
Abstract
Human cancers results in large part from the accumulation of multiple mutations. The progression of premalignant cells is an evolutionary process in which mutations provide the fundamental driving force for genetic diversity. The increased mutation rate in premalignant cells allows selection for increased proliferation and survival and ultimately leads to invasion, metastasis, recurrence, and therapeutic resistance. Therefore, it is important to understand the molecular determinants of the mutational processes. Recent genome-wide sequencing data showed that apolipoprotein B mRNA editing catalytic polypeptide-like 3B (APOBEC3B) is a key molecular driver inducing mutations in multiple human cancers. APOBEC3B, a DNA cytosine deaminase, is overexpressed in a wide spectrum of human cancers. Its overexpression and aberrant activation lead to unexpected clusters of mutations in the majority of cancers. This phenomenon of clustered mutations, termed kataegis (from the Greek word for showers), forms unique mutation signatures. In this review, we will discuss the biological function of APOBEC3B, its tumorigenic role in promoting mutational processes in cancer development and the clinical potential to develop novel therapeutics by targeting APOBEC3B.
Similar content being viewed by others

Background
It is well known that the accumulation of diverse mutations is closely linked to the development of carcinogenesis [1, 2]. Cancer genomic sequencing studies have identified a variety of mutational signatures that reflect the corresponding causes of these mutations.
Mutagenesis originates from exogenous sources found in the environment, and endogenous sources that reside intracellularly [3, 4]. Exogenous sources include radiation and chemical damage. An example is cytosine to thymine (C-to-T) transitions caused by ultraviolet light and oxidative damage, which ultimately form pyrimidine dimers [5, 6]. Endogenous sources can be further divided into passive and active sources of DNA damage. Passive alteration is characterized by an inability to repair the DNA damage after it has been triggered. The active endogenous sources of mutation are agents that impair DNA directly, including hydrolytic deamination of cytosine [7].
Previous studies have shown that normal enzymatic activity in DNA repair systems can also be a major endogenous source of DNA injury and mutation in cancer, which adds to the complexity of the mechanisms of carcinogenesis [8]. Analyses of whole-genome and exome-wide mutation data files in The Cancer Genome Atlas (TCGA) have revealed that the existence of apolipoprotein B mRNA editing catalytic polypeptide-like (APOBEC) cytidine deaminase mutagenesis patterns could have a role in somatic mutations of carcinogenesis and ultimately lead to genome instability [9, 10].
The biological function of the APOBEC family
A major contributor of mutations in many different tumor types is the APOBEC family of enzymatic DNA cytosine deaminases [11,12,13,14]. The APOBEC family came to light with the discovery that apolipoprotein B (apoB) mRNA included a cytosine to uracil (C-to-U) base modification that was not hereditarily encoded [15].
APOBEC family members normally functions as DNA mutators participating in the innate immune system that defends against their targets (retrovirus and retrotransposon) propagation. For instance, APOBEC proteins can inhibit human immunodeficiency virus type 1 (HIV-1) viral reverse transcription by DNA-editing dependent and independent processes [16,17,18,19]. The APOBEC family in most humans is composed of seven enzymes, each with conserved cytidine deaminase domains (CDAs). The human APOBEC family includes activation-induced cytosine deaminase (hAID), APOBEC1 (hA1), APOBEC2 (hA2), APOBEC3 (hA3A–hA3H) encoded in a tandem cluster on chromosome 22, and APOBEC4 on chromosome 1 [20, 21].
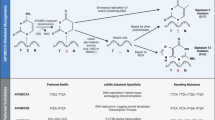
All enzymes of the AID/APOBEC family have at least one zinc-dependent catalytic domain, which contains the consensus amino acid sequence H-X-E-X23-28-P-C-X2-4-C (X stands for any amino acid) [22]. APOBEC3A, APOBEC3C, APOBEC3H, AID, and APOBEC1 have a single conserved zinc-dependent domain, while APOBEC3B, APOBEC3D, APOBEC3F, APOBEC3G have two conserved zinc-coordinating domains [14, 23] (Fig. 1a).

