
Abstract
Alternative splicing (AS) regulates gene expression patterns at the post-transcriptional level and generates a striking expansion of coding capacities of genomes and cellular protein diversity. RNA splicing could undergo modulation and close interaction with genetic and epigenetic machinery. Notably, during the adipogenesis processes of white, brown and beige adipocytes, AS tightly interplays with the differentiation gene program networks. Here, we integrate the available findings on specific splicing events and distinct functions of different splicing regulators as examples to highlight the directive biological contribution of AS mechanism in adipogenesis and adipocyte biology. Furthermore, accumulating evidence has suggested that mutations and/or altered expression in splicing regulators and aberrant splicing alterations in the obesity-associated genes are often linked to humans’ diet-induced obesity and metabolic dysregulation phenotypes. Therefore, significant attempts have been finally made to overview novel detailed discussion on the prospects of splicing machinery with obesity and metabolic disorders to supply featured potential management mechanisms in clinical applicability for obesity treatment strategies.
Similar content being viewed by others


Introduction
Alternative splicing (AS) is a crucial post-transcriptional mechanism to reprogram gene expression profiles and expand transcriptomic and proteomic diversity in eukaryotic organisms [1, 2]. This highly dynamic process alternatively removes introns from a transcribed precursor messenger RNA (pre-mRNA) and combines various exons to facilitate one single protein-coding gene to finally form variable forms of mature mRNA species [2]. About 95% of human multi-exon genes can undergo splicing process [3]; nevertheless, a large majority of alternatively spliced variants may not be translated into proteins, so very few annotated alternative isoforms can be detected in large-scale proteomics studies [4].
Numerous alternatively spliced transcripts are elicited in a cell-type-specific manner. Indeed, extensive AS programs are frequent during early embryonic development and function pivotal regulatory roles in cell fate determination and differentiation, organogenesis, and tissue development [5, 6]. The meticulous expression of AS profiles maintains tissue identity and function, while the temporal expression switch between splicing variants usually promotes cell differentiation and tissue development [7]. Importantly, dysregulated AS pathways often employ aberrant biological actions causing various disease conditions in the human body [8].
In this review, we accumulate comprehensive insights into the AS regulatory role in adipogenesis based on the available studies from 2002 to 2020. We put a large emphasis on the reported adipose developmental-stage-specific alternative splicing events (ASEs) and the integrated regulatory role of different splicing regulators during adipogenic differentiation processes in white, brown, and beige adipocytes, respectively. We also explore recent advances in the association of the splicing program with the pathophysiological obese condition and metabolic disorders. In summary, our review is an exclusive rendition of three main aspects: how AS exerts a profound influence on adipocyte development, and how the pathologic roles of alternative RNA splicing variations operate in obesity and related metabolic disorders, and great emphasis on discussing the application of AS in the form of viable therapeutic option for combating obesity and obesity-associated harmful chronic diseases.
Alternative RNA splicing and adipogenesis
Regulatory mechanism of cellular alternative splicing process
Different RNA splicing modes give rise to different mRNA transcripts with varying coding potential, untranslated regions (UTRs), or RNA stability. Substantially, the premRNA splicing outcomes can be determined under complex mechanisms involving splicing factors, transcriptional machinery, epigenetic modifications, and genomic mutations/single-nucleotide polymorphisms (SNPs) (Fig. 1).

The paradigmatic splicing regulation mechanisms in precursor messenger RNA (pre-mRNA). Generally, AS has complex correlations with splicing factors, transcriptional machinery (RNA polymerase II elongation rates), and epigenetic modifications (histone marks, epigenetic modifiers, and DNA methylation) at the post- and co-transcriptional levels. During AS process, spliceosome, a highly dynamic ribonucleoprotein complex mainly composed of five different small nuclear ribonucleoprotein (snRNP) complexes (U1, U2, and the tri-snRNP U4/U6. U5 structure), can function in splice sites recognition and reaction on pre-mRNA molecules. The spliceosome assembly process starts with the recognition of the initial 5′ splice site by E complex containing U1 snRNP at the GU motif and the identification of 3′ splice site by three interacting proteins-U2 small nuclear RNA auxiliary factor 1 (U2AF1), U2 small nuclear RNA auxiliary factor 2 (U2AF2), and splicing factor 1 (SF1); then the A complex of U2 snRNP binds to the branch site whose key protein component is splicing factor 3 subunit B1 (SF3B1); finally, the U4/U6. U5 tri-snRNP, forming B complex, triggers the core catalytic reaction of spliceosome. During this process, splicing factors target and interplay with spliceosome components to regulate 5′ and 3′ splice-site recognition flanking the alternative exon, such as serine/arginine-rich (SR) proteins and heterogeneous ribonucleoproteins (hnRNPs). The SR proteins are general splicing activators via binding to exonic/intronic splicing enhancers (E/ISEs) to facilitate exon formation, whereas hnRNPs are general splicing inhibitors via binding to exonic/intronic splicing silencers (E/ISSs) to interfere with the splice site recognition
Splicing-site selection regulatory mechanism
AS decisions are influenced by global interactions between cisacting RNA sequences and the surrounding sequence contexts (including exonic/intronic splicing enhancers (E/ISEs) or silencers (E/ISSs), 5′ and 3′ splice sites) and their binding transregulatory factors (e.g., splicing factors, etc.) [9, 10]. Indeed, RNA-binding proteins (RBPs), accounting for most splicing factors, can exert different regulatory functions in splice-site choices under a cooperative or antagonistic manner through their expression levels, nuclear localization, mRNA stability, and their own splicing regulated by other RBPs. This process usually exhibits a mechanistic interplay between RBPs with other multiple splicing regulators, such as epigenetic and transcriptional machinery, at the post- and co-transcriptional levels. Moreover, the outcome prediction and identification of genome-wide splicing pattern in specific cell conditions have been proposed to be determined by a “splicing code” (large combinations of RNA features in AS regulation), combing cis-regulatory features and splicing factor binding [11], whose depth and complexity remain to be deciphered.
Epigenetic splicing regulation mechanisms
Histone modifications, epigenetic modifiers, and DNA methylation can function broadly in RNA splice site recognition, spatially and temporally [12,13,14,15,16]. A set of underlying mechanistic modes of particular histone post-translational modifications (hPTMs) in modulating alternative exon splicing (exon inclusion/exclusion) are: (1) through direct recruitment or sequestration of splicing factors [14]; (2) and adaptor proteins [17]; (3) or through indirect transcriptional elongation regulation of the RNA polymerase II (Pol II) [18, 19]. Besides, RNA modifications, such as N6-methyladenosine (m6A), also have functional relevance with the AS regulation process [20, 21]. It was first reported that m6A peaks exhibited more considerable enrichment in multi-isoform genes than in single-isoform ones and in alternatively spliced exons than in constitutive ones [22]. Subsequent results further revealed the main m6A modification-related regulatory mechanisms, involving the binding modulation of splicing factors throughout RNA conformation [23], the binding of m6A readers near the splice sites that affects the recruitment of splicing factors [24], and the splicing-factors-mediated recognition of m6A motifs [25].
Other AS regulation mechanisms
Transcription initiation and Pol II elongation rates greatly influence the RBPs’ recognition to nascent pre-mRNA splice sites of various strengths in a time-dependent manner [26,27,28,29]. Works have underscored the determinative role of kinetic coupling in co-transcriptional splicing reactions that the upstream site events have a competitive advantage over the downstream, especially when elongation rates are reduced [29, 30]. Under the slow Pol II control, weak splice site recognition is favored, and the alternative up-and down-regulation of exon inclusion can be allowed [28]; by contrast, fast Pol II attenuates weak splice site recognition and promotes alternative exon-skipping events [31].
Additionally, AS patterns could also be tightly affected by genetic mutations [32]. According to the Human Gene Mutation Database, SNPs that fall within critical regions of cis-acting splicing elements and splicing factors may display mis-splicing phenotypes [33]. Of note, 22% of disease-causing SNPs, initially identified as missense mutations, appear to be more likely to disrupt the pre-mRNA splicing process than canonical variations; together with the known splicing mutations category, they suggest that about one-third of disease-causing mutations could affect pre-mRNA splicing patterns [33].
Regulatory mechanisms of adipogenesis process
Adipose tissues, including white adipose tissue (WAT), brown adipose tissue (BAT), and beige adipose tissue, lie in the regulatory center of metabolic functions and systemic energy homeostasis, linking closely with multiple physiological processes, such as lipid metabolism, insulin sensitivity, satiety, thermoregulation, and inflammation [34, 35]. Adipose tissues exhibit high plasticity to undergo a two-phase adipogenesis process (early commitment and terminal differentiation) that multipotent mesenchymal precursors restrict their fate to the committed adipocyte lineage (Fig. 2), surging newly insulin-responsive and profoundly influencing metabolic health and energy homeostasis [36,37,38].

