
Abstract
Angiotensin II type 1 receptor (AT1R) is a promising therapeutic target for cardiovascular diseases. Compared with orthosteric ligands, allosteric modulators attract considerable attention for drug development due to their unique advantages of high selectivity and safety. However, no allosteric modulators of AT1R have been applied in clinical trials up to now. Except for the classical allosteric modulators of AT1R such as antibody, peptides and amino acids, cholesterol and biased allosteric modulators, there are non-classical allosteric modes including the ligand-independent allosteric mode, and allosteric mode of biased agonists and dimers. In addition, finding the allosteric pockets based on AT1R conformational change and interaction interface of dimers are the future of drug design. In this review, we summarize the different allosteric mode of AT1R, with a view to contribute to the development and utilization of drugs targeting AT1R allostery.
Similar content being viewed by others


Introduction
G protein-coupled receptors (GPCRs) are the largest family of membrane receptors with seven transmembrane (TM) α-helices. GPCR-tageting Drugs account for approximately 34% of all FDA-approved drugs [1]. Angiotensin II type 1 receptor (AT1R) is a member of the rhodopsin family of GPCRs. It possesses an extracellular N terminus, three extra- and intracellular loops (ECL1-3 and ICL1-3), an amphipathic helix 8 (H8), and an intracellular C terminus. Abnormal activation of AT1R leads to various diseases, such as hypertension, coronary artery disease, arrhythmia, and diabetic nephropathy [2, 3]. Therefore, AT1R is becoming a target for drug development. However, functional diversity exists due to the signaling diversity of AT1R, which poses a challenge for the application in diseases.
As an important mechanism of the signal diversity, allosteric modulation occurs when a variety of extracellular stimuli (ions, lipids, peptides, proteins, autoantibodies, etc.) interact with allosteric sites of GPCR, which affects the orthosteric ligands [4,5,6]. In addition to classical allostery, allosteric regulation has many other forms, such as the activation mode of biased ligands and dimers. The phenomenon that GPCRs preferentially form a certain conformation and select intracellular sensors to produce different signal activation is termed “biased agonism” or “functional selectivity”, which is a natural result of allosteric selection. In other words, biased activation implies the cytosol-directed allostery [7]. Besides, GPCR dimerization is a more subtle form of allosteric regulation, breaking the traditional concept of allosteric activators, that the initiator is a monomer in the dimer or its ligand, and the allosteric information is transmitted laterally [8,9,10].
As a valuable target for drug development, allosteric modulation of AT1R is still in its infancy. To better understand the mechanism behind the functional diversity of AT1R and expedite the development of novel allosteric drugs, this paper reviews the current status of AT1R allosteric regulation, focusing on the classical and non-classical allostery, and the allosteric pockets of AT1R.
Classical allostery and classical allosteric modulators of AT1R
Classical allostery suggests that binding an allosteric modulator to an allosteric site can modulate the properties of an orthosteric ligand in the same protein (enzyme, receptor, etc.) [10]. The allosteric modulators bind to the allosteric sites and subsequently regulate the binding of the orthosteric ligands to the orthosteric sites. Classically, most GPCR drugs target the orthosteric sites. Orthosteric ligands of GPCR are divided into three categories: (1) agonists that can activate GPCR; (2) inverse agonists that inhibit the constitutive activity of GPCR; and (3) antagonists that occupy the orthosteric site without effect) [11]. Except for the orthosteric sites, the binding sites are called allosteric sites or allosteric pockets, which bind to allosteric ligands [12]. Allosteric ligands transmit allosteric signals to functional sites through atomic fluctuations, amino acid residue networks, or domain movements, thereby increasing, maintaining, or decreasing the affinity and/or efficacy of orthosteric ligands [5, 13, 14]. During biological evolution, the orthosteric sites remained highly conserved. In contrast, the allosteric sites are less conserved and have structural diversity, not driven by evolutionary pressures. Meanwhile, allosteric modulators have higher target specificity, better spatiotemporal specificity (synergism with endogenous ligands), and functional specificity, thereby providing higher selectivity and better control of receptor dynamics [15, 16]. Moreover, allosteric modulators are saturable and do not disrupt signaling networks with increasing doses. This feature reduces the risk of side effects and offers potential therapeutic advantages [17].
Allosteric modulators trigger the allosteric influence of the receptor activated by orthosteric modulators. Allosteric modulators are classified into three types based on their effects: positive allosteric modulators (PAM), negative allosteric modulators (NAM), and neutral allosteric ligands (NAL, also known as silent allosteric modulators (SAM)) [16, 18]. In the absence of orthosteric modulators, some PAM, known as ago-allosteric modulators (ago-PAM), can activate the receptor alone [19]. Biased allosteric modulators (BAM) alter the activation mode of receptors through specific signaling [20]. Here, we summarize the classical allosteric modulators of AT1R.
Antibody
Antibodies produced against self-antigen are known as autoantibodies (AAs). The majority of AAs against GPCR (GPCR-AAs) are allosteric modulators that enhance or reduce the effect of orthosteric ligands and alter the endogenous biological properties of the receptor. They possess unique and complex pharmacological properties [21]. AT1R autoantibodies (AT1R-AAs) acted as ago-PAM and were first identified in patients with preeclampsia [22] (Fig. 1). AT1R-AAs are deemed to exert pathogenic effects and have been implicated in various diseases such as metabolic syndrome [23], malignant hypertension [24], unstable angina (UA) [25], renal transplant rejection [26], frailty [27], Alzheimer’s disease [28], and COVID-19 [29,30,31]. Angiotensin II (AngII), the orthosteric ligand of AT1R, activates G protein and β-arrestin signaling cascades [32, 33]. Unlike AngII, AT1R-AAs bind to the ECL2 of AT1R and independently activate the receptor for a long period of time by inhibiting β-arrestin1/2 recruitment and AT1R internalization. They can cause sustained vasoconstriction, tissue fibrosis, and migration of immune regulatory cells [34, 35]. This may be due to the fact that AT1R forms a conformation that does not easily bind to β-arrestin1/2 after being targeted by AT1R-AAs. Therefore, the effect of AT1R-AAs may be related to a type II hypersensitivity response, similar to autobodies of thyroid-stimulating hormone receptor (TSHR) that can overactivate TSHR in Graves’ disease [36].

The protein structure of human AT1R. Secondary structure of human AT1R with depiction of different motifs and important sites. Cys18–Cys274, the disulfide bond, stabilizes the N terminus and ECL3. And Cys101–Cys180 stabilizes TM3 and ECL2. The DRY motif and NPxxY motif, the microswitches of AT1R, are considered to participate in receptor activation. The interaction, N1113.35 hydrogen bonds with N2957.46, stabilizes the inactive state of AT1R.