a The spatial location of genes encoding 11 members of APOBEC family in human. AID, APOBEC1, APOBEC3A, APOBEC3C, and APOBEC3H have single zinc-coordinating domains, whereas APOBEC3B, APOBEC3D, APOBEC3F, and APOBEC3G are double domain enzymes. The colors represent the different categories of catalytic domains in APOBECs. Green represents a Z1 catalytic domain, yellow represents a Z2 catalytic domain, and violet represents a Z3 catalytic domain. AID is represented by pink, and the rest is represented by grey. b APOBEC family enzymes catalyze the hydrolytic reaction of cytosine to uracil (C-to-U) in single-strand DNA (ssDNA) substrate
The intron/exon organization of the APOBEC3A to APOBEC3G genes was established by DNA sequencing and restriction enzyme mapping of the bacterial artificial chromosome (BAC) and P1-derived artificial chromosome (P1) clones. These genes include two eight-exon genes (APOBEC3B and 3G), one seven-exon gene (APOBEC3F), one five-exon gene (APOBEC3A), two four-exon genes (APOBEC3C and 3E), and one three-exon gene (APOBEC3D).
The primary biochemical reaction induced by APOBEC family proteins is cytosine to uracil (C-to-U) deamination (Fig. 1b). However, cytosine to guanine (C-to-G) transitions and other mutations can be induced by these enzymes [13, 14].
All of the APOBEC enzymes, except for APOBEC2 and APOBEC4, are capable of converting cytosine in ssDNA through a deamination reaction to uracil (C-to-U). The enzymatic deamination occurs at much faster rates on unprotected ssDNA substrates. However, different APOBEC enzymes with DNA editing activity can have independent physiological functions [24].
AID, emerging as one member of the first APOBECs, is a key enzyme in adaptive immunity for antibody diversity and affinity maturation. AID can initiate the somatic hypermutation and class-switch recombination of immunoglobulin genes. In addition, it can mutate chromosomal DNA at a limited number of secondary targets. This function of AID has been implicated in carcinogenesis [25, 26].
APOBEC1 is the first APOBEC family member to be identified and characterized as an RNA editor, which specifically deaminates mRNA in ApoB at cytosine6666 to uracil [27]. Other mRNA targets of APOBEC1 have been depicted recently, where the reciprocal action occurs at AU-rich sequence in 3′ untranslated regions (3′ UTRs) of diverse genes and modulates mRNA stability [28]. These physiological functions of APOBEC1 help explain mechanisms by which overexpression of APOBEC1 can initiate cancer [14].
APOBEC2 expression is well defined in the heart, skeletal muscle and tumor necrosis factor alpha (TNF-α) activated liver cells, however its precise physiological activity has yet to be determined [29,30,31]. As for APOBEC4, early and recent research has suggested that it may have a natural role in regulating host promoters or endogenous long terminal repeat (LTR) promoters [32].
The family members of genes encoding APOBEC3 proteins is positioned within a 200 kb APOBEC3 genomic cluster on human chromosome 22q13.1, and the corresponding protein function is to protect human cells against retroviruses and endogenous mobile retroelements as potent mutators of viral DNA [33]. Whereas the fundamental function of AID is in adaptive immunity, APOBEC3 members play an important part in innate immunity. Thus, APOBEC3 proteins are powerful forces against both endogenous and exogenous viruses. Nonetheless, they are closely involved in immunity in multiple ways. For example, DNA editing can be induced by A3G in adaptive immunity. Previous study designed to identify a host cell suppressor of the HIV-1 accessory protein, viral infectivity factor (VIF), reported its function as an antiviral host factor [19, 34]. A3G has also been shown to promote CD8+ cytotoxic T lymphocytes (CTL) recognition of infected T lymphatic cells and restrict marginal zone B cells, possibly resulting in a shift from a prompt immune response to a much more sustained germinal center B cell response [35]. Recent studies have shown that A3A induced by inflammation-related factors edits the mRNAs of thousands of genes, some associated with viral pathogenesis in macrophages and monocytes [36, 37]. Besides editing nuclear DNA or mitochondrial DNA and some transfected plasmids, A3A can also be involved in a novel G-to-A form of mRNA editing [38, 39].