Molecular mechanisms of the two-phase adipogenesis process. A common two-phase adipogenesis process is described: early determination and terminal differentiation phases, involving an intricate integration of cytoarchitecture, transcription factors and co-regulators, and signaling pathways. In the first commitment step, mesenchymal progenitors commit their fate to certain preadipocytes exclusively under the restriction control of bone morphogenetic protein (BMP) signaling. Subsequently, during the second adipogenic differentiation step in both white and brown adipocytes, the master regulator of adipogenesis-peroxisome proliferator-activated receptor-γ (PPARγ) is stimulated and synergizes with CCAAT/enhancer-binding protein α (C/EBPα) to fully activate a transcriptional cascade contributing to and maintaining stable maturation of functional adipocytes and to further engage in adipose biology modulation. Significantly, the zinc-finger transcriptional co-regulator PR domain-containing 16 (PRDM16), cooperating with PPARγ and C/EBPs, and PPARG coactivator 1α (PGC-1α) are of fundamental importance to induce mitochondrial biogenesis and BAT-specific genes expression in the brown adipocyte terminal differentiation and white adipocytes browning process. MYF5 myogenic factor 5, BMP2,4,7 bone morphogenetic protein 2,4,7, EBF2 early B-cell factor 2, ZFP423 zinc finger protein 423, TCF7L1 T cell-specific transcription factor 7-like 1, CREB cAMP-response element-binding protein, KLFs Krüppel-like factors
Distinguished from the WAT adipogenesis that mainly occurs in the postnatal period, BAT mainly develops early during embryonic development before birth [39]. Nevertheless, the nature and origin of active beige adipocytes with improved thermogenesis ability is an intensely ongoing debate. Upon environmental cold or high-fat-diet exposure or sympathetic stimulation, white adipocytes can undergo trans-differentiation into brown-like adipocytes dependent on PPARγ [40, 41], or that dormant beige adipocytes in WAT are recruited and activated [42], or that mesenchyme-derived resident precursors are differentiated de novo [43]. Notably, the process of beige adipogenesis to increase the number of metabolically active adipocytes ameliorating metabolism-related profiles is highlighted as a promising therapeutic strategy for obesity and metabolic disorders.
Alternative splicing events implicated in adipogenesis of different adipose tissues
Within the past decades, high-throughput RNA sequencing techniques, such as next-generation sequencing (NGS) of RNA, have become popular tools for studying pre-mRNA ASEs through unbiased assessments. These technologies, generating abundant transcriptomic data, help to identify widespread ASEs and also a new class of adipogenesis-specific splicing profiles. Indeed, annotation of different splicing choices and splicing transitions occurred during the time course of adipocyte development is a common theme in deciphering the AS regulatory mechanism in adipogenesis.
Alternative splicing events in WAT adipogenesis
Mechanistic investigations clearly indicate that distinct AS-originated transcripts of critical adipogenic regulators contribute to the regulation of divergent cell fate determination pathways. A typical example is tension-induced/inhibited proteins (TIPs), known stretch-responsive factors with three alternative spliced isoforms-tension-induced/inhibited protein 1 (TIP-1), 2 (TIP-2), and 3 (TIP-3), which function differently in the cell fate selection of adipogenesis or myogenesis [44]. The TIP-3 isoform recruited by PPARγ2 promoter can stimulate the selection into the adipogenic program, while TIP-1 promotes myogenic differentiation [44]. Moreover, the past decades have also profusely addressed AS to extend the action modes of adipogenic regulators that exert different effects on adipocyte terminal differentiation mediated by switches or alterations in splicing patterns, as mainly summarized in Fig. 3a. Among them, some of the splicing switches can appear with slower kinetics in a developmentally regulated manner during adipogenesis, such as protein kinase C βII (PKCβII) [45] and melanocortin 2 receptor (MC2-R) [46]. Further transcriptomic characterization of differentiating human mesenchymal stem cells (hMSCs) has led to the finding of time-specific AS profiles of this process, which reveals multiple AS types in genes of adipogenic regulators, including cassette exon, alternative to 3′ splice site, and alternative to 5′ splice site, and topological distribution patterns on potential key ASEs [47].

Examples of modulated alternative splicing events implicated in white, brown, and beige adipogenesis
Actually, some of the previously unidentified alternatively spliced variants in specific tissues or cells can be found in extensive analyses of related gene locus through combined approaches of oligonucleotides, cloning for various isoforms, Sanger sequencing, as well as re-analysis on published RNA-sequencing datasets or Genotype-Tissue Expression supported information. Based on these methods, numerous findings exist of the inhibitory effects that splicing alterations in adipogenic genes have on the adipocyte differentiation process. A series of spliced variants in genes of peroxisome proliferator-activated receptor gamma (PPARG) [48,49,50,51], preadipocyte factor-1 (Pref-1) [52], and nuclear factor erythroid 2-related factor 1 (NRF1) [53] in adipogenic differentiation offer excellent examples for this case. Above all, for PPARγ, the necessary “master regulator” of adipogenesis, alternative RNA splicing often serves as a dominant-negative mechanism antagonizing PPARγ biological activity and function. Apart from four main canonical splice transcripts of PPARG (PPARG1, PPARG2, PPARG3, and PPARG4), the PPARG gene regions which encode for DNA-binding domain (DBD) and ligand-binding domain (LBD) containing multiple cis-regulatory elements can generate several inhibitory truncated isoforms-PPARγ1(tr) [48], γORF4 [49, 50], and PPARγ∆5 [51]-by alternative promoter usage and splicing processes (Fig. 4). Substantially, in adipose tissues, these alternative PPARG splice variants show differential ligand specificity and functional relevance during adipogenesis: γORF4, in lack of LBD, suppresses adipogenesis in a stage-specific expression manner [49, 50]; PPARγ∆5, another naturally occurring negative isoform, undergoes exon skipping event modulated by serine and arginine rich splicing factor 1 (SRSF1) leading to a lack of the entire LBD and impairs the PPARγ transcriptional network in a negative feedback mechanism during adipogenesis [51] (Fig. 3a). In addition, AS determines Pref-1 function by regulating the production of the biologically negative soluble isoform during adipogenesis. The alternatively spliced transcripts of Pref-1 could suppress adipogenesis in differentiating 3T3-L1 cells either in a juxtacrine/paracrine or in an endocrine manner that Pref-1A and Pref-1B, producing two large soluble forms, can biologically activate anti-adipogenic signaling, whereas Pref-1C and Pref-1D, generating only smaller fragments lacking in-frame juxtamembrane, have no biological effectiveness in adipogenesis [52].

Schematic illustration of canonical and alternative spliced transcripts of peroxisome proliferator activated receptor gamma (PPARG) gene. In the central part, the PPARG gene structure is depicted. Four main PPARG canonical splice transcripts-PPARG1, PPARG2, PPARG3, PPARG3, and PPARG4-and alternative splice variants-PPARγ1(tr), γ1ORF4, γ2ORF4, γ3ORF4, and PPARγ∆5-are shown in the upper and below part, respectively. The corresponding encoded protein isoforms with functional domains are sketched in the right panel with divergent distribution patterns. The orange columns represent exons; the grey lines represent introns; the blue columns represent untranslated regions (UTRs); the grey column in PPARγ2 represents the 30 additional amino acids at N-terminus; the dark column in γORF4 represents the 21 amino acids at C terminus; the red rectangles represent stop codons; the purple rectangles represent premature termination codons (PTCs)
It is worth noting that the programmed AS alterations in transcriptional corepressors can contribute to the conversion in the affinity of target gene panels, transcriptional environment, and biological program during adipogenesis. Such mechanism has been implicated in the nuclear receptor co-repressor 1 (NCoR1)/silencing mediator of retinoid and thyroid hormone receptor (SMRT) [54,55,56]. Beyond the influence of NCoR1/ SMRT on the adipogenic differentiation phenomenon detected in 3T3-L1 cells of NCoR1/SMRT pan-specific knockdown or site-specific mutagenesis in mice models [57, 58], AS pathway generates a set of correlated splice variants with disparate tissue distribution characteristics and numbers and sequences of receptor interaction domains (RIDs) (associated with different PPARγ affinity in adipocytes) that serve to diversify their transcriptional and biological effects on adipogenic differentiation process [55, 56]. One study overexpressed different NCoR splice variants in 3T3-L1 cells to introduce adipogenesis ex vivo and revealed that NCoRω suppressed, whereas NCoRδ with fewer RIDs accelerated adipogenesis, and the decrease in their relative abundance could promote the adipogenic terminal differentiation [54] (Fig. 3a).
Additionally, the programmed splicing process of apoptotic-resistant factors is a determinate switch in adipogenesis, allowing preadipocytes to differentiate into mature adipocytes of highly pro-survival characteristics [59]. Through the utilization of differentiating preadipocytes in vitro, it was identified that protein kinase C δ (PKCδ), Bclx, and caspase9 could be alternatively spliced to convert to their pro-survival variants, producing dramatic biological effects on adipogenic development [59]; the naturally occurring splice variants-protein kinase C δII (PKCδII) of pro-survival activity and protein kinase C δI (PKCδI) of pro-apoptosis effect-were expressed increasingly and decreasingly, respectively, to facilitate an anti-apoptosis phenotype in mature adipocytes through the Bcl2 pathway [60, 61].
