]. In plants, the Cl atom in metazachlor is replaced by glutathione, which can be decomposed by oxidation and hydroxylation [2] to form ethanesulfonic acid (ESA) and oxanillic acid (OA) in the environment [10]. The major metabolites of metazachlor are 479M04 [N-(2,6-dimethylphenyl)-N-(1H-pyrazol-1-ylmethyl)oxalamide], 479M08 [N-(2,6-dimethylphenyl)-N-(1H-pyrazol-1-ylmethyl)aminocarbonylmethylsulfonic acid], and 479M16 [3-[N-(2,6-dimethylphenyl)-N-(1H-pyrazol-1-ylmethyl)aminocarbonylmethylsulfinyl]-2-hydroxypropanoic acid], the structures of which are shown in Fig. 1 [8]. In the European Community, maximum residue limits (MRLs) of 0.01–0.4 mg/kg have been established for metazachlor in more than 378 products, including oranges and eggs. The residue definition for foods of plant origin was proposed as the sum of metabolites 479M04, 479M08, and 479M16, expressed as metazachlor, whereas for foods of animal origin, the sum of metazachlor and its metabolites containing a 2,6-DMA moiety, expressed as metazachlor, was used [8, 11]. Therefore, similar definitions should be used to establish MRLs in Korea.

Structures of metazachlor and its metabolites
Although no prior analytical method for metazachlor determination has been reported in Korea, various chloroacetamide herbicides (alachlor, metolachlor, metazachlor, etc.) are included in an official multiresidue analytical method (Ministry of Food and Drug Safety (MFDS) code 7.1.2) [12]. In the case of acetochlor, metabolites extracted from agricultural products using acetonitrile and then purified by dispersive-solid phase extraction (MFDS code 7.1.4.216) are also included [13]. Outside of Korea, chloroacetamide herbicides and their acidic metabolites (–ESA, –OA) in water have been purified using C18 cartridges and analyzed simultaneously using liquid chromatography–tandem mass spectrometry (LC–MS/MS) [10, 14, 15]. Recently, sample preparation using a quick, easy, cheap, effective, rugged, and safe (QuEChERS) method has been reported for studying metazachlor in soil, fruit, and kohlrabi [6, 16, 17]. In addition, Valls-Cantenys et al. [18] and Karier et al. [5] developed an analytical method for the determination of metazachlor and its metabolites (479M04 and 479M08), which was applied to monitor river water samples. Notably, 479M04 was detected in all samples, whereas 479M08 was detected in a drinking water reservoir in Luxembourg. Owing to this serious contamination, there is a necessity for safety management in consideration of the hazards to consumers, even though the metabolites have lower toxicities than the parent compound [5]. Therefore, the purpose of this study was to develop an analytical method for the determination of metazachlor metabolites in crops.
Materials and methods
Reagents, standard compounds and crop samples
479M04 (metazachlor-OA, 96.5%), 479M08 (metazachlor-ESA, 99.1%), and 479M16 (94.2%) standards were supplied from Kyung Nong Co. Ltd. (Seoul, Korea). Acetonitrile and methanol were HPLC grade (Merck, Darmstadt, Germany). Formic acid and ammonium formate were ACS grade (Sigma-Aldrich, Buchs, Switzerland). Sodium chloride (NaCl) was obtained from Junsei Chemical Co. Ltd. (Tokyo, Japan). Hydrophilic–lipophilic balance (HLB) cartridges (6 cm3, 500 mg) were purchased from Waters (Milford, MA, USA) and syringe filters (PVDF, 0.2 μm × 13 mm) were obtained from Teknokroma (Barcelona, Spain). Hulled rice, potato, soybean, mandarin, and green pepper of “residue-free grade” were purchased from a local market. The agricultural samples were homogenized and kept at a temperature below −50 °C in sealed containers until use.
Stock and working solution
The stock solutions of 479M04, 479M08, and 479M16 at a concentration of 1000 μg/mL were prepared dissolving the appropriate amount of each compound in methanol. The working solutions were prepared by appropriate dilution of the stock solutions with methanol. For the matrix-matched calibration curves, the concentrations of 0.002–0.2 μg/mL were obtained by mixing with blank extracts and standard solutions were mixed at a ratio of 9:1. All the standard solutions were stored at 4 °C in amber vials and the matrix-matched standards were newly prepared before each analysis.