The biological function of APOBEC3B
In general, all APOBEC3 family members can lead to hypermutation of viral genomes, which are replicated via syntheses of ssDNA intermediates. The intron/exon boundaries of APOBEC3B, APOBEC3G, and APOBEC3F are in identical positions, except APOBEC3F terminates after exon 7. In APOBEC3B, APOBEC3G, and APOBEC3F exons 2, 3, and 4 are duplicated in exons 5, 6, and 7, so that introns 1–4 are in the same position as introns 5–7 [40].
On the basis of their structure, the APOBEC3 proteins are divided into two groups. APOBEC3B, APOBEC3D, APOBEC3F, and APOBEC3G contain two zinc-dependent cytidine deaminase domains (ZD-CDAs), instead of one in APOBEC3A, APOBEC3C, and APOBEC3H [23]. Although these deaminase domains are usually conserved, they can function and evolve independently. Thus these variations can promote evolutionary flexibility [23] (Fig. 1a).
It’s well known that APOBEC3B plays a crucial role in retrovirus and endogenous retrotransposon restriction by hyperediting complementary DNA (cDNA) intermediates [41]. A3B contains two CDAs, and there are controversial reports about whether both domains are required for full editing activity in restricting HIV-1, whereas only carboxyl-terminal CDA is required for blocking HBV replication and editing bacterial DNA [2, 42]. A recent study has demonstrated that only the carboxyl-terminal CDA has C deamination activity, and N-terminal CDA is inactive [43].
Since the discovery of the APOBEC DNA mutating features in 2002, the APOBEC proteins have been linked to cancer [17]. APOBEC3 cytidine deaminase activity has been proved being involved with tumor evolution and metastasis [44, 45]. Research has shown that three human APOBEC3 members are strictly cytoplasmic (APOBEC3D, APOBEC3F and APOBEC3G) because of selection for paralogs. Previous researches have shown that APOBEC3A, APOBEC3C and APOBEC3H exhibit both cytoplasmic and nuclear localizations, but APOBEC3B is expressed almost exclusively in the nucleus. APOBEC3A and APOBEC3B can deaminate nuclear DNA as well as 5-methyl-deoxycytidine (5-MeC) residues in ssDNA, with APOBEC3A being the more efficient [2, 43, 46,47,48,49,50,51]. Furthermore, AID and APOBEC3H also have been shown to deaminate MeC [52,53,54,55,56]. It has been reported that nuclear DNA editing caused by APOBEC3A up-regulation can lead to double stranded DNA (dsDNA) breaks and apoptosis [57, 58].
The increased expression of APOBEC3B in human cancers
Increasing evidence have shown that APOBEC3B may be a predominant mutagenic agent having effects on the genesis and evolution of various cancers [4, 8, 48]. This DNA mutator hypothesis is supported by studies indicating that APOBEC3B expression is elevated in diverse forms of cancer tissues and cell lines [40, 48, 59], in contrast to its comparatively low levels in the corresponding normal human tissues spanning all major organs [8, 48, 59]. This hypothesis is also supported by its unique localization to nucleus, which can serve as a unique driving force for mutagenesis promoting tumor development [48, 60].
An in-depth analysis has shown that the APOBEC3B mutation signature is specifically enriched in at least six types of cancers, including those of the cervix, breast, lung (adeno and squamous cell), head and neck, and bladder [8, 61].
Recent observations linked DNA cytosine deaminase APOBEC3B to the mutational process driving breast carcinogenesis. These studies have demonstrated that APOBEC3B is a biomarker of poor prognosis and poor outcomes for estrogen receptor (ER)+ breast cancer, strongly indicating that genetic aberrations induced by APOBEC3B contribute to breast cancer progression [62,63,64]. Genetic, cellular and biochemical studies have demonstrated that APOBEC3B-catalyzed genomic uracil lesions are responsible for a large proportion of both dispersed and clustered mutations in multiple distinct cancers [8, 48, 61, 63, 65,66,67,68,69,70,71,72,73,74,75,76,77,78,79].
The observations of APOBEC3B overexpression in different forms of cancers are shown in Table 1.
The mutational process induced by APOBEC3B
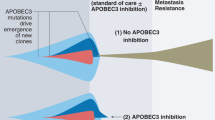
Whether APOBEC3B mutagenic activity is a potential cancer driver or a downriver effector remains an open question, and the mechanism of APOBEC3B upregulation in cancer cells needs further evidence. The collective studies suggest that the up-regulation of APOBEC3B in developing tumors promotes cancer progression [12] (Fig. 2).