It has been shown that AT1R-AAs amplify AngII-induced vasoconstrictor response and increase the sensitivity of AngII to AT1R, but the mechanism is unclear [37,38,39]. The two disulfide bonds of AT1R, Cys18-Cys274 and Cys101-Cys180, stabilize the conformation of the N terminus and ECL2 [40, 41] (Fig. 1). AT1R-AAs may affect the strain on the disulfide bond by binding to the ECL2 epitope. Cascade reactions of the receptor core alter the movement of the transmembrane helix, thereby affecting the recruitment of intracellular sensors. Alternatively, AT1R-AAs may have a microeffect on the binding residues of AngII to AT1R, thereby promoting the binding affinity and efficacy of AngII to the receptor or hindering the removal of AngII from the pocket.
Unexpectedly, AT1R-AAs were found to exert protective effects on COVID-19 possibly by interfering with the action of AngII and reducing the inflammatory response in the acute phase response of COVID-19 [42]. Uncertainty regarding the antigenic epitopes of the antibodies is a possible reason for the different effects of AT1R-AAs. It has been shown that unlike the agonistic effect of autoantibodies against ECL2, a monoclonal antibody against the N-terminal of AT1R, 6313/G2, inhibited AngII-induced cell proliferation [43]. Therefore, antibodies generated against different domains of a receptor can exert different modulating effects.
Researchers often use antibodies to stabilize the receptor and obtain the active crystal structure. These antibodies, always act as PAM for the receptor. To stabilize the AT1R-activated conformation, the nanobody AT110i1 was used to target ICL2 of AT1R by a synthetic yeast-displayed library. AT110i1 increased the binding affinity of the partial agonist S1I8, the full agonist AngII, and β-arrestin-biased agonists TRV026 and TRV023 to AT1R, and made the ligand-receptor complex more stable [44, 45] (Fig. 2A). Although the nanobody AT110i1 fixes the activated AT1R by simulating the binding of Gq, it is possibly differ from the activated conformation in the physiological state. Identification of the structure of the true complexity of AT1R with sensors such as GPCR kinase (GRK) and β-arrestin can help better understand receptor conformational change. Even the new conformational states can be unfolded, which is crucial for the design of allosteric modulators.

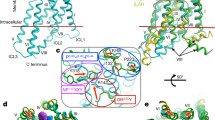
Allosteric modulators and pockets of AT1R. A AT110il and CLR are allosteric modulators of AT1R. B The active and inactive states of AT1R shows significant structural change in the binding site of AT1R-AAs. C A cryptic allosteric pocket is formed during MD simulation. D1) S1I8-bound AT1R with allosteric compound DCP1 and AT1R-AAs binding epitope are highlighted. D2) The cartoon picture of D1). E1) A potential cryptic allosteric pocket P6 is observed by using Fpocket. P6 is only identified during the movement of H8. E2) The cartoon picture of E1).
Peptides and amino acids
The hemoglobin-derived peptides LVV-hemorphin-7 (LVV-H7) and homocysteine (Hcy) are newly identified PAM of AT1R. LVV-H7 was reported to enhance AT1R-mediated Gq and β-arrestin signaling produced by AngII [46]. Conformation changes are prerequisites for alterations in signaling and effects. Molecular docking and molecular dynamics (MD) simulations indicated that LVV-H7 targeted residues on the second and third intracellular loops of AT1R, thereby allosterically enhancing the binding affinity of AngII. In the presence of LVV-H7, AngII binds deeper in the orthosteric pocket, forming more hydrogen bonds, and hydrophobic and polar interactions. Moreover, the side chain of Arg1263.50 slightly orients toward TM6, which facilitates the opening of the G protein-binding groove and enhances G protein binding to AT1R [47]. Hcy is a sulfur-containing non-essential amino acid and the high serum level of Hcy is a risk factor for cardiovascular diseases. It has been shown that Hcy can activate AT1R directly via Arg1674.64 and Cys2897.40. Hcy allosterically interacts with AT1R through Cys2897.40, triggering a unique conformation of AT1R ICL2, which synergistically activates the receptor with AngII and aggravates vascular injury in the abdominal aortic aneurysm [48].
Cholesterol
GPCR functions in a cell membrane environment where cholesterol (CLR), as a sterol-like type of lipid, is highly abundant and can directly bind to the receptor, thereby allosterically regulating the affinity and efficacy of the ligand as well as the spontaneous activity of the receptor. CLR may also indirectly affect GPCR and its signal transduction by altering the fluidity of the cell membrane. In the crystal structure of AT1R with the biased agonist TRV023 (6OS1), cholesterol interacts with the receptor at Phe391.43, Phe441.48, and Ser471.51 of TM1 and H8 [15] (Fig. 2A). Six cholesterol molecules were identified in the AT1R-Gq structure(49). Cholesterol may directly or indirectly affect the binding of drugs to their receptors. Cholesterol can prevent the antagonistic effect of losartan on AT1R by preventing its access to AT1R [50]. Understanding the role of cholesterol in the allosteric activation of GPCR is an important step in the treatment of hypercholesterolemia.
Biased allosteric modulators (BAM) of AT1R
The advent of BAM led to a new breakthrough in GPCR drug discovery. Unlike biased agonists that bind to the orthosteric site, BAM bind to the allosteric site, and exert pathway-specific effects, with the potential to selectively stimulate relevant signals and avoid side effects [20]. Cartilage oligomeric matrix protein (COMP) is an endogenous biased inhibitory ligand of AT1R that directly interacts with the N-terminal of AT1R via the structural domain of epidermal growth factor (EGF). COMP allosterically regulated receptor conformation and selectively inhibited AT1R/β-arrestin2 signaling in mice [51]. COMP can be used as a biased allosteric target for developing drugs for cardiovascular diseases in the future. BAM can help develop more effective and selective treatments.
Non-classical allostery of AT1R
In addition to classical activation, AT1R can be activated by non-classical allosteric patterns, including ligand-independent allosteric mode, and allosteric mode of biased agonists and dimers. GPCRs can be spontaneously activated in the absence of ligands, known as constitutive activity [52, 53]. It has been found that allosteric modulation can occur through the constitutive activity of receptors because of constitutively active mutants (CAMs) and mechanical stretch [52, 54]. “Biased agonists” or “biased ligands” mainly activate one of the receptor-mediated downstream pathways, such as G protein-dependent or non-G protein-dependent β-arrestin pathway for biased activation [55,56,57]. The allostery of GPCR depends on the type of ligand and signaling protein. Different ligands can control sensor coupling and biased signal selection through allostery [58]. Allostery provides a mechanistic explanation for dimers. Compared to monomers, the allosteric process of dimers broadens the range of cellular signaling pathways and increases the complexity of GPCR signaling [59]. The obvious functional advantage of dimers is that they can function as allosteric machines. Changes in the spatial conformation of one receptor can allosterically regulate the function of another receptor, affecting the selectivity of the downstream signaling pathway of GPCR dimers and triggering a series of functional changes with various pharmacological properties [60, 61].