Alternative splicing events associated with BAT adipogenesis and beige adipocyte development
During the adipocyte differentiation of brown and beige adipocytes, programmed splicing shifts also exert broad influences on these processes and energy expenditure signatures. Through performing transcriptome analyses in embryonic BATs and postnatal BATs [62], discriminative splicing profiles have been identified, including several adipogenic regulators, such as PRDM16 [63] and muscleblind-like 1 (MBNL1) [64] (Fig. 3b). Regarding the specific splicing transition of PRDM16, the determinant transcriptional regulator in both brown and beige adipocyte development [65], a gradual alternatively spliced isoforms shift from an exon inclusion isoform (PRDM16+ex16-PRDM16L) to an exon exclusion one (PRDM16−ex16-PRDM16S) has been proved to contribute to a more prominent effect on the differentiation gene program and energy expenditure in the development of brown adipocytes [63]. Results from co-immunoprecipitation tests identified that the splicing factor RNA-binding motif protein 4a (RBM4a) enabled the regulated splicing switch determining the relative expression of the two PRDM16 transcripts, which could constitute a feed-forward circuit with the RBM4a abundance [63]. Besides, RBM4a also participates in the regulated generation of a splicing mode change of MBNL1 (from MBNL1+ex5 isoform 7 to MBNL1−ex5 isoform 1) through an autoregulatory mechanism, bringing about a more robust simulation in beige cell-selective splicing program throughout BAT development and during the in vitro beige adipogenesis [64] (Fig. 3b).
However, the AS regulatory mechanism in brown and beige adipocyte development has not been comprehensively investigated. Significant to our understanding is whether AS networks modulate brown and beige adipogenesis through undefined mechanisms and what biological roles they act during this process. Additionally, global transcriptome surveys identifying the spectrum of genes with differentially expressed ASEs at the exonic resolution have been conducted in white adipogenesis, whereas much remains to be completed to characterize precise and extensive genome-wide analysis during brown and beige adipogenesis.
Splicing regulators involved in adipogenesis
Emerging evidence combined functional mechanistic studies in specific adipocytes, transgenic mice models, and high-throughput methods has extensively identified the physiological roles of distinct RNA splicing regulators in splicing networks for white-, brown-, and beige-adipocyte differentiation and maturation process. Among them, are splicing factors-Src-associated substrate during mitosis of 68 kDa (Sam68), serine and arginine rich splicing factor 10 (SRSF10), and RNA binding motif 4 (RBM4), splicing actions of which function significantly during adipogenesis (Table 1). Moreover, multiple other types of molecules with various biological functions, such as fat mass and obesity-associated (FTO), have been studied and demonstrated to bring about functional consequences of AS in adipogenic regulators. Hence, changes in their target splicing networks often exert a vital influence on adipogenesis process.
Splicing regulators functioning in WAT adipogenesis
The functional AS outcomes of different splicing regulators have been convincingly documented in WAT adipogenesis. Sam68, highly involved in cellular RNA processing events and signal transduction pathways [70, 89], has been demonstrated in adipocyte development to increase the number of early adipocyte progenitors by controlling AS of the mechanistic target of mTOR signaling [66, 68]. The Sam68 binding sites often localize near AS junctions within pre-mRNAs, so they often engage in regulating neighboring alternative exon usage and splice site selection as either splicing enhancers or silencers [69]. In adipocytes, Sam68 can recognize the 5′ splice site of mTOR intron5 through recruitment and interaction with U1 snRNP and bind to intronic splice elements where an in-frame premature termination codon (PTC) exists, which helps to promote an efficient intron exclusion [66, 70]; however, in Sam68 deficiency adipocytes, mTOR intron5 retention occurs, generating a short and unstable transcript degraded by nonsense-mediated decay (NMD), thus causing impaired insulin-stimulated S6 and Akt phosphorylation pathway [66] (Fig. 3a). Additionally, another independent study conducted cross-linking and immunoprecipitation and showed that Sam68 could also counteract the splicing effects of SRSF1 in splicing regulation of ribosomal S6 kinase (Rps6kb1) gene during adipogenesis, resulting in a decrease in the abnormal inhibitory short isoform-Rps6kb-002 [71].
Moreover, another axis linking one splicing factor with specific ASEs and clear functional outcomes in adipocyte development has also been demonstrated. The RBP SRSF10 is an atypical SR protein with distinguished functions whose activity is positively or negatively determined by cycles of phosphorylation and dephosphorylation-SRSF10 acts as a general splicing repressor by dephosphorylation [90], while functions as a sequence-specific splicing activator under phosphorylation conditions [91]. Besides, in controlling exon splicing, SRSF10 functions positively or negatively depending on the binding locations that the binding on cassette exon leads to exon inclusion, while the binding to downstream exon promotes exon skipping [92, 93]. For adipocyte biology, SRSF10 can directly repress exon inclusion in LPIN1, forming an adipogenic exon-skipping isoform LPIN1α to promote initial adipocyte differentiation [80] (Fig. 3a), and in PPARG coactivator 1α (PGC-1α), causing an NMD in the PGC-1α pre-mRNA to affect gluconeogenesis and glucose metabolism [81].
As mentioned above, the evidence lines that splicing machinery and epigenetic modification factors have close interactions can be identified in the adipogenic differentiation modulation process. FTO is the first identified gene providing the strongest genetic association with human non-syndromic obesity [75, 76]. In one study combining transcriptome analyses with m6A-seq, it has been revealed that FTO can mediate m6A demethylation mechanism extended in AS process of one adipogenic factor-runt-related transcription factor 1 (RUNX1T1) during adipogenesis [77, 78]. FTO antagonizes the RNA binding ability of another splicing factor-serine and arginine rich splicing factor 2 (SRSF2) and inhibits target exon inclusion through demethylating of m6A-RRACHs (R = G or A; H = A, C or U) near exonic splice sites which are spatially overlapped the ESE binding regions of SRSF2 [77] to produce more exon-skipped isoform RUNX1T1-S with pro-adipogenic activity and decrease negative constitutive isoform RUNX1T1-L, thus leading to an active effect on adipocyte differentiation [77] (Fig. 3a).
Of note, accumulating evidence has reported that other types of the molecule and signaling pathway can play roles in splicing decisions through interacting with or controlling the splicing activity of splicing machinery present in adipocytes, which are sufficient to modulate adipogenesis via the relative abundance regulation of adipogenesis-specific isoforms, such as molecular scaffold protein 14-3-3 proteins [94, 95] and zinc finger protein 638 (ZNF638) [96] in the recruitment and sequestration of different trans-acting splicing factors, Clk/STY (cdc2-Like Kinase 1) in proper phosphorylation of serine/arginine-rich protein 40 (SRp40) [45, 97], and long non-coding RNA NEAT1 associated with SRp40 phosphorylation [98].
Splicing factors involved in BAT adipogenesis and beige adipocyte development
The spectrum of splicing factors identified in brown adipocyte differentiation process and intricate physiological functions of BAT mainly includes RBM4, serine and arginine rich splicing factor 6 (SRSF6), serine and arginine rich splicing factor 3 (SRSF3), and serine and arginine rich splicing factor protein kinase 1 (SRPK1). Given that the physiological roles of splicing factors functioning in brown- and beige-adipocyte formation processes are recognized still at an early stage, further identification for the underlying mechanism is needed.
The splicing factor RBM4, firstly identified in tissue-specific splicing network of muscles and pancreatic β-islets, can multifunctionally participate in post-transcriptional regulation and lies in the center hub of AS decisions in brown and beige fat development and functions (Table 1). The ablation of RBM4a in mice (RBM4a−/−) has been found to cause impaired development of interscapular BAT and abnormal triglyceride clearance in serum [84]. Moreover, in RBM4-deficiency or-overexpression adipocytes where gene-splicing profiles are reprogrammed, the expression level of BAT development inhibitors (neuro-oncological ventral antigen 1 (Nova1), polypyrimidine tract-binding protein 1 (PTBP1), and 2 (PTBP2)) or adipogenic factors (insulin receptor (IR), PPARγ, Pref-1, fibroblast growth factor receptor 2 (FGFR2), PRDM16, and bone morphogenetic protein 7 (BMP7)) can be affected differently [84, 85], modulating brown and beige adipogenesis, mitochondrial activity, and energy expenditure process. In particular, RBM4 mostly interacts with intronic splicing regulatory elements and inhibits exon inclusion, functioning either as a splicing activator or an inhibitor in different genes [99]. Further, a set of RBM4-governed splicing cascades are indicated through deep RNA-sequencing during the differentiation of brown adipocytes, including RBM4-PRDM16 network [63], RBM4-Nova1-SRSF6 pathway [62], RBM4a-SRSF3-mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) pathway [83], RBM4-myocyte enhancer factor 2c (MEF2C) [86], RBM4-SRPK1 [87], and RBM4-apoptotic chromatin condensation inducer 1 (Acin1)-SRSF3 [88], generating different splice isoforms to function distinct roles in transcriptional regulation and form feedback circuits associated with RBM4 abundance.