The simplified process of A3B-induced tumor development
Many studies have demonstrated a positive correlation between a defined mutation signature and overexpression of APOBEC3B in many tumor types [8, 46, 53, 55, 57,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. Furthermore, the cancer types expressing the highest levels of APOBEC3B are likely to have the highest frequency of mutations. It is essential for us to obtain a general understanding of the main mutations resulting from APOBEC3B cytosine deamination to uracil.
Based on the previous studies on AID, it is established that U:G mispairs resulting from cytosine deamination can result in all six base substitution mutations [80]. While many U:G lesions are likely repaired in an error-free manner by the canonical base excision repair pathway, lesions that escape this process have multiple distinct mutagenic potentials [81]. Simple DNA replication across uracilated DNA results in C-to-T transitions. Mutagenic mismatch repair (MMR) at U:G mispairs may result in transitions and/or transversions. Translesion DNA synthesis across abasic sites can result in transition mutations. The repair process may generate nicks on both strands of the DNA double helix that are relatively close to one another potentially resulting in double-stranded breaks.
In breast cancer, APOBEC3B upregulation correlated with increased levels of transition mutations, suggesting that a proportion of the genomic uracils created by APOBCE3B either persist through DNA synthesis or are generated at a high enough rate that they are detectable in non-replicated DNA [48]. If a uracil is not excised by a DNA glycosylase prior to DNA replication, it will template as a thymine and base pair with adenosine. After a subsequent round of DNA replication, the result is a C-to-T transition mutation.
C → T transitions in multiple human cancers have been suggested to be caused by APOBEC3B. A uracil residue results from APOBEC3B cytosine deamination can be excised by uracil DNA glycosylase and then generates an abasic site (AP site) leading to insertion of adenine opposite the AP site [82]. Thus APOBEC3B editing results in C → T transitions in carcinogenesis. Other processes like spontaneous or chemical-induced cytosine deamination, error-prone bypass can also create AP site and C → T transitions. APOBEC3B preferentially deaminates cytosine residues when it is adjacent to a 5′ thymine and a 3′ thymine or adenine [83]. Current studies have shown that only cytosine substitutions that occur within the trinucleotide TCA or TCT sequence context are attributed to APOBEC3B mutagenesis [4].
In addition to C deamination of APOBEC3B contributes to mutagenesis, recently studies have shown that a methionine residue at the joint of the carboxyl-terminal CDA and the N-terminal CDA has been proved to play a role in high mutagenicity [51, 84]. It has been established that the A3B’s capability of 5-MeC deamination is much less efficient than that of APOBEC3A [2, 43, 46,47,48,49,50,51]. Although the carboxyl-terminal CDA of APOBEC3B have been shown to comparatively weakly convert some 5-MeC into T in ssDNA substrates, the C-to-U deamination of APOBEC3B is much more efficient than that of APOBEC3A [51]. Multiple factors contributing to the 5-MeC deamination activity and specificity by APOBEC3B may promote mutagenesis [43, 51].
Studies have shown that a significantly large subset of Asian (37%), Amerindian (58%), and Oceania (93%) populations have a deletion in the APOBEC3B gene, which is associated with an approximate 20-fold increase in the expression of an APOBEC3A from an mRNA variant containing the 3′-UTR of APOBEC3B [85]. This 29.5 kB deletion between exon 5 in APOBEC3A and exon 8 in APOBEC3B is linked to increased risk for breast cancer, hepatocellular carcinoma (HCC) and epithelial ovarian cancer, whereas this deletion polymorphism is not involved with clinical outcome of mammary cancer regardless of APOBEC3B mRNA levels [13, 86,87,88,89].
Conclusion
Above all, APOBEC3B may represent an important marker for various human cancers and a strong candidate for targeted intervention, especially given its essential nature to tumor progression and heterogeneity. Therefore APOBEC3B inhibition may decrease the rate of cancer progression and keep the stability of the targeted genome [48]. Future in-depth research is demanded to understand APOBEC3B protein regulation and the potential interaction with many other oncogenes and tumor suppressors. All studies of APOBEC3B in the last decade show that APOBEC3B will be a promising target for cancer prevention and therapy.