Sample preparation
For homogenization of grains and soybeans, approximately 1 kg of sample pulverized to pass through a standard sieve of 420 μm. For fruits and vegetables, approximately 1 kg of sample were ground. Then, 5 g of each homogenized sample was accurately weighed and placed in a 50 mL centrifuge tube. For grains and soybeans, 5 mL of distilled water was added and the mixture was left to stand for 10 min, whereas this step was not necessary for the other samples. Following the addition of 25 mL of acetonitrile, the mixture was shaken for 1 min and then the pH was adjusted to 3 using formic acid (for grains and soybeans, 4 g of NaCl was also added). After shaking for 10 min, the extract was centrifuged for 5 min (4000g, 4 °C), the supernatant was collected, and 25 mL of acetonitrile was added. Subsequently, the above procedure was repeated twice and the extracts were combined. The extract was evaporated to dryness at 40 °C and redissolved in 12.5 mL of methanol. After loading 5 mL of the extract on an HLB cartridge, which was conditioned with 3 mL of methanol and 3 mL of distilled water, 5 mL of the eluate was collected. Subsequently, the cartridge was eluted with 5 mL of methanol, which was then mixed with the previous eluate and the total volume was adjusted to 10 mL. This solution was then filtered through a syringe filter (PVDF, 0.2 μm × 13 mm).
LC–MS/MS conditions
LC–MS/MS analysis was performed using an Acquity UPLC/Xevo TQ-S system (Waters, Milford, MA, USA) equipped with a Capcell Core ADME column (100 mm × 2.1 mm i.d., 2.7 μm, Shiseido, Tokyo, Japan) at a temperature of 40 °C. The injection volume and flow rate were 5.0 μL and 0.3 mL/min, respectively. Separation was performed by gradient elution using methanol (mobile phase A) and 5 mM ammonium formate in water (mobile phase B). The initial mobile phase of 10/90 (A/B, v/v) was retained for 3 min, changed to 90/10 (A/B, v/v) over 5 min and then held for 2 min, and finally changed to 10/90 (A/B, v/v) over 0.1 min and then retained until 10 min. MS/MS was conducted using positive electrospray ionization (ESI) and multiple reaction monitoring (MRM). The ion source temperature and the desolvation temperature were 150 °C and 500 °C, respectively, and the capillary voltage was 1.0 kV. The MRM conditions are summarized in Table 1.
Method validation
To validate the developed analytical method, the recovery (accuracy and repeatability), linearity, selectivity, limit of detection (LOD), and limit of quantification (LOQ) in agricultural samples were assessed based on the MFDS guidelines on standard procedures for preparing analysis methods and the CODEX guidelines [19, 20]. The selectivity was compared using chromatograms of blank samples and blank samples spiked with metazachlor metabolites to confirm whether interfering peaks occurred at the retention times of the standards. The linearity of the matrix-matched calibration curves at concentrations of 0.002–0.2 μg/mL were assessed by the coefficients of determination (r2). The LOD and LOQ values of the instrument were determined as 3 and 10 times, respectively, the signal to noise (s/n) ratio of the chromatogram. Subsequently, the LOQ of the analytical method was calculated by considering the LOQ of the instrument, the weight of the sample, and the volume of solvent. Recovery tests were conducted at three fortification levels (0.01 (LOQ), 0.1 (10 LOQ), and 0.5 mg/kg (50 LOQ), n = 5) in the five representative crops. The accuracy and repeatability of the analytical method were assessed by calculating the average and relative standard deviation (RSD) values. Finally, inter-laboratory testing was conducted at Chonnam National University to confirm the validity of the developed method.