Alternative splicing machinery has correlations with obesity and metabolic disorders
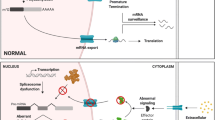
Obesity and overweight, a severe growing health concern worldwide, dramatically elevates human mortality risk factors along with different comorbidities, such as metabolic syndrome, type 2 diabetes, cardiovascular disorders, dementia, and cancers [100]. As reviewed in detail below, emerging evidence constitutes a mechanistic understanding that both alternative mRNA mis-splicing patterns and aberrant expression or splicing efficiency of splicing factors, contributing to transcriptome changes, have profound associations with human obesity and metabolic disorders, as illustrated in Fig. 5.

Schematic diagram of alternative splicing mechanism in adipogenesis and metabolic health
Aberrant splicing landscapes associated with obesity and metabolic diseases
First and foremost, accumulating evidence has revealed that a set of adipogenesis regulators exhibit abnormal splicing patterns in adipose tissues from obese patients compared with control subjects, suggesting a vital link of AS with adiposity and metabolic phenotypes-usually the susceptibility to extreme body weight gain and serious metabolic illness [50, 51, 101]. One classical example is PPARγ, the master modulator in whole-body crosstalk of metabolic organs, whose canonical transcripts and alternatively spliced negative isoforms exhibit remarkably different enrichment patterns in overweight or obese patients with diabetes: in subcutaneous adipose tissue, the specific expression of PPARγΔ5, a naturally occurring negative PPARG isoform, can display a positive correlation with body mass index (BMI) in the patient’s population of these diseases [51]; γORF4 with dominant-negative activity has also been found to be physiologically expressed in tissues related to complications of metabolic syndrome in humans [50]. Another example involves apoptosis-pathway genes, which can undergo crucial splicing switches to generate prosurvival splice variants (PKCδ, Bclx, and caspase9), promoting the shift to apoptosis-resistant mature adipocytes; while a series of alternatively spliced transcripts are abnormally overexpressed in obese patient-derived adipose tissues [59]. Moreover, other cohort studies revealed quantitative AS changes in the transcriptome in WAT where gene splicing profiles displayed abnormal features in the contexts of nutritional changes, obesity, and dysmetabolism [102], including the genes of Lamin A/C (LMNA) [103], cannabinoid type I receptor (CB1R) [104], and T cell-specific transcription factor 7-like 2 (TCF7L2) [105]. Importantly, their splicing alterations in WAT suggest strong associations with the adipose tissue biology and related metabolic parameters of obesity and type 2 diabetes, such as insulin action, fatty acid metabolism, and systemic and local chronic inflammation. Besides, not limited to adipose tissues, the splicing patterns have also been found to be influenced by hormonal and nutritive events (dietary carbohydrates changes) as well as obese and metabolic states [106], for example, the IR gene in insulin target tissues [107]. Nevertheless, the underlying physiology relevance between these molecular profiles and obesity remains unexplained, thus it will be of particular importance to further investigate and decipher the detailed mechanisms.
Significantly, several groups also identified that imbalances of different splice isoforms could directly lead to the dysfunction of white- and brown-adipose tissues as well as the initial development of diabesity and metabolic disturbances [107,108,109]. Studies used the mouse mutant engineered to express one particular splice variant of genes, for example, the PGC-1α gene and the synaptosomal-associated protein of 25 kDa (SNAP-25) gene [108, 109], through knockout/knockin replacement, and revealed how these different splice variants functioned in adipose biology of WAT and BAT as well as energy and metabolism homeostasis under the HFD condition, and what effect they exerted on the susceptibility and predisposition to obesity and metabolic diseases. Further, given the heterogeneity and complexity of these related disorders in humans, particularly noteworthy is that evaluating whether and how splicing profile changes have correlations with different obese and dysmetabolic features among patient’s population should be based on seriously comprehensive analysis with different factors, mainly involving the matched age, target tissues, other complications, and medical therapies.
Finally, it may also need to concern that obesity and related chronic metabolic inflammation link intimately to unfolded protein response (UPR) and interfere with endoplasmic reticulum (ER) homeostasis, where the non-conventional splicing process of the X-box–binding protein 1 (XBP1) mRNA mediated by the ER sensor-inositol requiring enzyme 1α (IRE1α) is disrupted [110]. This defective IRE1α-mediated XBP1 splicing process has been proved in the liver in conditions of both genetic and diet-induced obesity; and these ER-remodeling-associated interactions are worth further examination in adipocytes in the obesity setting.
Connection of splicing regulators in obesity and metabolic dysregulation
Representative works have demonstrated that splicing machinery components can be associated with the adipogenesis process and further energy expenditure regulation and body metabolic state. In fact, the conditions of dysregulated metabolic state or high-fat diet can bring about altered expression of genes encoding trans-acting splicing factors [102], or alterations in splicing activity [111], both contributing to widespread mis-spliced pre-mRNAs; and abnormal splicing factors-mediated AS program can also contribute to the development of metabolic disorders and diet-induced obesity. For example, the obese and metabolic impact on the reduced expression of transformer 2β homolog (TRA2B/SFRS10) which belongs to the SR-like protein family of splicing factors was observed in liver and muscle tissues from obese patients; the SFRS10 down-regulation can alter the splicing pattern of LPIN1 to induce lipid accumulation, which therefore causes aberrant metabolic phenotypes and obesity [112]. In another example, the vital role of neuro-oncological ventral antigen (NOVA) splicing factors in the pathogenesis of obesity has been confirmed in the NOVA-deficient mice that NOVA-regulated splicing program participates in glycemia increase and thermogenesis suppression in adipocytes [102].
Apart from these expression changes associated with metabolic phenotypes, polymorphisms in the splicing regulator genes detected by amounts of genome-wide association studies in humans, such as FTO, could consistently exert a strong influence on body fat mass and the risk of developing diabesity and type 2 diabetes [113,114,115].
Splicing-related SNPs contribute to obesity and metabolic disorders
Genetic association studies that detect the link of SNPs affecting splicing-related process with obesity and metabolic disorders have made it possible to provide candidate diagnostic tools and potential targets for clinical intervention. Evidence from the sequencing-based approach and large-scale analysis in obese populations has highlighted that the SNPs connected with the aberrant splicing patterns in obesity-associated genes could represent possible biomarkers for extreme BMI susceptibility [116]. Substantially, broad obesity-related mutation spectrums of synonymous and nonsynonymous SNPs are located near splice junctions and are prone to affect the cis-regulatory elements of exons with weak splice sites allowing the shift from constitutive exon patterns to alternative ones and thus give rise to multiple RNA isoforms [116]. Moreover, collective evidence has displayed the tight link between splicing related SNPs and human body metabolic state, involving the genes of uncoupling protein-3 (UCP3) [117], glucose-6-phosphatase catalytic unit 2 (G6PC2) [118], insulin-like growth factor 2 (IGF2) [119], adenylate cyclase 3 (ADCY3) [120], and an intragenic variant of insulin gene (IVS1-6A/T (-23HphI ±)) [121]. For example, a novel genetic variation with a disruption in canonical splice-site acceptor of the IGF2 gene among the Mexican population has been identified to function as a protective factor to reduce the type 2 diabetes risk at ∼ 20% through repressing splicing between the exons 1 and 2 and reducing the expression of IGF2 isoforms [119]. Additionally, one loss-of-function genetic variant of ADCY3 with a disruption of a splice acceptor site causing exon skip or intron retention was detected among the Greenlandic population and positively correlated with increased adiposity and predisposition of type 2 diabetes [120]. Besides, one genome-wide association study among European women has evaluated the physiological relevance of the polymorphisms in the leptin receptor (LEPR) in human diabetes-related traits, which appear to modulate the expression level of plasma soluble leptin receptor-one alternative isoform of LEPR gene correlated inversely with BMI and diabetes risk factors [122].
Conclusions and future perspectives
Findings provide the mechanistic role of AS in multiple facets of adipocyte development and function and the broad influences on metabolic health. A variety of research efforts have compiled the comprehensive crosstalk between AS and the adipocyte differentiation processes of different adipose tissues and whole-body energy and metabolic homeostasis. Substantially, profiling alternative splicing alterations in obesity and metabolic disorders might provide possible biomarkers and designs for novel diagnostic strategies. Noteworthy, high-throughput RNA sequencing techniques and transcriptome bioinformatics analysis fully support the identification of the massive ASEs amounts and the regulatory role of splicing factors during adipogenesis and facilitate sufficient clarity into how adipogenesis is modulated at different stages on a genome-wide scale.