Abbreviations
- C-to-T:
-
cytosine to thymine
- TCGA:
-
The Cancer Genome Atlas
- APOBEC:
-
apolipoprotein B mRNA editing catalytic polypeptide-like
- AID:
-
activation-induced cytidine deaminase
- C-to-U:
-
cytosine to uracil
- HIV-1:
-
human immunodeficiency virus type 1
- CDAs:
-
cytidine deaminase domains
- C-to-G:
-
cytosine to guanine
- ssDNA:
-
single-strand DNA
- BAC:
-
bacterial artificial chromosome
- P1:
-
P1-derived artificial chromosome
- 3′ UTRs:
-
3′ untranslated regions
- TNF-α:
-
tumor necrosis factor alpha
- LTR:
-
long terminal repeat
- VIF:
-
viral infectivity factor
- CTL:
-
cytotoxic T lymphocytes
- G-to-A:
-
guanine to adenine
- ZD-CDAs:
-
zinc-dependent cytidine deaminase domains
- dsDNA:
-
double stranded DNA
- ER:
-
estrogen receptor
- HCC:
-
hepatocellular carcinoma
- NSCLC:
-
non-small cell lung cancer
- EGFR:
-
epidermal growth factor receptor
- KRAS:
-
kirsten rat sarcoma viral oncogene
- OSCC:
-
oral squamous cell carcinomas
- MMR:
-
mismatch repair
- AP site:
-
abasic site
- 5-MeC:
-
5-methyl-deoxycytidine
References
Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002;347(20):1593–603.
Shinohara M, Io K, Shindo K, Matsui M, Sakamoto T, Tada K, Kobayashi M, Kadowaki N, Takaori-Kondo A. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci Rep. 2012;2:806.
Luch A. Nature and nurture—lessons from chemical carcinogenesis. Nat Rev Cancer. 2005;5(2):113–25.
Kuong KJ, Loeb LA. APOBEC3B mutagenesis in cancer. Nat Genet. 2013;45(9):964–5.
Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485(7399):502–6.
Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, Davis S, Stemke-Hale K, Davies MA, Gershenwald JE, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43(5):442–6.
Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–76.
Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45(9):977–83.
Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–9.
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–60.
Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014;14(12):786–800.
Burns MB, Leonard B, Harris RS. APOBEC3B: pathological consequences of an innate immune DNA mutator. Biomed J. 2015;38(2):102–10.
Henderson S, Fenton T. APOBEC3 genes: retroviral restriction factors to cancer drivers. Trends Mol Med. 2015;21(5):274–84.
Swanton C, McGranahan N, Starrett GJ, Harris RS. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 2015;5(7):704–12.
Powell LM, Wallis SC, Pease RJ, Edwards YH, Knott TJ, Scott J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell. 1987;50(6):831–40.
Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4(11):868–77.
Harris RS, Petersen-Mahrt SK, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10(5):1247–53.
Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418(6893):99–103.
Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–50.
Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008;9(6):229.
Macduff DA, Harris RS. Directed DNA deamination by AID/APOBEC3 in immunity. Curr Biol. 2006;16(6):R186–9.
Vieira VC, Soares MA. The role of cytidine deaminases on innate immune responses against human viral infections. Biomed Res Int. 2013;2013:683095.
LaRue RS, Jonsson SR, Silverstein KA, Lajoie M, Bertrand D, El-Mabrouk N, Hotzel I, Andresdottir V, Smith TP, Harris RS. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol Biol. 2008;9:104.
Ehrlich M, Norris KF, Wang RY, Kuo KC, Gehrke CW. DNA cytosine methylation and heat-induced deamination. Biosci Rep. 1986;6(4):387–93.
Pavri R, Nussenzweig MC. AID targeting in antibody diversity. Adv Immunol. 2011;110:1–26.
Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HC 3rd, Nussenzweig MC, Dalla-Favera R. AID is required for germinal center-derived lymphomagenesis. Nat Genet. 2008;40(1):108–12.
Chester A, Scott J, Anant S, Navaratnam N. RNA editing: cytidine to uridine conversion in apolipoprotein B mRNA. Biochim Biophys Acta. 2000;1494(1–2):1–13.