Results and discussion
Optimization of LC–MS/MS conditions
Currently, there are no analytical methods for the determination of metazachlor and its metabolites in the MFDS food code, although metazachlor has been included in a multiresidue analytical method using gas chromatography–tandem mass spectrometry (GC–MS/MS) [12]. However, metazachlor metabolites are difficult to analyze with GC, as these compounds are relatively polar (for 479M04 and 479M16, log Pow = − 0.54). Therefore, owing to the introduction of the positive list system, analysis methods are required for these analytes, and LC–MS/MS has been chosen as an appropriate analysis technique owing to its high selectivity and low LOQ (≤ 0.01 mg/kg) [5, 18]. ESI has a lower noise level and better sensitivity than atmospheric pressure chemical ionization (APCI) for pesticide analysis [21]. In particular, for metazachlor, ESI has been shown to have higher sensitivity than APCI [18]. When a mobile phase of 0.1% formic acid in water and 0.1% formic acid in acetonitrile was used, peak splitting was observed for the metazachlor metabolites, likely owing to the presence of diastereomers, as observed for the acidic metabolites of chloroacetamide [10, 22, 23]. Shortening the column length [10] and increasing the analysis temperature [22, 23] have been reported to provide improved performance for the analysis of metazachlor. In this study, the analysis conditions were further improved by using ammonium formate and changing from acetonitrile to methanol (Fig. 2). As peak tailing occurred when formic acid or acetic acid was added to the mobile phase, no acid was added. A comparison of the peak shapes with ammonium formate or ammonium acetate added to water (mobile phase B) showed that the tailing issue was resolved when formate was used. This result is consistent with the findings of Valls-Cantenys et al. [18], who found that the addition of formate improved the peak shape and increased the s/n ratio. For 479M04 (exact mass: 273.11), 479M08 (exact mass: 323.09), and 479M16 (exact mass: 379.12), the direct injection of standard solutions (0.1 μg/mL) into the mass spectrometer at a constant rate (10 μL/min) gave [M + H]+ peaks at m/z 274, 324, and 380, respectively. The product ion with the highest intensity was used as the quantification ion and the product ion with the second highest intensity was used as the qualification ion (Table 1). When operating LC–MS/MS, the extent of ionization suppression or enhancement for each compound due to the matrices was accounted for by matrix-matched calibration.

Optimization of peak shape according to mobile phases
Optimization of sample extraction
Acetonitrile and methanol, which have been used in previous studies [3, 23,24,25], were first considered as extraction solvents. The QuEChERS method, which has the advantages of requiring relatively low amounts of solvent and short extraction times, was evaluated using three different procedures (original, AOAC 2007.01, and CEN15662) that differ according to the reagents used in the extraction step [26,27,28]. Using the three QuEChERS methods, extraction with acetonitrile and methanol from mandarin and soybean gave poor recoveries (22.3%–74.0%). Therefore, the pHs of the acetonitrile and methanol extracts were adjusted to 1–7 using formic acid (Fig. 3). While methanol still exhibited low extraction efficiencies, all the metabolites exhibited excellent recovery with acetonitrile at pH 3. Although this extraction method was found to be inefficient for soybeans, this issue was resolved by repeating the extraction process twice (89.0% → 95.2%). Furthermore, when dried samples (hulled rice and soybean) were extracted, amount of sodium chloride was increased and then recovery results were improved (479M04 77.2% → 87.4%, 479M08 69.2% → 81.0%, 479M16 78.8% → 82.4%). Because it increases ionic strength and thus the distribution efficiency from the water layer to the organic solvent layer via the salting out effect [29].

pH dependence of the extraction efficiencies of metazachlor metabolites from the mandarin matrix using a acetonitrile and b methanol
Optimization of cartridge clean-up
Various purification methods, such as using a C18 cartridge [24, 30] or liquid–liquid partitioning [25], have been widely studied. However, as polar compounds, the metazachlor metabolites elute without adsorption to C18 and are difficult to collect following partitioning, as they move into the water layer. Recently, in recent studies on metazachlor monitoring in water sample, an HLB cartridge was applied for sample clean-up [5, 18]. Thus, an HLB cartridge was adopted for sample clean-up in this study. The HLB cartridge gave good recoveries of the metazachlor metabolites from a mixed standard solution at 0.05 μg/mL (479M04: 100.7%, 479M08: 109.8%, and 479M16: 128.5%).
Method validation
To validate the developed analytical method, the recovery (accuracy and repeatability), linearity, selectivity, LOD, and LOQ were assessed for the five agricultural samples. When analyzing the blank (nonspiked) samples, no interfering peaks appeared at the same retention times and mass-to-charge ratios (m/z) as those of the metabolites. Therefore, this analytical method provided high separation ability and selectivity for the metazachlor metabolites (Fig. 4). The standard solutions were diluted with a blank extract to obtain matrix-matched standards at concentrations of 0.002–0.2 μg/mL, and the linearity of each matrix-matched calibration curve was assessed based on the coefficient of determination (r2). From the analyses at various concentrations, LOQ values of ≤ 0.01 mg/kg were determined for all five products. Recovery tests for the five agricultural products were performed in triplicate at three fortification levels (0.01, 0.1, and 0.5 mg/kg), and the accuracy and repeatability of the analytical method were assessed based on the calculated average and RSD. The results of interlaboratory test satisfied the criteria of both the CODEX and MFDS guidelines [19, 20] (Table 2).