Ligand-independent allosteric mode
CAMs and mechanical stretch allosterically regulate AT1R signaling. N111G-AT1R preferentially couples to Gq and increases IP production [62]. However, D74N, DRY/AAY, and N298A mutants of AT1R have strong interaction with β-arrestin2 [63]. CAMs may change the impact of TMs on the allosteric pathway, which provides effective tools for screening inverse agonists.
Mechanical stress can allosterically promote the constitutive activation of AT1R. In the absence of AngII and G protein activation, mechanical stress can induce GRK5/6-dependent β-arrestin-biased signaling as downstream of AT1R [64]. Mechanical stretch increases the binding affinity and efficiency of the biased agonist TRV120023 by stabilizing the specific β-arrestin-activated conformation of AT1R [54]. Leu212, Gln257, and Cys289 are key sites for activating AT1R by mechanical stretch [65]. Non-ligand regulation enhances multidimensional activation mode of AT1R. However, it is unclear whether mechanical stretch works independently or through mechanical components such as ion channels. AT1R activation due to mechanical stimuli is likely the result of a combined effect.
Allosteric mode of biased agonists
AT1R-biased agonists are obtained by modifying AngII. Gq-biased agonists of AT1R include TRV055 and TRV056. β-arrestin-biased agonists include SII, TRV120023, TRV120027, etc. Interestingly, some of these β-arrestin-biased ligands exhibit unique beneficial properties. In 2002, Alice et al. identified the first β-arrestin-biased agonist of AT1R, [Sar1-Ile4-Ile8]-AngII (SII), where Asp1 of AngII was replaced by sarcosine, and Tyr4 and Phe8 were replaced by isoleucine [66]. In a rat model of cardiac ischemia-reperfusion, SII pretreatment reduced myocardial infarct size by 24% [67]. In isolated adult mouse cardiomyocytes, SII promoted positive inotropic and lusitropic responses via GRK6/β-arrestin2 [68]. And then the investigators developed TRV120023 and TRV120027 (KD = 12 nM) with higher affinity and efficiency. As a potential therapeutic agent for dilated cardiomyopathy (DCM), TRV120023 enhanced cardiac contractility by upregulating ventricular myosin light chain-2 phosphorylation, without mobilizing Ca2+ [69]. Similarly, TRV120027 not only reduced mean arterial pressure but also enhanced cardiomyocyte contractility, increased cardiac performance, and preserved cardiac stroke volume [70], which was demonstrated in heart failure canines [71]. A study in pediatric heart failure (PHF) found that TRV027 induced a long-acting, strong positive inotropic effect without affecting heart rate, reactive oxygen species production, and adrenal aldosterone secretion in neonatal mouse [72]. These results support the therapeutic potential of biased agonists over ARB. Trevena, Inc. announced a clinical trial to study the effect of TRV027 on patients with COVID-19. Data from 30 patients showed that TRV027 was well-tolerated and decreased circulating D-dimer in 70% of patients. TRV027 was associated with 92% probability of a potential beneficial treatment effect, with the potential to improve COVID-19 progression-related biologic markers and clinical endpoints [73].
However, the allosteric mechanism of the receptor for biased ligands is poorly understood. It is not clear how the ligand induces the corresponding conformation of the receptor and GPCR signal selection. The biased agonist binds to the orthosteric pocket of the receptor, activates a microswitch through allosteric pathways, triggers a conformational rearrangement of the receptor core, and initiates some intracellular sensors. Highly conserved residue motifs such as DRY and NPxxY act as microswitches. DRY motif (Asp1253.49-Arg1263.50-Tyr1273.51) in AT1R TM3, commonly referred to as ion lock, and NPxxY motif (Asn2987.49-Pro2997.50-Leu3007.51-Glu3017.52-Tyr3027.53) in TM7 are crucial for the recruitment of G-protein and receptor activation [74] (Fig. 1). Wingler et al. resolved the crystal structures of AT1R with AngII (2.9 Å), the β-arrestin-biased agonist TRV026 (2.7 Å), and TRV023 (2.8 Å) [45]. In line with previous findings, AngII adopts a vertical binding mode, reaching deep into the receptor core while contacting the extracellular surface [75]. Compared with the inactive state, in the active state, the extracellular changes of AT1R are the inward movement of TM5 and TM7, and intracellular changes of AT1R are the outward movement of TM5 and TM6, inward movement of TM7, and movement of H8 toward the cell membrane (Fig. 3A). In the AngII-AT1R structure, the phenylalanine at position 8 plays a decisive role in the initiation of allostery, triggering TM3 to rotate around the axis and Leu1123.34 to rotate inward to occupy position Tyr2927.43. Phe8 engaged Ile2887.39 of AT1R in van der Waals interaction, while pulling Tyr2927.43 [76]. To avoid spatial limitation, Tyr2927.43 turns downward and the flip of Asn1113.35 break the hydrogen bond formed with Asn2957.46, which is the main constraint in the inactive state. This is believed to adapt to the insertion of Gα subunit, which is essential for Gq signaling. Tyr3027.53 of the NPxxY motif and Arg1263.50 of the DRY motif are both involved in stabilizing the activation state. However, Asn1113.35 is maintained near the reoriented Asn2957.46 in the structure of the β-arrestin-biased ligand and AT1R. It restricts TM3 and TM7 to form a blocking conformation. The binding of the β-arrestin-biased agonist triggers the TM6 transition and the conformational rearrangement of Asn2957.46, which is a marker of GPCR activation and may be sufficient for the conformation required for β-arrestin coupling [77] (Fig. 3B).

Comparison of the allosteric structures of human AT1R bound to different ligands. A Overall conformational changes in human AT1R with blocker ZD7155 (PDB ID: 4YAY, green), β-arrestin biased ligand TRV026 (PDB ID: 60S2, yellow) and endogenous agonist AngII (PDB ID: 6OS0, pink). In the active state, TM5 and TM6 move out of AT1R whereas TM7 moves inward of the receptor. Helix 8 adopts a position parallel to the membrane compared to the inactive conformation bent away from the membrane. Viewing from the extracellular side, TM5 and TM7 move inward of inactive AT1R. B Superimposed structural details of AT1R induced by different ligands. The bulky phenylalanine of AngII at position 8 pushes L1123.36 inward and Y2927.43 in a relocation. A hydrogen bond between N2957.46 and N1113.35 breaks, and the two residuces move inward. However, due to TRV026 being less deeply into the binding pocket of AT1R, it has very little effect on movement of these residuces except for N2957.46. C The alternative conformation is chosen when TM7 points toward TM3, and the canonical active conformation is chosen when TM7 points toward TM2.