Blanc V, Park E, Schaefer S, Miller M, Lin Y, Kennedy S, Billing AM, Ben Hamidane H, Graumann J, Mortazavi A, et al. Genome-wide identification and functional analysis of Apobec-1-mediated C-to-U RNA editing in mouse small intestine and liver. Genome Biol. 2014;15(6):R79.
Liao W, Hong SH, Chan BH, Rudolph FB, Clark SC, Chan L. APOBEC-2, a cardiac- and skeletal muscle-specific member of the cytidine deaminase supergene family. Biochem Biophys Res Commun. 1999;260(2):398–404.
Lada AG, Krick CF, Kozmin SG, Mayorov VI, Karpova TS, Rogozin IB, Pavlov YI. Mutator effects and mutation signatures of editing deaminases produced in bacteria and yeast. Biochem (Mosc). 2011;76(1):131–46.
Sato Y, Probst HC, Tatsumi R, Ikeuchi Y, Neuberger MS, Rada C. Deficiency in APOBEC2 leads to a shift in muscle fiber type, diminished body mass, and myopathy. J Biol Chem. 2010;285(10):7111–8.
Marino D, Perkovic M, Hain A, Jaguva Vasudevan AA, Hofmann H, Hanschmann KM, Muhlebach MD, Schumann GG, Konig R, Cichutek K, et al. APOBEC4 enhances the replication of HIV-1. PLoS ONE. 2016;11(6):e0155422.
Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol. 2008;26:317–53.
Smith HC. APOBEC3G: a double agent in defense. Trends Biochem Sci. 2011;36(5):239–44.
Beck-Engeser GB, Winkelmann R, Wheeler ML, Shansab M, Yu P, Wunsche S, Walchhutter A, Metzner M, Vettermann C, Eilat D, et al. APOBEC3 enzymes restrict marginal zone B cells. Eur J Immunol. 2015;45(3):695–704.
Sharma S, Patnaik SK, Taggart RT, Kannisto ED, Enriquez SM, Gollnick P, Baysal BE. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat Commun. 2015;6:6881.
Shaban NM, Shi K, Li M, Aihara H, Harris RS. 1.92 angstrom zinc-free APOBEC3F catalytic domain crystal structure. J Mol Biol. 2016;428(11):2307–16.
Niavarani A, Currie E, Reyal Y, Anjos-Afonso F, Horswell S, Griessinger E, Luis Sardina J, Bonnet D. APOBEC3A is implicated in a novel class of G-to-A mRNA editing in WT1 transcripts. PLoS ONE. 2015;10(3):e0120089.
Kostrzak A, Henry M, Demoyen PL, Wain-Hobson S, Vartanian JP. APOBEC3A catabolism of electroporated plasmid DNA in mouse muscle. Gene Ther. 2015;22(1):96–103.
Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79(3):285–96.
Refsland EW, Harris RS. The APOBEC3 family of retroelement restriction factors. Curr Top Microbiol Immunol. 2013;371:1–27.
Bogerd HP, Wiegand HL, Doehle BP, Cullen BR. The intrinsic antiretroviral factor APOBEC3B contains two enzymatically active cytidine deaminase domains. Virology. 2007;364(2):486–93.
Fu Y, Ito F, Zhang G, Fernandez B, Yang H, Chen XS. DNA cytosine and methylcytosine deamination by APOBEC3B: enhancing methylcytosine deamination by engineering APOBEC3B. Biochem J. 2015;471(1):25–35.
Nowarski R, Kotler M. APOBEC3 cytidine deaminases in double-strand DNA break repair and cancer promotion. Cancer Res. 2013;73(12):3494–8.
Cescon DW, Haibe-Kains B, Mak TW. APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc Natl Acad Sci USA. 2015;112(9):2841–6.
Caval V, Suspene R, Vartanian JP, Wain-Hobson S. Orthologous mammalian APOBEC3A cytidine deaminases hypermutate nuclear DNA. Mol Biol Evol. 2014;31(2):330–40.
Suspene R, Aynaud MM, Guetard D, Henry M, Eckhoff G, Marchio A, Pineau P, Dejean A, Vartanian JP, Wain-Hobson S. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc Natl Acad Sci USA. 2011;108(12):4858–63.
Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494(7437):366–70.