Nevertheless, our understanding of the AS mechanism involved in brown and beige adipogenesis is still evolving. Further work on deciphering “splicing code” in brown and beige adipogenic differentiation to offer broader and more meticulous insights into the complex interplay of trans-splicing regulators with cis-acting elements is vital for an in-depth understanding of adipocytes physiology. Additionally, more investigations are required on the functional mechanism of specific splice variants from different sorts of adipose tissue in the pathogenesis of obesity. Accordingly, fixing splicing dysregulation emerges as a promising therapeutic option and provides more viable strategies for future management for obesity and metabolic disorders.
Code availability
Not applicable.
Abbreviations
- Acin1:
-
Apoptotic chromatin condensation inducer 1
- ADCY3:
-
Adenylate cyclase 3
- AS:
-
Alternative splicing
- ASE:
-
Alternative splicing event
- BAT:
-
Brown adipose tissue
- BMI:
-
Body mass index
- BMP:
-
Bone morphogenetic protein
- BMP7:
-
Bone morphogenetic protein 7
- CB1R:
-
Cannabinoid type I receptor
- Clk/STY:
-
Cdc2-like kinase 1
- CREB:
-
CAMP-response element-binding protein
- C/EBPα:
-
CCAAT/enhancer-binding protein α
- DBD:
-
DNA-binding domain
- EBF2:
-
Early B-cell factor 2
- ER:
-
Endoplasmic reticulum
- E/ISEs:
-
Exonic/intronic splicing enhancers
- E/ISSs:
-
Exonic/intronic splicing silencers
- FGFR2:
-
Fibroblast growth factor receptor 2
- FTO:
-
Fat mass and obesity-associated
- G6PC2:
-
Glucose-6-phosphatase catalytic unit 2
- hMSCs:
-
Human mesenchymal stem cells
- hnRNPs:
-
Heterogeneous ribonucleoproteins
- hPTMs:
-
Histone post-translational modifications
- LBD:
-
Ligand-binding domain
- IGF2:
-
Insulin-like growth factor 2
- IR:
-
Insulin receptor
- IRE1α:
-
Inositol requiring enzyme 1α
- KLFs:
-
Krüppel-like factors
- LBP:
-
Ligand-binding domain
- LEPR:
-
Leptin receptor
- LMNA:
-
Lamin A/C
- MAP4K4:
-
Mitogen-activated protein kinase kinase kinase kinase 4
- MBNL1:
-
Muscleblind-like 1
- MC2-R:
-
Melanocortin 2 receptor
- MEF2C:
-
Myocyte enhancer factor 2c
- m6A:
-
N6-methyladenosine
- MYF5:
-
Myogenic factor 5
- NCoR1:
-
Nuclear receptor co-repressor 1
- NGS:
-
Next-generation sequencing
- NMD:
-
Nonsense-mediated decay
- NOVA:
-
Neuro-oncological ventral antigen
- Nova1:
-
Neuro-oncological ventral antigen 1
- NRF1:
-
Nuclear factor erythroid 2-related factor 1
- PGC-1α:
-
PPARG coactivator 1α
- PKCβII:
-
Protein kinase C βII
- PKCδ:
-
Protein kinase C δ
- PKCδI:
-
Protein kinase C δI
- PKCδII:
-
Protein kinase C δII
- Pol II:
-
RNA polymerase II
- PPARG:
-
Peroxisome proliferator-activated receptor gamma
- PPARγ:
-
Peroxisome proliferator-activated receptor-γ
- PRDM16:
-
PR domain-containing 16
- pre-mRNA:
-
Precursor messenger RNA
- Pref-1:
-
Preadipocyte factor-1
- PTBP1:
-
Polypyrimidine tract-binding protein 1
- PTBP2:
-
Polypyrimidine tract-binding protein 2
- PTC:
-
Premature termination codon
- RBM4:
-
RNA binding motif 4
- RBM4a:
-
RNA-binding motif protein 4a
- RBPs:
-
RNA-binding proteins
- RIDs:
-
Receptor interaction domains
- Rps6kb1:
-
Ribosomal S6 kinase
- RUNX1T1:
-
Runt-related transcription factor 1
- Sam68:
-
Src-associated substrate during mitosis of 68 kDa
- SF1:
-
Splicing factor 1
- SF3B1:
-
Splicing factor 3 subunit B1
- SMRT:
-
Silencing mediator of retinoid and thyroid hormone receptor
- SNAP-25:
-
Synaptosomal-associated protein of 25 kDa
- SNPs:
-
Single-nucleotide polymorphisms
- snRNP:
-
Small nuclear ribonucleoprotein
- SR:
-
Serine/arginine-rich
- SRPK1:
-
Splicing factor protein kinase 1
- SRp40:
-
Serine/arginine-rich protein 40
- SRSF1:
-
Serine and arginine rich splicing factor 1
- SRSF2:
-
Serine and arginine rich splicing factor 2
- SRSF3:
-
Serine and arginine rich splicing factor 3
- SRSF6:
-
Serine and arginine rich splicing factor 6
- SRSF10:
-
Serine and arginine rich splicing factor 10
- TCF7L1:
-
T cell-specific transcription factor 7-like 1
- TCF7L2:
-
T cell-specific transcription factor 7-like 2
- TIP:
-
Tension-induced/inhibited protein
- TRA2B:
-
Transformer 2β homolog
- UCP1:
-
Uncoupling protein 1
- UCP3:
-
Uncoupling protein 3
- UPR:
-
Unfolded protein response
- UTRs:
-
Untranslated regions
- U2AF1:
-
U2 small nuclear RNA auxiliary factor 1
- U2AF2:
-
U2 small nuclear RNA auxiliary factor 2
- WAT:
-
White adipose tissue
- XBP1:
-
X-box–binding protein 1
- ZFP423:
-
Zinc finger protein 423
- ZNF638:
-
Zinc finger protein 638
References
Yang XP, Coulombe-Huntington J, Kang SL, Sheynkman GM, Hao T, Richardson A, Sun S, Yang F, Shen YA, Murray RR, Spirohn K, Begg BE, Duran-Frigola M, MacWilliams A, Pevzner SJ, Zhong Q, Trigg SA, Tam S, Ghamsari L, Sahni N, Yi S, Rodriguez MD, Balcha D, Tan G, Costanzo M, Andrews B, Boone C, Zhou XJ, Salehi-Ashtiani K, Charloteaux B, Chen AA, Calderwood MA, Aloy P, Roth FP, Hill DE, Iakoucheva LM, Xia Y, Vidal M. Widespread expansion of protein interaction capabilities by alternative splicing. Cell. 2016;164(4):805–17.
Koch L. Alternative splicing: a thermometer controlling gene expression. Nat Rev Genet. 2017;18(9):515.
Bonnal SC, López-Oreja I, Valcárcel J. Roles and mechanisms of alternative splicing in cancer-implications for care. Nat Rev Clin Oncol. 2020;17(8):457–74.
Tress ML, Abascal F, Valencia A. Alternative splicing may not be the key to proteome complexity. Trends Biochem Sci. 2017;42(2):98–110.
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6.
Gabut M, Samavarchi-Tehrani P, Wang X, Slobodeniuc V, O’Hanlon D, Sung HK, Alvarez M, Talukder S, Pan Q, Mazzoni EO, Nedelec S, Wichterle H, Woltjen K, Hughes TR, Zandstra PW, Nagy A, Wrana JL, Blencowe BJ. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell. 2011;147(1):132–46.
Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF, Black DL, Kharchenko PV, Sharp PA, Walsh CA. Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell. 2016;166(5):1147.e1115-1162.e1115.
Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136(4):777–93.
Hang J, Wan R, Yan C, Shi Y. Structural basis of pre-mRNA splicing. Science. 2015;349(6253):1191–8.
Plaschka C, Lin PC, Charenton C, Nagai K. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature. 2018;559(7714):419–22.
Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ. Deciphering the splicing code. Nature. 2010;465(7294):53–9.
Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479(7371):74–9.
Rahhal R, Seto E. Emerging roles of histone modifications and HDACs in RNA splicing. Nucleic Acids Res. 2019;47(10):4911–26.
Kim YE, Park C, Kim KE, Kim KK. Histone and RNA-binding protein interaction creates crosstalk network for regulation of alternative splicing. Biochem Biophys Res Commun. 2018;499(1):30–6.
Wong JJ, Gao D, Nguyen TV, Kwok CT, van Geldermalsen M, Middleton R, Pinello N, Thoeng A, Nagarajah R, Holst J, Ritchie W, Rasko JEJ. Intron retention is regulated by altered MeCP2-mediated splicing factor recruitment. Nat Commun. 2017;8:15134.
Hu Q, Greene CS, Heller EA. Specific histone modifications associate with alternative exon selection during mammalian development. Nucleic Acids Res. 2020;48(9):4709–24.
Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8(5):e1002717.
Xu Y, Wang Y, Luo J, Zhao W, Zhou X. Deep learning of the splicing (epi)genetic code reveals a novel candidate mechanism linking histone modifications to ESC fate decision. Nucleic Acids Res. 2017;45(21):12100–12.