Based on the crystal structures, Suomivuori et al. revealed the existence of two active conformations of AT1R by MD simulations: the classical conformation and the alternative conformation. The classical conformation can bind to both Gq and β-arrestin, while the alternative conformation only binds to β-arrestin. The main difference was the orientation of TM7: the classical conformation is preferred when TM7 points to TM2; the alternative conformation is preferred when TM7 points to TM3, forming an allosteric network (Fig. 3C). In the alternative conformation, TM7 is twisted counterclockwise above the proline kink and the intracellular portion is moved toward TM3, forming a hydrogen bond between Asn461.50 and Cys2967.47. The side chains of Tyr3027.53 and Arg1263.50 rotate downward, clashing with the α5 helix of Gq and the alternative conformation readily accommodates the β-arrestin finger loop. During the simulation, the Gq-biased agonist adopted the horizontal F8 orientation more frequently than AngII. In addition, the lack of positively charged residues in the second position prevented binding to negatively charged pocket residues Asp2636.58 and Asp2817.32 with less restriction on TM6 and greater tendency for the extracellular end of TM6 to move outward [77]. Structural allostery in AT1R-biased agonists provides a plausible explanation for their effects.
Allosteric mode of dimers
GPCRs are known to form homodimers and heterodimers [8]. Homodimers consist of receptor subtypes of the same family. Heterodimers consist of two different types of receptors or different subtypes of the same receptor [78]. It has been shown that a negative allosteric regulation occurs between the promoters of AT1R homodimer [79]. Heterodimers, with at least two orthosteric sites and two allosteric sites, are more complex than monomers or homodimers. The formation of heterodimers provides an opportunity for their respective ligands to exert reinforcing/antagonistic effects, or even generate new signaling pathways [80].
Some receptors that form heterodimers with AT1R can allosterically enhance the potency of AT1R signaling (Fig. 4A). The first receptor to be found to heterodimerize with AT1R was the bradykinin B2 receptor (B2R). The formation of AT1R-B2R enhanced the effect of AngII and triggered the symptoms of preeclampsia in pregnant mice [81, 82]. In addition, heterodimer formation by AT1R and alpha 1D adrenoceptors was barely detected in healthy pregnant rats, but was abundantly found in preeclamptic rats [83]. AngII and thrombin are two key regulators of vascular homeostasis. It has been found that AT1R can interact with the prothrombin receptor (PAR1). Simultaneous activation of the two receptors produced synergistic effects, suggesting positive allosteric interactions. AT1R-PAR1 can be a therapeutic target for coagulation disorder in patients with essential hypertension [84]. Using the appropriate technology and developing drugs that target the reinforcing effect of dimers can greatly improve clinical efficacy and reduce side effects.

Allostery of AT1R dimers. A The heterodimers formed by AT1R with other 7TM receptors could enhance or B decrease signaling capabilities. C New signaling of AT1R/α2CAR heterodimer. D Asymmetry of AT1R/FP heterodimer. E The interface between the homodimer of AT1R (PDB ID: 6do1) is constituted by ECL1, TM1, TM2, TM3, and Helix 8.
Some receptors form heterodimers with AT1R and allosterically antagonize receptor function (Fig. 4B). Angiotensin II type 2 receptor (AT2R) inhibits AngII-induced signaling and antagonizes AT1R in terms of vascular tone and proliferative migration [85, 86]. Ang [1,2,3,4,5,6,7] exerts vasodilatory and anti-proliferative effects by binding to MAS receptors encoded by mas proto-oncogenes. MAS receptors inhibit the fuction of AngII by forming heterodimers with AT1R [87]. As an endogenous negative regulator of RAAS, the Apelin-angiotensin receptor-like (APJ) form a heterodimer with AT1R and exert a negative allosteric effect on AngII signaling [88].
Cross-antagonism is a specific form of dimeric allosteric regulation. Single receptor antagonists effectively inhibit both downstream signaling and receptor trafficking. This effect does not interfere with the binding efficacy of the ligand to the receptor, but diminishes the efficacy of one monomer by binding to the other [89]. A dimer was found between AT1R and dopamine D2 receptor (D2R) in the striatum, and antagonists of AT1R inhibited D2R-mediated signaling [90]. Similarly, in the striatum, the combined use of two receptor antagonists inhibited the effects produced by the heterodimerization of adenosine A2A receptor (A2AR) and AT1R. These findings can be helpful for the treatment of tardive dyskinesia (TD) [91]. However, AT1R antagonist losartan enhanced the interaction between AT1R and dopamine D1 receptor (D1R). It might exert anti-hypertensive effects by allosteric enhancement of D1R signaling [92]. After clarifying the allosteric antagonistic function of the dimer, certain drugs may be clinically re-purposed for their unexpected therapeutic effects.
New signaling pathways different from those of monomers or asymmetric signals may form for dimers. In most cases, signaling regulation of dimerization refers to enhanced or diminished activation of the G protein originally coupled to each receptor, but it is possible to form new G protein couplings. A study showed that α2C-adrenergic receptor (α2CAR) with NE as its ligand and AT1R with AngII as its ligand could form a heterodimer. However, the concurrent presence of both agonists promoted a new conformation of the dimer, triggering a new form of Gs/cAMP/PKA signaling. It can be a potential new pharmacological target for the treatment of arterial hypertension (HT) and heart failure (HF) [93] (Fig. 4C). AT1R and prostaglandin receptor (FP) formed a heterodimeric complex in HEK293 and vascular smooth muscle cells, forming a new allosteric signaling. It displayed the symmetric and asymmetric signaling behavior depending on how each of the two receptors was affected by agonists or antagonists. Symmetric signaling refers to the fact that one antagonist attenuates the agonistic effect induced by the agonist of another receptor, and one agonist increases the agonistic effect induced by the agonist of another receptor. Asymmetric signaling indicated that antagonists of AT1R greatly enhanced FP-dependent ERK1/2 signaling but had no effect on prostaglandin F2α (PGF2α)-induced cell growth. In addition, antagonists of FP increased the affinity of AngII to AT1R, but inhibited AngII-induced cell growth, and MAPK signaling remained unchanged [94] (Fig. 4D). The emergence of new pathways means the emergence of new therapeutic targets.