Carpenter MA, Li M, Rathore A, Lackey L, Law EK, Land AM, Leonard B, Shandilya SM, Bohn MF, Schiffer CA, et al. Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J Biol Chem. 2012;287(41):34801–8.
Wijesinghe P, Bhagwat AS. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 2012;40(18):9206–17.
Siriwardena SU, Guruge TA, Bhagwat AS. Characterization of the catalytic domain of human APOBEC3B and the critical structural role for a conserved methionine. J Mol Biol. 2015;427(19):3042–55.
Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279(50):52353–60.
Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci USA. 2003;100(7):4102–7.
Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463(7284):1101–5.
Larijani M, Frieder D, Sonbuchner TM, Bransteitter R, Goodman MF, Bouhassira EE, Scharff MD, Martin A. Methylation protects cytidines from AID-mediated deamination. Mol Immunol. 2005;42(5):599–604.
Gu J, Chen Q, Xiao X, Ito F, Wolfe A, Chen XS. Biochemical characterization of APOBEC3H variants: implications for their HIV-1 restriction activity and mC modification. J Mol Biol. 2016;428(23):4626–38.
Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011;12(5):444–50.
Mussil B, Suspene R, Aynaud MM, Gauvrit A, Vartanian JP, Wain-Hobson S. Human APOBEC3A isoforms translocate to the nucleus and induce DNA double strand breaks leading to cell stress and death. PLoS ONE. 2013;8(8):e73641.
Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38(13):4274–84.
Lackey L, Demorest ZL, Land AM, Hultquist JF, Brown WL, Harris RS. APOBEC3B and AID have similar nuclear import mechanisms. J Mol Biol. 2012;419(5):301–14.
Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45(9):970–6.
Sieuwerts AM, Willis S, Burns MB, Look MP, Meijer-Van Gelder ME, Schlicker A, Heideman MR, Jacobs H, Wessels L, Leyland-Jones B, et al. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm Cancer. 2014;5(6):405–13.
Tsuboi M, Yamane A, Horiguchi J, Yokobori T, Kawabata-Iwakawa R, Yoshiyama S, Rokudai S, Odawara H, Tokiniwa H, Oyama T, et al. APOBEC3B high expression status is associated with aggressive phenotype in Japanese breast cancers. Breast Cancer. 2016;23(5):780–8.
Periyasamy M, Patel H, Lai CF, Nguyen VT, Nevedomskaya E, Harrod A, Russell R, Remenyi J, Ochocka AM, Thomas RS, et al. APOBEC3B-mediated cytidine deamination is required for estrogen receptor action in breast cancer. Cell Rep. 2015;13(1):108–21.
Tokunaga E, Yamashita N, Tanaka K, Inoue Y, Akiyoshi S, Saeki H, Oki E, Kitao H, Maehara Y. Expression of APOBEC3B mRNA in primary breast cancer of Japanese women. PLoS ONE. 2016;11(12):e0168090.
Cescon DW, Haibe-Kains B. DNA replication stress: a source of APOBEC3B expression in breast cancer. Genome Biol. 2016;17(1):202.
Law EK, Sieuwerts AM, LaPara K, Leonard B, Starrett GJ, Molan AM, Temiz NA, Vogel RI, Meijer-van Gelder ME, Sweep FC, et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci Adv. 2016;2(10):e1601737.
Zhang J, Wei W, Jin HC, Ying RC, Zhu AK, Zhang FJ. The roles of APOBEC3B in gastric cancer. Int J Clin Exp Pathol. 2015;8(5):5089–96.
Jin Z, Han YX, Han XR. The role of APOBEC3B in chondrosarcoma. Oncol Rep. 2014;32(5):1867–72.
Luo X, Huang Y, Chen Y, Tu Z, Hu J, Tavis JE, Huang A, Hu Y. Association of hepatitis B virus covalently closed circular DNA and human APOBEC3B in hepatitis B virus—related hepatocellular carcinoma. PLoS ONE. 2016;11(6):e0157708.
Wu PF, Chen YS, Kuo TY, Lin HH, Liu CW, Chang LC. APOBEC3B: a potential factor suppressing growth of human hepatocellular carcinoma cells. Anticancer Res. 2015;35(3):1521–7.