Matveeva EA, Al-Tinawi QMH, Rouchka EC, Fondufe-Mittendorf YN. Coupling of PARP1-mediated chromatin structural changes to transcriptional RNA polymerase II elongation and cotranscriptional splicing. Epigenetics Chromatin. 2019;12(1):15.
Tian C, Huang Y, Li Q, Feng Z, Xu Q. Mettl3 regulates osteogenic differentiation and alternative splicing of vegfa in bone marrow mesenchymal stem cells. Int J Mol Sci. 2019;20(3):551.
Elman JS, Ni TK, Mengwasser KE, Jin D, Wronski A, Elledge SJ, Kuperwasser C. Identification of FUBP1 as a long tail cancer driver and widespread regulator of tumor suppressor and oncogene alternative splicing. Cell Rep. 2019;28(13):3435.e3435-3449.e3435.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45(10):6051–63.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, Wang X, Ma HL, Huang CM, Yang Y, Huang N, Jiang GB, Wang HL, Zhou Q, Wang XJ, Zhao YL, Yang YG. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–19.
Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–308.
Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, Reed R. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell. 2007;26(6):867–81.
Close P, East P, Dirac-Svejstrup AB, Hartmann H, Heron M, Maslen S, Chariot A, Soding J, Skehel M, Svejstrup JQ. DBIRD complex integrates alternative mRNA splicing with RNA polymerase II transcript elongation. Nature. 2012;484(7394):386–9.
Dujardin G, Lafaille C, de la Mata M, Marasco LE, Munoz MJ, Le Jossic-Corcos C, Corcos L, Kornblihtt AR. How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell. 2014;54(4):683–90.
Saldi T, Cortazar MA, Sheridan RM, Bentley DL. Coupling of RNA polymerase II transcription elongation with pre-mRNA splicing. J Mol Biol. 2016;428(12):2623–35.
de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell. 2003;12(2):525–32.
Herzel L, Ottoz DSM, Alpert T, Neugebauer KM. Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat Rev Mol Cell Biol. 2017;18(10):637–50.
Tian J, Wang Z, Mei S, Yang N, Yang Y, Ke J, Zhu Y, Gong Y, Zou D, Peng X, Wang X, Wan H, Zhong R, Chang J, Gong J, Han L, Miao X. CancerSplicingQTL: a database for genome-wide identification of splicing QTLs in human cancer. Nucleic Acids Res. 2019;47(D1):D909–16.
Lim KH, Ferraris L, Filloux ME, Raphael BJ, Fairbrother WG. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc Natl Acad Sci USA. 2011;108(27):11093–8.
Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156(1–2):20–44.
Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;20(4):242–58.
Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. 2013;19(10):1338–44.
Vishvanath L, Gupta RK. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J Clin Invest. 2019;129(10):4022–31.
Ehrlund A, Mejhert N, Bjork C, Andersson R, Kulyte A, Astrom G, Itoh M, Kawaji H, Lassmann T, Daub CO, Carninci P, Forrest AR, Hayashizaki Y, Sandelin A, Ingelsson E, Consortium F, Ryden M, Laurencikiene J, Arner P, Arner E. Transcriptional dynamics during human adipogenesis and its link to adipose morphology and distribution. Diabetes. 2017;66(1):218–30.
Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. 2013;19(10):1252–63.
Lee YH, Petkova AP, Konkar AA, Granneman JG. Cellular origins of cold-induced brown adipocytes in adult mice. FASEB J. 2015;29(1):286–99.
Wang QA, Tao C, Jiang L, Shao M, Ye R, Zhu Y, Gordillo R, Ali A, Lian Y, Holland WL, Gupta RK, Scherer PE. Distinct regulatory mechanisms governing embryonic versus adult adipocyte maturation. Nat Cell Biol. 2015;17(9):1099–111.
Rosenwald M, Perdikari A, Rulicke T, Wolfrum C. Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol. 2013;15(6):659–67.
Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, Huang K, Tu H, van Marken Lichtenbelt WD, Hoeks J, Enerback S, Schrauwen P, Spiegelman BM. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150(2):366–76.
Jakkaraju S, Zhe X, Pan D, Choudhury R, Schuger L. TIPs are tension-responsive proteins involved in myogenic versus adipogenic differentiation. Dev Cell. 2005;9(1):39–49.
Kleiman E, Carter G, Ghansah T, Patel NA, Cooper DR. Developmentally spliced PKCbetaII provides a possible link between mTORC2 and Akt kinase to regulate 3T3-L1 adipocyte insulin-stimulated glucose transport. Biochem Biophys Res Commun. 2009;388(3):554–9.
Noon LA, Bakmanidis A, Clark AJ, O’Shaughnessy PJ, King PJ. Identification of a novel melanocortin 2 receptor splice variant in murine adipocytes: implications for post-transcriptional control of expression during adipogenesis. J Mol Endocrinol. 2006;37(3):415–20.
Yi X, Yang Y, Wu P, Xu X, Li W. Alternative splicing events during adipogenesis from hMSCs. J Cell Physiol. 2020;235(1):304–16.
Kim HJ, Woo IS, Kang ES, Eun SY, Kim HJ, Lee JH, Chang KC, Kim JH, Seo HG. Identification of a truncated alternative splicing variant of human PPARgamma1 that exhibits dominant negative activity. Biochem Biophys Res Commun. 2006;347(3):698–706.
Sabatino L, Casamassimi A, Peluso G, Barone MV, Capaccio D, Migliore C, Bonelli P, Pedicini A, Febbraro A, Ciccodicola A, Colantuoni V. A novel peroxisome proliferator-activated receptor gamma isoform with dominant negative activity generated by alternative splicing. J Biol Chem. 2005;280(28):26517–25.
Aprile M, Ambrosio MR, D’Esposito V, Beguinot F, Formisano P, Costa V, Ciccodicola A. PPARG in human adipogenesis: differential contribution of canonical transcripts and dominant negative isoforms. PPAR Res. 2014;2014:537865.
Aprile M, Cataldi S, Ambrosio MR, D’Esposito V, Lim K, Dietrich A, Bluher M, Savage DB, Formisano P, Ciccodicola A, Costa V. PPARγΔ5, a naturally occurring dominant-negative splice isoform, impairs PPARγ function and adipocyte differentiation. Cell Rep. 2018;25(6):1577.e1576-1592.e1576.
Mei B, Zhao L, Chen L, Sul HS. Only the large soluble form of preadipocyte factor-1 (Pref-1), but not the small soluble and membrane forms, inhibits adipocyte differentiation: role of alternative splicing. Biochem J. 2002;364(Pt 1):137–44.
Xue P, Hou Y, Zuo Z, Wang Z, Ren S, Dong J, Fu J, Wang H, Andersen ME, Zhang Q, Xu Y, Pi J. Long isoforms of NRF1 negatively regulate adipogenesis via suppression of PPARγ expression. Redox Biol. 2020;30:101414.
Goodson ML, Mengeling BJ, Jonas BA, Privalsky ML. Alternative mRNA splicing of corepressors generates variants that play opposing roles in adipocyte differentiation. J Biol Chem. 2011;286(52):44988–99.
Faist F, Short S, Kneale GG, Sharpe CR. Alternative splicing determines the interaction of SMRT isoforms with nuclear receptor–DNA complexes. Biosci Rep. 2009;29(3):143–9.
Privalsky ML, Snyder CA, Goodson ML. Corepressor diversification by alternative mRNA splicing is species specific. BMC Evol Biol. 2016;16(1):221.
Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J Biol Chem. 2005;280(14):13600–5.
Nofsinger RR, Li P, Hong SH, Jonker JW, Barish GD, Ying H, Cheng SY, Leblanc M, Xu W, Pei L, Kang YJ, Nelson M, Downes M, Yu RT, Olefsky JM, Lee CH, Evans RM. SMRT repression of nuclear receptors controls the adipogenic set point and metabolic homeostasis. Proc Natl Acad Sci USA. 2008;105(20):20021–6.
Carter G, Apostolatos A, Patel R, Mathur A, Cooper D, Murr M, Patel NA. Dysregulated alternative splicing pattern of PKCδ during differentiation of human preadipocytes represents distinct differences between lean and obese adipocytes. ISRN Obes. 2013;2013:161345.
Patel R, Apostolatos A, Carter G, Ajmo J, Gali M, Cooper DR, You M, Bisht KS, Patel NA. Protein kinase C δ (PKCδ) splice variants modulate apoptosis pathway in 3T3L1 cells during adipogenesis: identification of PKCδII inhibitor. J Biol Chem. 2013;288(37):26834–46.
Patel RS, Carter G, Cooper DR, Apostolatos H, Patel NA. Transformer 2β homolog (Drosophila) (TRA2B) regulates protein kinase C δI (PKCδI) splice variant expression during 3T3L1 preadipocyte cell cycle. J Biol Chem. 2014;289(46):31662–72.
Lin JC, Chi YL, Peng HY, Lu YH. RBM4-Nova1-SRSF6 splicing cascade modulates the development of brown adipocytes. Biochim Biophys Acta. 2016;1859(11):1368–79.