The future of drug design based on AT1R: finding the allosteric sites
Different binding sites of allosteric modulators can lead to different conformational changes of receptor; therefore, the pharmacological effects will be different. A key step in the discovery of new allosteric modulators is the identification of efficient allosteric sites. Due to the lack of visibility of the crystal structure, it is difficult to identify the allosteric sites that are hidden during conformational changes [95]. The discovery of allosteric pockets at protein-protein interfaces is also challenging. The characterization and identification of potential allosteric sites have been considerably improved by rapid progress in kinetic studies and bioinformatics [13, 14].
Finding the allosteric pockets of AT1R during conformational change
Structural recognition of AT1R facilitates the study of allosteric pockets and the design of allosteric modulators. The transition of receptors from inactivated to an activated state exposes several hidden sites. MD simulations are powerful tools for finding these hidden sites [95]. Two allosteric pockets of AT1R have been identified. Firstly, ECL2 of AT1R is a good design target for allosteric modulators due to its high flexibility and variability [96, 97]. A significant conformational change in ECL2 was discovered from the inactivated to the activated state through MD simulations. Researchers also identified druggable allosteric pockets surrounding the AT1R-AAs epitope. Through high-throughput virtual screening, researchers identified DCP1, a small molecule negative conformational modulator targeting the allosteric pocket, providing a model for the search of other GPCR-AAs inhibitors to intervene in GPCR-AAs-related diseases [98] (Fig. 2B–D). Secondly, the Markov state model (MSM) revealed a mysterious allosteric pocket during AT1R activation and uncovered the kinetic nature of the transition between conformational states. P6 (F1.48, L1.52, I1.57 N7.49, P7.50, F7.55, K8.49, F8.50, K8.51, Y8.53, F8.54) is a hidden allosteric pocket that transiently exists during dynamic conformational changes. Hidden between TM7 and H8, P6 was only observed during the upward movement of H8. Mutation of this hidden allosteric site can allosterically impair the downstream G protein and β-arrestin signals, helping the design of AT1R allosteric modulators [99] (Fig. 2E). However, these allosteric pockets still require extensive in vivo experiments for validation.
Finding the allosteric pockets based on the interaction interface of AT1R dimers
In order to successfully design and screen therapeutic agents targeting specific dimers, it is critical to elucidate structural details, such as dimer interaction interfaces and residues [59]. The dimer interaction interface propagates energy perturbations at certain sites of the receptor to neighboring receptors, mediating the synergistic effects of receptors [100]. Allosteric agonists/inhibitors modulate the activation of receptors by acting on the allosteric pocket at the dimer interaction interface [101]. Currently, there is no report of allosteric modulators based on AT1R dimers. Therefore, it is important to identify the interface of specific interactions. Due to the complexity of heterodimers, there is no crystal structure of AT1R heterodimers for interaction interface studies. The interaction interfaces of AT1R homodimer have been well studied. TM4 was central to the study of AT1R homodimers, but mutations in one face of TM4 or both faces of TM4 were not sufficient to completely disrupt homologous AT1R interactions. It has been proposed that at least two interaction interfaces are essential for AT1R homodimer: TM4,5 and TM6,7 [102]. With the help of MD simulations, the four most reasonable mutual interfaces have been proposed: symmetric TM1,2,8, TM5, TM4, and TM4,5 [103]. The only report on AT1R homodimer structure (PDB ID: 6do1) indicates that the interface between individual protomers consists of hydrophobic and aromatic amino acid side chain contacts at ECL1, TM1, TM2, TM3, and H8 [104] (Fig. 4E). Interestingly, AT1R homodimers mediated β-arrestin signaling, but not Gq signaling [105]. The formation of homodimer interfaces may allosterically modulate the downstream signal. Some researchers proposed a “rolling dimer” interface model, in which several conformations of the dimer coexist and interconvert [106], providing a new concept for dimer interaction interface in physiological conditions.
Conclusion
Classical and non-classical allostery are critical pathways depending on GPCR activation, which creates unlimited possibilities of cellular and biological functions. Distinctively, biased agonists and dimers of AT1R both exert unique functions through allosteric modulation, alter ligand recognition patterns, and change receptor core cascade and downstream signal transduction. In particular, the discovery of allosteric regulators improved our understanding of the signaling and functional diversity of AT1R in physiological and pathological conditions. The discovery of allosteric sites provides new opportunities for developing allosteric drugs. Despite great progress in the structural identification of AT1R, the structures of non-antibody allosteric activators in complex with AT1R, especially the extracellular loops, are yet unknown and do not meet the current needs for allosteric drug development. In addition, the bidirectional communication in the allosteric regulation of AT1R needs more studies. Currently, the development of allosteric modulators of GPCR is in progress. Computational biology, bioinformatics, and experimental methods can accelerate the development of allosteric modulators targeting AT1R.
Availability of data and materials
All data are included in the manuscript.
Abbreviations
- AT1R:
-
Angiotensin II type 1 receptor
- ARB:
-
AT1R blocker
- GPCRs:
-
G protein-coupled receptors
- TM:
-
Transmembrane
- ECL1-3 and ICL1-3:
-
Extra- and intracellular loops
- H8:
-
Helix 8
- PAM:
-
Positive allosteric modulators
- NAM:
-
Negative allosteric modulators
- NAL:
-
Neutral allosteric ligands
- ago-PAM:
-
Ago-allosteric modulator
- BAM:
-
Biased allosteric modulators
- AT1R-AAs:
-
AT1R autoantibodies
- MD:
-
Molecular dynamics
- COMP:
-
Cartilage oligomeric matrix protein
- TSHR:
-
Thyroid-stimulating hormone receptor
- CAMs:
-
Constitutively active mutants
- FP:
-
Prostaglandin receptor
- α2CAR:
-
α2C-adrenergic receptor
References
Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16(12):829–42.
Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov. 2002;1(8):621–36.
Thomas WG, Mendelsohn FA. Angiotensin receptors: form and function and distribution. Int J Biochem Cell Biol. 2003;35(6):774–9.
Sleno R, Hebert TE. The Dynamics of GPCR oligomerization and their functional consequences. Int Rev Cell Mol Biol. 2018;338:141–71.
Grundmann M, Bender E, Schamberger J, Eitner F. Pharmacology of free fatty acid receptors and their allosteric modulators. Int J Mol Sci. 2021;22(4):1763.
Renault P, Giraldo J. Dynamical correlations reveal Allosteric Sites in G protein-coupled receptors. Int J Mol Sci. 2020;22(1):187.
Thal DM, Glukhova A, Sexton PM, Christopoulos A. Structural insights into G-protein-coupled receptor allostery. Nature. 2018;559(7712):45–53.
Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev. 2010;62(4):701–25.
Farran B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol Res. 2017;117:303–27.
Casado-Anguera V, Casado V. Unmasking allosteric-binding sites: novel targets for GPCR drug discovery. Expert Opin Drug Discov. 2022. https://doi.org/10.1080/17460441.2022.2085684.
Gurevich VV, Gurevich EV. GPCR Signaling Regulation: the role of GRKs and arrestins. Front Pharmacol. 2019;10:125.
May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51.
Lu S, He X, Ni D, Zhang J. Allosteric modulator discovery: from serendipity to structure-based design. J Med Chem. 2019;62(14):6405–21.
He X, Ni D, Lu S, Zhang J. Characteristics of allosteric proteins, sites, and modulators. Adv Exp Med Biol. 2019;1163:107–39.
Jakubik J, El-Fakahany EE. Allosteric modulation of GPCRs of class A by cholesterol. Int J Mol Sci. 2021;22(4):1953.
Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov. 2013;12(8):630–44.
Lu S, Zhang J. Small molecule allosteric modulators of G-Protein-coupled receptors: drug-target interactions. J Med Chem. 2019;62(1):24–45.
Wakefield AE, Bajusz D, Kozakov D, Keseru GM, Vajda S. Conservation of allosteric ligand binding sites in G-Protein coupled receptors. J Chem Inf Model. 2022. https://doi.org/10.1021/acs.jcim.2c00209.
Cong Z, Chen LN, Ma H, Zhou Q, Zou X, Ye C, et al. Molecular insights into ago-allosteric modulation of the human glucagon-like peptide-1 receptor. Nat Commun. 2021;12(1):3763.
Slosky LM, Caron MG, Barak LS. Biased allosteric modulators: New Frontiers in GPCR Drug Discovery. Trends Pharmacol Sci. 2021;42(4):283–99.
Skiba MA, Kruse AC. Autoantibodies as endogenous modulators of GPCR Signaling. Trends Pharmacol Sci. 2021;42(3):135–50.
Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103(7):945–52.
Zhang S, Zhang X, Yang L, Yan Z, Yan L, Tian J, et al. Increased susceptibility to metabolic syndrome in adult offspring of angiotensin type 1 receptor autoantibody-positive rats. Antioxid Redox Signal. 2012;17(5):733–43.
Fu ML, Herlitz H, Schulze W, Wallukat G, Micke P, Eftekhari P, et al. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens. 2000;18(7):945–53.
Tian M, Sheng L, Huang P, Li J, Zhang CH, Yang J, et al. Agonistic autoantibodies against the angiotensin AT1 receptor increase in unstable angina patients after stent implantation. Coron Artery Dis. 2014;25(8):691–7.
Taniguchi M, Rebellato LM, Cai J, Hopfield J, Briley KP, Haisch CE, et al. Higher risk of kidney graft failure in the presence of anti-angiotensin II type-1 receptor antibodies. Am J Transplant. 2013;13(10):2577–89.
Abadir PM, Jain A, Powell LJ, Xue QL, Tian J, Hamilton RG, et al. Discovery and Validation of Agonistic angiotensin receptor autoantibodies as biomarkers of adverse outcomes. Circulation. 2017;135(5):449–59.
Giil LM, Kristoffersen EK, Vedeler CA, Aarsland D, Nordrehaug JE, Winblad B, et al. Autoantibodies toward the angiotensin 2 type 1 receptor: a Novel Autoantibody in Alzheimer’s Disease. J Alzheimers Dis. 2015;47(2):523–9.
Jiang Y, Duffy F, Hadlock J, Raappana A, Styrchak S, Beck I, et al. Angiotensin II receptor I auto-antibodies following SARS-CoV-2 infection. PLoS ONE. 2021;16(11):e0259902.
Labandeira CM, Pedrosa MA, Suarez-Quintanilla JA, Cortes-Ayaso M, Labandeira-Garcia JL, Rodriguez-Perez AI. Angiotensin system autoantibodies correlate with routine prognostic indicators for COVID-19 severity. Front Med 2022;9:840662.
Cabral-Marques O, Halpert G, Schimke LF, Ostrinski Y, Vojdani A, Baiocchi GC, et al. Autoantibodies targeting GPCRs and RAS-related molecules associate with COVID-19 severity. Nat Commun. 2022;13(1):1220.
Lefkowitz RJ. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J Biol Chem. 1998;273(30):18677–80.
Shenoy SK, Lefkowitz RJ. Angiotensin II-stimulated signaling through G proteins and beta-arrestin. Sci STKE. 2005;2005(311):cm14.
Bian J, Lei J, Yin X, Wang P, Wu Y, Yang X, et al. Limited AT1 receptor internalization is a novel mechanism underlying sustained Vasoconstriction Induced by AT1 receptor Autoantibody from Preeclampsia. J Am Heart Assoc. 2019;8(6):e011179.
Philogene MC, Johnson T, Vaught AJ, Zakaria S, Fedarko N. Antibodies against angiotensin II type 1 and endothelin A receptors: relevance and pathogenicity. Hum Immunol. 2019;80(8):561–7.
Bajwa SF, Mohammed RHA. Type II Hypersensitivity Reaction. StatPearls. Treasure Island (FL)2022.
Zhang W, Zheng Y, Liu F, Wang X, Jin Z, Zhi J. Mechanism of agonistic angiotensin II type I receptor autoantibody-amplified contractile response to Ang II in the isolated rat thoracic aorta. Acta Biochim Biophys Sin (Shanghai). 2015;47(10):851–6.
Wenzel K, Rajakumar A, Haase H, Geusens N, Hubner N, Schulz H, et al. Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension. 2011;58(1):77–84.
Brewer J, Liu R, Lu Y, Scott J, Wallace K, Wallukat G, et al. Endothelin-1, oxidative stress, and endogenous angiotensin II: mechanisms of angiotensin II type I receptor autoantibody-enhanced renal and blood pressure response during pregnancy. Hypertension. 2013;62(5):886–92.
Zhang H, Luginina A, Mishin A, Baidya M, Shukla AK, Cherezov V. Structural insights into ligand recognition and activation of angiotensin receptors. Trends Pharmacol Sci. 2021;42(7):577–87.
Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, et al. Angiotensin II Signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98(3):1627–738.
Papola F, Biancofiore V, Angeletti C, Grimaldi A, Carucci AC, Cofini V, et al. Anti-AT1R autoantibodies and prediction of the severity of Covid-19. Hum Immunol. 2022;83(2):130–3.
Xiao F, Puddefoot JR, Barker S, Vinson GP. Changes in angiotensin II type 1 receptor signalling pathways evoked by a monoclonal antibody raised to the N-terminus. J Endocrinol. 2008;197(1):25–33.