Xu L, Chang Y, An H, Zhu Y, Yang Y, Xu J. High APOBEC3B expression is a predictor of recurrence in patients with low-risk clear cell renal cell carcinoma. Urol Oncol. 2015;33(8):340 e341–348.
Gwak M, Choi YJ, Yoo NJ, Lee S. Expression of DNA cytosine deaminase APOBEC3 proteins, a potential source for producing mutations, in gastric, colorectal and prostate cancers. Tumori. 2014;100(4):112e–7e.
Lamy P, Nordentoft I, Birkenkamp-Demtroder K, Thomsen MB, Villesen P, Vang S, Hedegaard J, Borre M, Jensen JB, Hoyer S, et al. Paired exome analysis reveals clonal evolution and potential therapeutic targets in urothelial carcinoma. Cancer Res. 2016;76(19):5894–906.
Yan S, He F, Gao B, Wu H, Li M, Huang L, Liang J, Wu Q, Li Y. Increased APOBEC3B predicts worse outcomes in lung cancer: a comprehensive retrospective study. J Cancer. 2016;7(6):618–25.
Sasaki H, Suzuki A, Tatematsu T, Shitara M, Hikosaka Y, Okuda K, Moriyama S, Yano M, Fujii Y. APOBEC3B gene overexpression in non-small-cell lung cancer. Biomed Rep. 2014;2(3):392–5.
Kosumi K, Baba Y, Ishimoto T, Harada K, Nakamura K, Ohuchi M, Kiyozumi Y, Izumi D, Tokunaga R, Taki K, et al. APOBEC3B is an enzymatic source of molecular alterations in esophageal squamous cell carcinoma. Med Oncol. 2016;33(3):26.
Fanourakis G, Tosios K, Papanikolaou N, Chatzistamou I, Xydous M, Tseleni-Balafouta S, Sklavounou A, Voutsinas GE, Vastardis H. Evidence for APOBEC3B mRNA and protein expression in oral squamous cell carcinomas. Exp Mol Pathol. 2016;101(3):314–9.
Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL, et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013;73(24):7222–31.
Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22.
Krokan HE, Saetrom P, Aas PA, Pettersen HS, Kavli B, Slupphaug G. Error-free versus mutagenic processing of genomic uracil—relevance to cancer. DNA Repair (Amst). 2014;19:38–47.
Sousa MM, Krokan HE, Slupphaug G. DNA-uracil and human pathology. Mol Asp Med. 2007;28(3–4):276–306.
Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol. 2010;17(2):222–9.
Chen J, Miller BF, Furano AV. Repair of naturally occurring mismatches can induce mutations in flanking DNA. Elife. 2014;3:e02001.
Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007;3(4):e63.
Qi G, Xiong H, Zhou C. APOBEC3 deletion polymorphism is associated with epithelial ovarian cancer risk among Chinese women. Tumour Biol. 2014;35(6):5723–6.
Liu J, Sieuwerts AM, Look MP, van der Vlugt-Daane M, Meijer-van Gelder ME, Foekens JA, Hollestelle A, Martens JW. The 29.5 kb APOBEC3B deletion polymorphism is not associated with clinical Ooutcome of breast cancer. PLoS One. 2016;11(8):e0161731.
Han Y, Qi Q, He Q, Sun M, Wang S, Zhou G, Sun Y. APOBEC3 deletion increases the risk of breast cancer: a meta-analysis. Oncotarget. 2016;7(46):74979-86.
Rezaei M, Hashemi M, Hashemi SM, Mashhadi MA, Taheri M. APOBEC3 deletion is associated with breast cancer risk in a sample of southeast iranian population. Int J Mol Cell Med. 2015;4(2):103–8.
Authors’ contributions
JZ, CW, XM, ED and GP planed the manuscript outline. JZ and CW wrote the draft manuscript, EW and XM revised the manuscript, GP finalized the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was also supported by Cancer Center Support Grant CA016672 to The University of Texas MD Anderson Cancer Center and by, Susan G. Komen for the Cure Foundation CCR14300500 to G.P.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data are fully available without restriction.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zou, J., Wang, C., Ma, X. et al. APOBEC3B, a molecular driver of mutagenesis in human cancers. Cell Biosci 7, 29 (2017). https://doi.org/10.1186/s13578-017-0156-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-017-0156-4