Chi YL, Lin JC. RBM4a modulates the impact of PRDM16 on development of brown adipocytes through an alternative splicing mechanism. Biochim Biophys Acta Mol Cell Res. 2018;1865(11 Pt A):1515–25.
Hung CS, Lin JC. Alternatively spliced MBNL1 isoforms exhibit differential influence on enhancing brown adipogenesis. Biochim Biophys Acta Gene Regul Mech. 2020;1863(1):194437.
Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, Wu J, Gunawardana SC, Banks AS, Camporez JP, Jurczak MJ, Kajimura S, Piston DW, Mathis D, Cinti S, Shulman GI, Seale P, Spiegelman BM. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell. 2014;156(1–2):304–16.
Huot ME, Vogel G, Zabarauskas A, Ngo CT, Coulombe-Huntington J, Majewski J, Richard S. The Sam68 STAR RNA-binding protein regulates mTOR alternative splicing during adipogenesis. Mol Cell. 2012;46(2):187–99.
Li N, Hebert S, Song J, Kleinman CL, Richard S. Transcriptome profiling in preadipocytes identifies long noncoding RNAs as Sam68 targets. Oncotarget. 2017;8(47):81994–2005.
Zhou J, Cheng M, Boriboun C, Ardehali MM, Jiang C, Liu Q, Han S, Goukassian DA, Tang YL, Zhao TC, Zhao M, Cai L, Richard S, Kishore R, Qin G. Inhibition of Sam68 triggers adipose tissue browning. J Endocrinol. 2015;225(3):181–9.
Sumithra B, Saxena U, Das AB. A comprehensive study on genome-wide coexpression network of KHDRBS1/Sam68 reveals its cancer and patient-specific association. Sci Rep. 2019;9(1):11083.
Subramania S, Gagne LM, Campagne S, Fort V, O’Sullivan J, Mocaer K, Feldmuller M, Masson JY, Allain FHT, Hussein SM, Huot ME. SAM68 interaction with U1A modulates U1 snRNP recruitment and regulates mTor pre-mRNA splicing. Nucleic Acids Res. 2019;47(8):4181–97.
Song J, Richard S. Sam68 regulates S6K1 alternative splicing during adipogenesis. Mol Cell Biol. 2015;35(11):1926–39.
Klein ME, Younts TJ, Cobo CF, Buxbaum AR, Aow J, Erdjument-Bromage H, Richard S, Malinow R, Neubert TA, Singer RH, Castillo PE, Jordan BA. Sam68 enables metabotropic glutamate receptor-dependent LTD in distal dendritic regions of CA1 hippocampal neurons. Cell Rep. 2019;29(7):1789.e6-1799.e6.
Naro C, Pellegrini L, Jolly A, Farini D, Cesari E, Bielli P, de la Grange P, Sette C. Functional interaction between U1snRNP and Sam68 insures proper 3′ end pre-mRNA processing during germ cell differentiation. Cell Rep. 2019;26(11):2929.e2925-2941.e2925.
Huot ME, Richard S. Stay lean without dieting: lose Sam68. Adipocyte. 2012;1(4):246–9.
Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, Abdennur NA, Liu J, Svensson PA, Hsu YH, Drucker DJ, Mellgren G, Hui CC, Hauner H, Kellis M. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. 2015;373(10):895–907.
Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, Wells S, Bruning JC, Nolan PM, Ashcroft FM, Cox RD. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42(12):1086–92.
Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, Jin KX, Wang X, Huang CM, Fu Y, Ge XM, Song SH, Jeong HS, Yanagisawa H, Niu Y, Jia GF, Wu W, Tong WM, Okamoto A, He C, Rendtlew Danielsen JM, Wang XJ, Yang YG. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24(12):1403–19.
Deng K, Ren C, Liu Z, Gao X, Fan Y, Zhang G, Zhang Y, Ma ES, Wang F, You P. Characterization of RUNX1T1, an adipogenesis regulator in ovine preadipocyte differentiation. Int J Mol Sci. 2018;19(5):1300.
Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, Ruther U. Inactivation of the Fto gene protects from obesity. Nature. 2009;458(7240):894–8.
Li H, Cheng Y, Wu W, Liu Y, Wei N, Feng X, Xie Z, Feng Y. SRSF10 regulates alternative splicing and is required for adipocyte differentiation. Mol Cell Biol. 2014;34(12):2198–207.
Wei N, Cheng Y, Wang Z, Liu Y, Luo C, Liu L, Chen L, Xie Z, Lu Y, Feng Y. SRSF10 plays a role in myoblast differentiation and glucose production via regulation of alternative splicing. Cell Rep. 2015;13(8):1647–57.
Feng Y, Valley MT, Lazar J, Yang AL, Bronson RT, Firestein S, Coetzee WA, Manley JL. SRp38 regulates alternative splicing and is required for Ca(2+) handling in the embryonic heart. Dev Cell. 2009;16(4):528–38.
Peng HY, Liang YC, Tan TH, Chuang HC, Lin YJ, Lin JC. RBM4a-SRSF3-MAP4K4 splicing cascade constitutes a molecular mechanism for regulating brown adipogenesis. Int J Mol Sci. 2018;19(9):2646.
Lin JC, Tarn WY, Hsieh WK. Emerging role for RNA binding motif protein 4 in the development of brown adipocytes. Biochim Biophys Acta. 2014;1843(4):769–79.
Lin JC, Lu YH, Liu YR, Lin YJ. RBM4a-regulated splicing cascade modulates the differentiation and metabolic activities of brown adipocytes. Sci Rep. 2016;6:20665.
Lin JC. RBM4-MEF2C network constitutes a feed-forward circuit that facilitates the differentiation of brown adipocytes. RNA Biol. 2015;12(2):208–20.
Lin JC. Multi-posttranscriptional regulations lessen the repressive effect of SRPK1 on brown adipogenesis. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863(5):503–14.
Lin YC, Lu YH, Lee YC, Hung CS, Lin JC. Altered expressions and splicing profiles of Acin1 transcripts differentially modulate brown adipogenesis through an alternative splicing mechanism. Biochim Biophys Acta Gene Regul Mech. 2020;1863(9):194601.
Frisone P, Pradella D, Di Matteo A, Belloni E, Ghigna C, Paronetto MP. SAM68: signal transduction and RNA metabolism in human cancer. Biomed Res Int. 2015;2015:528954.
Shin C, Feng Y, Manley JL. Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature. 2004;427(6974):553–8.
Feng Y, Chen M, Manley JL. Phosphorylation switches the general splicing repressor SRp38 to a sequence-specific activator. Nat Struct Mol Biol. 2008;15(10):1040–8.
Zhou X, Wu W, Li H, Cheng Y, Wei N, Zong J, Feng X, Xie Z, Chen D, Manley JL, Wang H, Feng Y. Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucleic Acids Res. 2014;42(6):4019–30.
Wu W, Zong J, Wei N, Cheng J, Zhou X, Cheng Y, Chen D, Guo Q, Zhang B, Feng Y. CASH: a constructing comprehensive splice site method for detecting alternative splicing events. Brief Bioinform. 2018;19(5):905–17.
Lim GE, Albrecht T, Piske M, Sarai K, Lee JTC, Ramshaw HS, Sinha S, Guthridge MA, Acker-Palmer A, Lopez AF, Clee SM, Nislow C, Johnson JD. 14-3-3ζ coordinates adipogenesis of visceral fat. Nat Commun. 2015;6:7671.
Mugabo Y, Sadeghi M, Fang NN, Mayor T, Lim GE. Elucidation of the 14-3-3ζ interactome reveals critical roles of RNA-splicing factors during adipogenesis. J Biol Chem. 2018;293(18):6736–50.
Du C, Ma X, Meruvu S, Hugendubler L, Mueller E. The adipogenic transcriptional cofactor ZNF638 interacts with splicing regulators and influences alternative splicing. J Lipid Res. 2014;55(9):1886–96.
Li P, Carter G, Romero J, Gower KM, Watson J, Patel NA, Cooper DR. Clk/STY (cdc2-like kinase 1) and Akt regulate alternative splicing and adipogenesis in 3T3-L1 pre-adipocytes. PLoS ONE. 2013;8(1):e53268.
Cooper DR, Carter G, Li P, Patel R, Watson JE, Patel NA. Long non-coding RNA NEAT1 associates with SRp40 to temporally regulate PPARγ2 splicing during adipogenesis in 3T3-L1 cells. Genes (Basel). 2014;5(4):1050–63.
Wang Y, Ma M, Xiao X, Wang Z. Intronic splicing enhancers, cognate splicing factors and context-dependent regulation rules. Nat Struct Mol Biol. 2012;19(10):1044–52.
Ward ZJ, Bleich SN, Cradock AL, Barrett JL, Giles CM, Flax C, Long MW, Gortmaker SL. Projected U.S. state-level prevalence of adult obesity and severe obesity. N Engl J Med. 2019;381(25):2440–50.