Representative MRM (quantification ion) recovery chromatograms (fortification concentration: 0.1 mg/kg) of a 479M04, b 479M08 and c 479M16 in (1) mandarin control, (2) matrix-matched standard, and (3) mandarin test sample
In conclusion, the analytical methods were established for the determination of the residues of metazachlor metabolites in five agricultural commodities using LC–MS/MS. Through method validation, the method achieved good accuracy and repeatability, and the results were confirmed with chromatogram of LC–MS/MS (MRM). Finally, this study was can be used as an analytical method for the determination of metazachlor metabolites in agricultural products.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 2,6-DMA:
-
2,6-dimethylaniline
- APCI:
-
Atmospheric pressure chemical ionization
- ESA:
-
Ethanesulfonic acid
- ESI:
-
Electrospray ionization
- GC–MS/MS:
-
Gas chromatography–tandem mass spectrometry
- HLB:
-
Hydrophilic–lipophilic balance
- LC–MS/MS:
-
Liquid chromatography–tandem mass spectrometry
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- MFDS:
-
Ministry of Food and Drug Safety
- MRL:
-
Maximum residue limit
- MRM:
-
Multiple reaction monitoring
- OA:
-
Oxanillic acid
- QuEChERS:
-
Quick, easy, cheap, effective, rugged, and safe
- RSD:
-
Relative standard deviation
- s/n :
-
Signal to noise
References
Baćmaga M, Kucharski J, Wyszkowska J, Borowik A, Tomkiel M (2014) Responses of microorganisms and enzymes to soil contamination with metazachlor. Environ Earth Sci 72:2251–2262
Australian Pesticides and Veterinary Medicines Authority (APVMA) (2016) Public release summary: on the evaluation of the new active metazachlor in the product Bustisan Herbicide, APVMA Product Number 80664. Kingston0
Valverde S, Ares AM, Bernal JL, Nozal MJ, Bernal J (2017) Simultaneous determination of thiamethoxam, clothianidin and metazachlor residues in soil by ultrahigh performance liquid chromatography coupled to quadarupole time-of-flight mass spectrometry. J Sep Sci 40:1083–1090
Food and Agriculture Organization of the United Nations (FAO) (1999) FAO specifications and evaluations for plant protection products, metazachlor. Rome
Karier P, Kraus G, Kolber I (2017) Metazachlor traces in the main drinking water reservoir in Luxembourg: a scientific and political discussion. Environ Sci Eur 29(1):1–5
Jursík M, Kočárek M, Suchanová M, Kolářová M, Šuk J (2019) Effect of irrigation and adjuvant on residual activity of pendimethalin and metazachlor in kohlrabi and soil. Plant Soil Environ 65:387–394
MacBean C (2012) The pesticide manual (2012), 16th edn. British Crop Protection Council, Alton, pp 741–742
European Food Safety Authority (EFSA) (2009) Prepared by the Pesticides Unit (PRAPeR). Reasoned opinion of EFSA. Modification of the existing MRLs of metazachlor for certain products of animal origin. EFSA J 7:1–32
Jung YH, Kim JE, Kim JH, Lee YD, Lim CH, Hur JH (2004) The newest pesticide science. Sigmapress, Seoul, pp 293–294
Yokley RA, Mayer LC, Huang SB, Vargo JD (2002) Analytical method for the determination of metolachlor, acetochlor, alachlor, dimethenamid, and their corresponding ethanesulfonic and oxanillic acid degradates in water using SPE and LC/ESI-MS/MS. Anal Chem 74:3754–3759
European Commission (EC) (2016) EU –Pesticides database. https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/public/?event=pesticide.residue.CurrentMRL&language=EN. Accessed 15 Oct 2019
Ministry of Food and Drug Safety (MFDS) (2019) Korea food code. Seoul: pp 321-360
Ministry of Food and Drug Safety (MFDS) (2019) Korea food code. Seoul: pp 853-855
Zimmerman LR, Schneider RJ, Thurman EM (2002) Analysis and detection of the herbicides dimethenamid and flufenacet and their sulfonic and oxanilic acid degradates in natural water. J Agric Food Chem 50:1045–1052
de Oliveira Arias JL, Schneider A, Batista-Andrade JA, Vieira AA, Gehrke VR, Camargo ER, Caldas SS, Primel EG (2017) Evaluation of dilute-and-shoot and solid-phase extraction methods for the determination of S-metolachlor and metolachlor-OA in runoff water samples by liquid chromatography tandem mass spectrometry. Anal Methods 9:5777–5783
