
Abstract
The yeast Saccharomyces cerevisiae is widely used as a cell factory to produce recombinant proteins. However, S. cerevisiae naturally secretes only a few proteins, such as invertase and the mating alpha factor, and its secretory capacity is limited. It has been reported that engineering protein anterograde trafficking from the endoplasmic reticulum to the Golgi apparatus by the moderate overexpression of SEC16 could increase recombinant protein secretion in S. cerevisiae. In this study, the retrograde trafficking in a strain with moderate overexpression of SEC16 was engineered by overexpression of ADP-ribosylation factor GTP activating proteins, Gcs1p and Glo3p, which are involved in the process of COPI-coated vesicle formation. Engineering the retrograde trafficking increased the secretion of α-amylase but did not induce production of reactive oxygen species. An expanded ER membrane was detected in both the GCS1 and GLO3 overexpression strains. Physiological characterizations during batch fermentation showed that GLO3 overexpression had better effect on recombinant protein secretion than GCS1 overexpression. Additionally, the GLO3 overexpression strain had higher secretion of two other recombinant proteins, endoglucanase I from Trichoderma reesei and glucan-1,4-α-glucosidase from Rhizopus oryzae, indicating overexpression of GLO3 in a SEC16 moderate overexpression strain might be a general strategy for improving production of secreted proteins by yeast.
Similar content being viewed by others


Introduction
The yeast Saccharomyces cerevisiae is a well-known eukaryal cell factory for producing many valuable chemicals and proteins (Huang et al. 2014), due to its fast growth and robustness (Hong and Nielsen 2012) and being Generally Recognized As Safe (GRAS) (Shusta et al. 1998). Furthermore, the ease of doing genetic manipulation, the availability of many molecular tools, datasets and databases facilitate work with S. cerevisiae (Cox and Mann 2011; Hawkins et al. 2010; Reaves and Rabinowitz 2011; Snyder and Gallagher 2009). Like other Eukarya, S. cerevisiae has a secretory pathway, which means that recombinant proteins can undergo folding, disulfide bond formation, glycosylation, and be transported out of the cell (Hou et al. 2012b). However, the secretory capacity of wild type S. cerevisiae is limited (Idiris et al. 2010), and improving the secretory capacity through engineering of this pathway would reduce the cost of downstream processes (Nielsen 2013). Over the past few years, we have successfully improved recombinant protein secretion by S. cerevisiae (Hou et al. 2012a; Huang et al. 2015; Liu et al. 2012; Martínez et al. 2016). Liu et al. reported that different signal peptides have different effects on the secretion of recombinant proteins (Liu et al. 2012). The signal peptide is recognized by the signal recognition particle on the endoplasmic reticulum (ER), and the recombinant protein is co-translationally translocated into the ER (Plath et al. 1998). The nascent peptide undergoes disulfide bond formation in the ER with the assistance of the chaperone Pdi1p. Schröder and Robinson reported that overexpressing PDI1 improves recombinant protein secretion in yeast (Robinson et al. 1994; Schröder 2008). High level of recombinant protein expression may, however, cause ER stress due to protein misfolding. When unfolded proteins accumulate in the ER, the so-called unfolded protein response (UPR) will be triggered to relieve ER stress (Hou et al. 2012b). Overexpression of the transcription factor Hac1p, which is one of the key regulators of the UPR, enhances the secretion of α-amylase (Valkonen et al. 2003). Additionally, engineering the vesicle trafficking successfully increases heterologous protein secretion. Hou et al. found that overexpression of Sly1p, an SM (Sec1/Munc-18) family protein that regulates trafficking from the ER to the Golgi, enhances α-amylase secretion but not for human insulin precursor or invertase, whereas overexpression of Sec1p, which also belongs to the SM family, improves the secretion of all three proteins (Hou