Snyder CA, Goodson ML, Schroeder AC, Privalsky ML. Regulation of corepressor alternative mRNA splicing by hormonal and metabolic signaling. Mol Cell Endocrinol. 2015;413(C):228–35.
Vernia S, Edwards YJ, Han MS, Cavanagh-Kyros J, Barrett T, Kim JK, Davis RJ. An alternative splicing program promotes adipose tissue thermogenesis. Elife. 2016;5:e17672.
Miranda M, Chacon MR, Gutierrez C, Vilarrasa N, Gomez JM, Caubet E, Megia A, Vendrell J. LMNA mRNA expression is altered in human obesity and type 2 diabetes. Obesity (Silver Spring). 2008;16(8):1742–8.
Sarzani R, Bordicchia M, Marcucci P, Bedetta S, Santini S, Giovagnoli A, Scappini L, Minardi D, Muzzonigro G, Dessi-Fulgheri P, Rappelli A. Altered pattern of cannabinoid type 1 receptor expression in adipose tissue of dysmetabolic and overweight patients. Metabolism. 2009;58(3):361–7.
Kaminska D, Kuulasmaa T, Venesmaa S, Kakela P, Vaittinen M, Pulkkinen L, Paakkonen M, Gylling H, Laakso M, Pihlajamaki J. Adipose tissue TCF7L2 splicing is regulated by weight loss and associates with glucose and fatty acid metabolism. Diabetes. 2012;61(11):2807–13.
Preussner M, Goldammer G, Neumann A, Haltenhof T, Rautenstrauch P, Muller-McNicoll M, Heyd F. Body temperature cycles control rhythmic alternative splicing in mammals. Mol Cell. 2017;67(3):433.e434-446.e434.
Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30(6):586–623.
Valladolid-Acebes I, Daraio T, Brismar K, Harkany T, Ogren SO, Hokfelt TG, Bark C. Replacing SNAP-25b with SNAP-25a expression results in metabolic disease. Proc Natl Acad Sci USA. 2015;112(31):E4326–35.
Jun HJ, Joshi Y, Patil Y, Noland RC, Chang JS. NT-PGC-1α activation attenuates high-fat diet-induced obesity by enhancing brown fat thermogenesis and adipose tissue oxidative metabolism. Diabetes. 2014;63(11):3615–25.
Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, Yalcin A, Fu S, Hotamisligil GS. Metabolism. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science. 2015;349(6247):500.
Nutter CA, Jaworski EA, Verma SK, Deshmukh V, Wang Q, Botvinnik OB, Lozano MJ, Abass IJ, Ijaz T, Brasier AR, Garg NJ, Wehrens XHT, Yeo GW, Kuyumcu-Martinez MN. Dysregulation of RBFOX2 is an early event in cardiac pathogenesis of diabetes. Cell Rep. 2016;15(10):2200–13.
Pihlajamaki J, Lerin C, Itkonen P, Boes T, Floss T, Schroeder J, Dearie F, Crunkhorn S, Burak F, Jimenez-Chillaron JC, Kuulasmaa T, Miettinen P, Park PJ, Nasser I, Zhao Z, Zhang Z, Xu Y, Wurst W, Ren H, Morris AJ, Stamm S, Goldfine AB, Laakso M, Patti ME. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab. 2011;14(2):208–18.
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJ, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889–94.
Bakhashab S, Filimban N, Altall RM, Nassir R, Qusti SY, Alqahtani MH, Abuzenadah AM, Dallol A. The effect sizes of PPARγ rs1801282, FTO rs9939609, and MC4R rs2229616 variants on type 2 diabetes mellitus risk among the western Saudi population: a cross-sectional prospective study. Genes (Basel). 2020;11(1):98.
Al-Serri A, Alroughani R, Al-Temaimi RA. The FTO gene polymorphism rs9939609 is associated with obesity and disability in multiple sclerosis patients. Sci Rep. 2019;9(1):19071.
Goren A, Kim E, Amit M, Bochner R, Lev-Maor G, Ahituv N, Ast G. Alternative approach to a heavy weight problem. Genome Res. 2008;18(2):214–20.
Argyropoulos G, Brown AM, Willi SM, Zhu J, He Y, Reitman M, Gevao SM, Spruill I, Garvey WT. Effects of mutations in the human uncoupling protein 3 gene on the respiratory quotient and fat oxidation in severe obesity and type 2 diabetes. J Clin Invest. 1998;102(7):1345–51.
Baerenwald DA, Bonnefond A, Bouatia-Naji N, Flemming BP, Umunakwe OC, Oeser JK, Pound LD, Conley NL, Cauchi S, Lobbens S, Eury E, Balkau B, Lantieri O, Investigators M, Dadi PK, Jacobson DA, Froguel P, O’Brien RM. Multiple functional polymorphisms in the G6PC2 gene contribute to the association with higher fasting plasma glucose levels. Diabetologia. 2013;56(6):1306–16.
Mercader JM, Liao RG, Bell AD, Dymek Z, Estrada K, Tukiainen T, Huerta-Chagoya A, Moreno-Macias H, Jablonski KA, Hanson RL, Walford GA, Moran I, Chen L, Agarwala V, Ordonez-Sanchez ML, Rodriguez-Guillen R, Rodriguez-Torres M, Segura-Kato Y, Garcia-Ortiz H, Centeno-Cruz F, Barajas-Olmos F, Caulkins L, Puppala S, Fontanillas P, Williams AL, Bonas-Guarch S, Hartl C, Ripke S, Diabetes Prevention Program Research G, Diable, Tooley K, Lane J, Zerrweck C, Martinez-Hernandez A, Cordova EJ, Mendoza-Caamal E, Contreras-Cubas C, Gonzalez-Villalpando ME, Cruz-Bautista I, Munoz-Hernandez L, Gomez-Velasco D, Alvirde U, Henderson BE, Wilkens LR, Le Marchand L, Arellano-Campos O, Riba L, Harden M, Broad Genomics P, Gabriel S, Consortium TDG, Abboud HE, Cortes ML, Revilla-Monsalve C, Islas-Andrade S, Soberon X, Curran JE, Jenkinson CP, DeFronzo RA, Lehman DM, Hanis CL, Bell GI, Boehnke M, Blangero J, Duggirala R, Saxena R, MacArthur D, Ferrer J, McCarroll SA, Torrents D, Knowler WC, Baier LJ, Burtt N, Gonzalez-Villalpando C, Haiman CA, Aguilar-Salinas CA, Tusie-Luna T, Flannick J, Jacobs SBR, Orozco L, Altshuler D, Florez JC, Consortium STDG. A loss-of-function splice acceptor variant in IGF2 is protective for type 2 diabetes. Diabetes. 2017;66(11):2903–14.
Grarup N, Moltke I, Andersen MK, Dalby M, Vitting-Seerup K, Kern T, Mahendran Y, Jorsboe E, Larsen CVL, Dahl-Petersen IK, Gilly A, Suveges D, Dedoussis G, Zeggini E, Pedersen O, Andersson R, Bjerregaard P, Jorgensen ME, Albrechtsen A, Hansen T. Loss-of-function variants in ADCY3 increase risk of obesity and type 2 diabetes. Nat Genet. 2018;50(2):172–4.
Kralovicova J, Gaunt TR, Rodriguez S, Wood PJ, Day IN, Vorechovsky I. Variants in the human insulin gene that affect pre-mRNA splicing: is − 23HphI a functional single nucleotide polymorphism at IDDM2? Diabetes. 2006;55(1):260–4.
Sun Q, Cornelis MC, Kraft P, Qi L, van Dam RM, Girman CJ, Laurie CC, Mirel DB, Gong H, Sheu CC, Christiani DC, Hunter DJ, Mantzoros CS, Hu FB. Genome-wide association study identifies polymorphisms in LEPR as determinants of plasma soluble leptin receptor levels. Hum Mol Genet. 2010;19(9):1846–55.
Acknowledgements
This work was supported by grants from: The Key R & D Projects of Zhejiang Provincial Department of Science and Technology (2021 C03094), National Natural Science Foundation (81371215 and 81670786), and Zhejiang Medical and Health Scientific Research Fund (2016 KYA127). Figures in the article are created with BioRender.com.
Funding
This work was supported by grants from: The Key R & D Projects of Zhejiang Provincial Department of Science and Technology (2021 C03094), National Natural Science Foundation (81371215 and 81670786), and Zhejiang Medical and Health Scientific Research Fund (2016 KYA127).
Author information
Authors and Affiliations
Contributions
YC completed the conceptualization and original draft preparation. YC, MZ, KW, CH, YQ, YZ, LY, ZS reviewed the manuscript; YJ and CZ supervised and revised the work critically for important intellectual content. All authors have approved the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Availability of data and material
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chao, Y., Jiang, Y., Zhong, M. et al. Regulatory roles and mechanisms of alternative RNA splicing in adipogenesis and human metabolic health. Cell Biosci 11, 66 (2021). https://doi.org/10.1186/s13578-021-00581-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-021-00581-w