Stachniuk A, Fornal E (2013) Analytical considerations on the use of a fruit-specific and representative matrix in pesticide residue analysis by LC-ESI-MS/MS. Cent Eur J Chem 11:1112–1131
Mei W, Li Q, Fang H, Geng H, Chen X (2017) Determination of six amide herbicide residues in soil by QuEChERS-gas chromatography-mass spectrometry. Chin J Chromatogr 35:1317–1321
Valls-Cantenys C, Scheurer M, Iglesias M, Sacher F, Brauch HJ, Salvadó V (2016) A sensitive multi-residue method for the determination of 35 micropollutants including pharmaceuticals, iodinated contrast media and pesticides in water. Anal Bioanal Chem 408:6189–6200
Ministry of Food and Drug Safety (MFDS) (2016). Guidelines on standard procedures for preparing analysis method Cheongju, Korea
Codex Alimentarius Commission (2003). Guidelines on good laboratory practice in residue analysis. CAC/GL 40-1993. Rome
Wick A, Fink G, Ternes TA (2017) Comparison of electrospray ionization and atmospheric pressure chemical ionization for multi-residue analysis of biocides, UV-filters and benzothiazoles in aqueous matrices and activated sludge by liquid chromatography–tandem mass spectrometry. J Chromatogr A 1217:2088–2103
Dagnac T, Jeannot R, Mouvet C, Baran N (2002) Determination of oxanilic and sulfonic acid metabolites of acetochlor in soils by liquid chromatography–electrospray ionisation mass spectrometry. J Chromatogr A 957:69–77
Heberle SA, Aga DS, Hany R, Müller SR (2000) Simultaneous quantification of acetanilide herbicides and their oxanilic and sulfonic acid metabolites in natural waters. Anal Chem 72:840–845
Vryzas Z, Tsaboula A, Papadopoulou-Mourkidou E (2007) Determination of alachlor, metolachlor, and their acidic metabolites in soils by microwave-assisted extraction (MAE) combined with solid phase extraction (SPE) coupled with GC-MS and HPLC-UV analysis. J Sep Sci 30:2529–2538
Lazartigues A, Fratta C, Baudot R, Wiest L, Feidt C, Thomas M, Cren-Olivé C (2011) Multiresidue method for the determination of 13 pesticides in three environmental matrices: water, sediments and fish muscle. Talanta 85:1500–1507
AOAC Official Method (2007) Pesticide residues in food by acetonitrile extraction and partitioning with magnesium sulfate. Gas chromatography/mass spectrometry and liquid chromatography/tandem mass spectrometry. Rockville: AOAC International
European Commission for Standardization (CEN), Technical Committee CEN/TC 275. EN 15662:2008 (2008) Foods of plant origin—Determination of pesticide residues using GC-MS and/or LC-MS/MS following acetonitrile extraction/partitioning and clean-up by dispersive SPE–QuEChERS-method. Brussels
Seo EK, Kim TK, Hong SM, Kwon HY, Kwon JH, Son KA, Kim JE, Kim DH (2013) Analysis of systemic pesticide imidacloprid and its metabolites in pepper using QuEChERS and LC-MS/MS. Korean J Pestic Sci 17:264–270
Ko AY, Abd El-Alty AM, Rahman MM, Jang J, Kim SW, Choi JH, Shim JH (2014) A modified QuEChERS method for simultaneous determination of flonicamid and its metabolites in paprika using tandem mass spectrometry. Food Chem 157:413–420
Kucharski M, Sadowdki J (2014) Metazachlor residues in soil and rape seed. J Plant Prot Res 54:74–77
Acknowledgements
Not applicable.
Funding
This research was supported by a Grant (19161MFDS020) from the Ministry of Food and Drug Safety, Republic of Korea, in 2019.
Author information
Authors and Affiliations
Contributions
JSP and SSY conceived and designed the experiments. HSL, SMC, and SJL reviewed the literature and elucidated the compound structures. JSP performed the LC–MS/MS measurements and wrote the manuscript. JSP, HSL, SMC, SJL, HSS and SSY analyzed the experimental data. Also, JHS conducted the validation of analytical method. JHO and YHJ provided guidance and supervised the study. All authors helped prepare the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, JS., Lee, H.S., Cho, S.M. et al. Simultaneous determination of the metabolites of the herbicide metazachlor in agricultural crops by LC–MS/MS. Appl Biol Chem 63, 29 (2020). https://doi.org/10.1186/s13765-020-00513-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-020-00513-1