Wingler LM, McMahon C, Staus DP, Lefkowitz RJ, Kruse AC. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell. 2019;176(3):479–90.
Wingler LM, Skiba MA, McMahon C, Staus DP, Kleinhenz ALW, Suomivuori CM, et al. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science. 2020;367(6480):888–92.
Ali A, Palakkott A, Ashraf A, Al Zamel I, Baby B, Vijayan R, et al. Positive modulation of angiotensin II type 1 receptor-mediated signaling by LVV-Hemorphin-7. Front Pharmacol. 2019;10:1258.
Ali A, Johnstone EKM, Baby B, See HB, Song A, Rosengren KJ, et al. Insights into the interaction of LVV-Hemorphin-7 with angiotensin II type 1 receptor. Int J Mol Sci. 2020;22(1):209.
Li T, Yu B, Liu Z, Li J, Ma M, Wang Y, et al. Homocysteine directly interacts and activates the angiotensin II type I receptor to aggravate vascular injury. Nat Commun. 2018;9(1):11.
Zhang D, Liu Y, Zaidi SA, Xu L, Zhan Y, Chen A, et al. Structural insights into angiotensin receptor signaling modulation by balanced and biased agonists. EMBO J. 2023. https://doi.org/10.15252/embj.2022112940.
Kiriakidi S, Kolocouris A, Liapakis G, Ikram S, Durdagi S, Mavromoustakos T. Effects of cholesterol on GPCR function: insights from computational and experimental studies. Adv Exp Med Biol. 2019;1135:89–103.
Fu Y, Huang Y, Yang Z, Chen Y, Zheng J, Mao C, et al. Cartilage oligomeric matrix protein is an endogenous beta-arrestin-2-selective allosteric modulator of AT1 receptor counteracting vascular injury. Cell Res. 2021;31(7):773–90.
Gao N, Liang T, Yuan Y, Xiao X, Zhao Y, Guo Y, et al. Exploring the mechanism of F282L mutation-caused constitutive activity of GPCR by a computational study. Phys Chem Chem Phys. 2016;18(42):29412–22.
Wang Q, Dong X, Lu J, Hu T, Pei G. Constitutive activity of a G protein-coupled receptor, DRD1, contributes to human cerebral organoid formation. Stem Cells. 2020;38(5):653–65.
Tang W, Strachan RT, Lefkowitz RJ, Rockman HA. Allosteric modulation of beta-arrestin-biased angiotensin II type 1 receptor signaling by membrane stretch. J Biol Chem. 2014;289(41):28271–83.
Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28(8):416–22.
Liu Y, Yang Y, Ward R, An S, Guo XX, Li W, et al. Biased signalling: the instinctive skill of the cell in the selection of appropriate signalling pathways. Biochem J. 2015;470(2):155–67.
Chen H, Zhang S, Zhang X, Liu H. QR code model: a new possibility for GPCR phosphorylation recognition. Cell Commun Signal. 2022;20(1):23.
Bock A, Bermudez M. Allosteric coupling and biased agonism in G protein-coupled receptors. FEBS J. 2021;288(8):2513–28.
Schonenbach NS, Hussain S, O’Malley MA. Structure and function of G protein-coupled receptor oligomers: implications for drug discovery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7(3):408–27.
Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165(6):1659–69.
Goupil E, Laporte SA, Hebert TE. GPCR heterodimers: asymmetries in ligand binding and signalling output offer new targets for drug discovery. Br J Pharmacol. 2013;168(5):1101–3.
Unal H, Karnik SS. Constitutive activity in the angiotensin II type 1 receptor: discovery and applications. Adv Pharmacol. 2014;70:155–74.
Bonde MM, Hansen JT, Sanni SJ, Haunso S, Gammeltoft S, Lyngso C, et al. Biased signaling of the angiotensin II type 1 receptor can be mediated through distinct mechanisms. PLoS ONE. 2010;5(11):e14135.
Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3(125):ra46.
Jiang G, Gong H, Niu Y, Yang C, Wang S, Chen Z, et al. Identification of amino acid residues in angiotensin II type 1 receptor sensing mechanical stretch and function in cardiomyocyte hypertrophy. Cell Physiol Biochem. 2015;37(1):105–16.
Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, et al. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61(4):768–77.
Hostrup A, Christensen GL, Bentzen BH, Liang B, Aplin M, Grunnet M, et al. Functionally selective AT(1) receptor activation reduces ischemia reperfusion injury. Cell Physiol Biochem. 2012;30(3):642–52.
Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, et al. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA. 2006;103(44):16284–9.
Tarigopula M, Davis RT, Mungai PT, Ryba DM, Wieczorek DF, Cowan CL, et al. Cardiac myosin light chain phosphorylation and inotropic effects of a biased ligand, TRV120023, in a dilated cardiomyopathy model. Cardiovasc Res. 2015;107(2):226–34.
Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, et al. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335(3):572–9.
Boerrigter G, Lark MW, Whalen EJ, Soergel DG, Violin JD, Burnett JC. Jr. Cardiorenal actions of TRV120027, a novel ss-arrestin-biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ Heart Fail. 2011;4(6):770–8.
Kashihara T, Kawagishi H, Nakada T, Numaga-Tomita T, Kadota S, Wolf EE, et al. beta-arrestin-biased AT1 agonist TRV027 causes a neonatal-specific sustained positive Inotropic Effect without increasing Heart Rate. JACC Basic Transl Sci. 2020;5(11):1057–69.
Wu H, Sun Q, Yuan S, Wang J, Li F, Gao H, et al. AT1 receptors: their actions from hypertension to cognitive impairment. Cardiovasc Toxicol. 2022;22(4):311–25.
Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, et al. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100(19):10782–7.
Fillion D, Cabana J, Guillemette G, Leduc R, Lavigne P, Escher E. Structure of the human angiotensin II type 1 (AT1) receptor bound to angiotensin II from multiple chemoselective photoprobe contacts reveals a unique peptide binding mode. J Biol Chem. 2013;288(12):8187–97.
Singh KD, Unal H, Desnoyer R, Karnik SS. Mechanism of hormone peptide activation of a GPCR: angiotensin II activated state of AT1R initiated by van der Waals Attraction. J Chem Inf Model. 2019;59(1):373–85.
Suomivuori CM, Latorraca NR, Wingler LM, Eismann S, King MC, Kleinhenz ALW, et al. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science. 2020;367(6480):881–7.
Kenakin T. G protein coupled receptors as allosteric proteins and the role of allosteric modulators. J Recept Signal Transduct Res. 2010;30(5):313–21.
Szalai B, Barkai L, Turu G, Szidonya L, Varnai P, Hunyady L. Allosteric interactions within the AT(1) angiotensin receptor homodimer: role of the conserved DRY motif. Biochem Pharmacol. 2012;84(4):477–85.
Kleinau G, Muller A, Biebermann H. Oligomerization of GPCRs involved in endocrine regulation. J Mol Endocrinol. 2016;57(1):R59–80.
AbdAlla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature. 2000;407(6800):94–8.
Quitterer U, Fu X, Pohl A, Bayoumy KM, Langer A, AbdAlla S. Beta-Arrestin1 prevents preeclampsia by downregulation of mechanosensitive AT1-B2 receptor heteromers. Cell. 2019;176(1–2):318–33.
Gonzalez-Hernandez Mde L, Godinez-Hernandez D, Bobadilla-Lugo RA, Lopez-Sanchez P. Angiotensin-II type 1 receptor (AT1R) and alpha-1D adrenoceptor form a heterodimer during pregnancy-induced hypertension. Auton Autacoid Pharmacol. 2010;30(3):167–72.
Zamel IA, Palakkott A, Ashraf A, Iratni R, Ayoub MA. Interplay between angiotensin II type 1 receptor and thrombin receptor revealed by Bioluminescence Resonance Energy transfer assay. Front Pharmacol. 2020;11:1283.
AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem. 2001;276(43):39721–6.
Inuzuka T, Fujioka Y, Tsuda M, Fujioka M, Satoh AO, Horiuchi K, et al. Attenuation of ligand-induced activation of angiotensin II type 1 receptor signaling by the type 2 receptor via protein kinase C. Sci Rep. 2016;6:21613.
Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, Sexton PM, et al. G-protein-coupled receptor mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation. 2005;111(14):1806–13.
Siddiquee K, Hampton J, McAnally D, May L, Smith L. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br J Pharmacol. 2013;168(5):1104–17.
Barki-Harrington L, Luttrell LM, Rockman HA. Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation. 2003;108(13):1611–8.
Martinez-Pinilla E, Rodriguez-Perez AI, Navarro G, Aguinaga D, Moreno E, Lanciego JL, et al. Dopamine D2 and angiotensin II type 1 receptors form functional heteromers in rat striatum. Biochem Pharmacol. 2015;96(2):131–42.
Oliveira PA, Dalton JAR, Lopez-Cano M, Ricarte A, Morato X, Matheus FC, et al. Angiotensin II type 1/adenosine a 2A receptor oligomers: a novel target for tardive dyskinesia. Sci Rep. 2017;7(1):1857.
Li D, Scott L, Crambert S, Zelenin S, Eklof AC, Di Ciano L, et al. Binding of losartan to angiotensin AT1 receptors increases dopamine D1 receptor activation. J Am Soc Nephrol. 2012;23(3):421–8.
Bellot M, Galandrin S, Boularan C, Matthies HJ, Despas F, Denis C, et al. Dual agonist occupancy of AT1-R-alpha2C-AR heterodimers results in atypical Gs-PKA signaling. Nat Chem Biol. 2015;11(4):271–9.
Goupil E, Fillion D, Clement S, Luo X, Devost D, Sleno R, et al. Angiotensin II type I and prostaglandin F2alpha receptors cooperatively modulate signaling in vascular smooth muscle cells. J Biol Chem. 2015;290(5):3137–48.
Rehman AU, Lu S, Khan AA, Khurshid B, Rasheed S, Wadood A, et al. Hidden allosteric sites and de-novo drug design. Expert Opin Drug Discov. 2022;17(3):283–95.
Woolley MJ, Conner AC. Understanding the common themes and diverse roles of the second extracellular loop (ECL2) of the GPCR super-family. Mol Cell Endocrinol. 2017;449:3–11.
Khoury E, Clement S, Laporte SA. Allosteric and biased g protein-coupled receptor signaling regulation: potentials for new therapeutics. Front Endocrinol (Lausanne). 2014;5:68.
Singh KD, Jara ZP, Harford T, Saha PP, Pardhi TR, Desnoyer R, et al. Novel allosteric ligands of the angiotensin receptor AT1R as autoantibody blockers. Proc Natl Acad Sci USA. 2021;118:33.
Lu S, He X, Yang Z, Chai Z, Zhou S, Wang J, et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat Commun. 2021;12(1):4721.
Guidolin D, Tortorella C, Marcoli M, Cervetto C, Maura G, Agnati LF. Receptor-receptor interactions and glial cell functions with a special focus on G protein-coupled receptors. Int J Mol Sci. 2021;22(16):8656.
Liu L, Fan Z, Rovira X, Xue L, Roux S, Brabet I, et al. Allosteric ligands control the activation of a class C GPCR heterodimer by acting at the transmembrane interface. Elife. 2021. https://doi.org/10.7554/eLife.70188.
Young BM, Nguyen E, Chedrawe MAJ, Rainey JK, Dupre DJ. Differential contribution of transmembrane domains IV, V, VI, and VII to human angiotensin II type 1 receptor homomer formation. J Biol Chem. 2017;292(8):3341–50.
Erol I, Cosut B, Durdagi S. Toward understanding the impact of dimerization interfaces in angiotensin II type 1 receptor. J Chem Inf Model. 2019;59(10):4314–27.
Speck D, Kleinau G, Szczepek M, Kwiatkowski D, Catar R, Philippe A, et al. Angiotensin and endothelin receptor structures with implications for signaling regulation and pharmacological targeting. Front Endocrinol (Lausanne). 2022;13:880002.
Hansen JL, Theilade J, Haunso S, Sheikh SP. Oligomerization of wild type and nonfunctional mutant angiotensin II type I receptors inhibits galphaq protein signaling but not ERK activation. J Biol Chem. 2004;279(23):24108–15.
Dijkman PM, Castell OK, Goddard AD, Munoz-Garcia JC, de Graaf C, Wallace MI, et al. Dynamic tuneable G protein-coupled receptor monomer-dimer populations. Nat Commun. 2018;9(1):1710.
Acknowledgements
Not applicable.
Funding
This work was supported by the Major Program of the National Natural Science Foundation of China [91539205] and the National Natural Science Foundation of China [81900415].
Author information
Authors and Affiliations
Contributions
XZ wrote the manuscript. SZ discussed and revised this manuscript. MW supplied materials. HC provided writing assistance for this article. HL conceived and revised the article.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, X., Zhang, S., Wang, M. et al. Advances in the allostery of angiotensin II type 1 receptor. Cell Biosci 13, 110 (2023). https://doi.org/10.1186/s13578-023-01063-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-023-01063-x