et al. 2012a). Recently, we found that engineering the anterograde trafficking between the ER to the Golgi by moderate overexpression of SEC16 increases recombinant protein secretion (Bao et al. 2017). Sec16p, an ER peripheral protein, accumulates at the ER exit sites (ERESs) and serves as a scaffold for the formation of coat protein complex II (COPII) vesicles which transport the cargo protein from the ER to the Golgi (Jensen and Schekman 2011; Supek et al. 2002). In S. cerevisiae, the native level of Sec16p is lower than that of the other COPII vesicle proteins, which may cause a limitation in the COPII vesicle formation (Feizi et al. 2013). Moderate expression of SEC16 generates more ERESs facilitating the formation of COPII vesicles. A less expanded ER combined with the result of reactive oxygen species (ROS) staining indicates a reduced level of ER stress in SEC16 overexpression strain (Bao et al. 2017). The early secretory pathway is bidirectional: anterograde trafficking mediates the transportation of cargo protein from the ER to the Golgi, and the retrograde trafficking mediated by coat protein complex I (COPI) vesicles retrieves the necessary components for continued anterograde trafficking (Pelham 1995; Poon et al. 1999). The process of COPI-coated vesicle formation is similar to that of COPII-coated vesicles. GDP bounded Arf1p is activated by guanine exchange factor (ArfGEF) by exchanging GDP with GTP, which triggers the formation of COPI-coated vesicles (Poon et al. 1999). Following this the coatomers are recruited by the activated Arf1p-GTP followed by recruitment of the cargo proteins. The COPI-coated vesicles are formed subsequently. The mature COPI vesicles are prompted by the disassembly of coatomers, which is caused by GTP hydrolysis via the GTP activating proteins (GAPs) Gcs1p and Glo3p (Poon et al. 1999).
In our previous study, we found that moderate overexpression of SEC16 increases secretion of a range of heterologous proteins by S. cerevisiae (Bao et al. 2017). Overexpression of SEC16 provides more ERESs for cargo protein export. However, the enhanced flux of ER-to-Golgi transportation may bring excess lipids and ER associated proteins, such as v-SNARE proteins, to the Golgi via COPII vesicles (Szul and Sztul 2011). Sequential coupling between COPII and COPI vesicles is important to coordinate and direct bi-directional vesicular trafficking between the ER and the Golgi apparatus (Aridor et al. 1995), suggesting that improving the recycling of these components would further increase the trafficking flow to enhance protein secretion.
Here we used the recombinant protein α-amylase from Aspergillus oryzae as a reporter to evaluate the secretory capacity in yeast. We amplified the retrograde trafficking pathway by overexpressing GAPs, Gcs1p and Glo3p, in a SEC16-overexpression strain to further increase the secretion of heterologous proteins in yeast.
Materials and methods
Strains and media
The strains and plasmids used in this study are listed in Table 1. The primers used in this study are listed in Additional file 1: Table S1. Yeast strain CEN.PK530-6CK (MATa URA3 HIS3 LEU2 TRP1 SUC2 MAL2-8c tpi1(41-707)::loxP P GPD -SEC16) was used as a heterologous protein secretion host. Plasmid pAlphaAmyCPOT, which contains α-amylase from Aspergillus oryzae with α-factor leader was transformed into CEN.PK530-6CK, named YIGS16 (Bao et al. 2017). The yeast strains CEN.PK530-6CK + EG and CEN.PK530-6CK + AGL were constructed in the same way as YIGS16, that is the plasmids pAlphaTrEGCPOT and pCP-aGLA were transformed into CEN.PK530-6CK respectively, resulting CEN.PK530-6CK + EG and CEN.PK530-6CK + AGL (Bao et al. 2017).
The promoter replacement cassette “UP(GCS1)-amdSYM(GCS1)-TEF(GCS1)-DW(GCS1)” for GCS1 was constructed by the following steps: The “UP(GCS1)” fragment is the upstream flanking region, which was amplified from the yeast genome by using primers GCS1-up-100-F and GCS1-up-100-R-amds; the “amdSYM(GCS1)” is the selection marker, which was amplified from plasmid pamdSYM by primers amdS-F and REC-amdSYM-R(gcs1); The “TEF(GCS1)” fragment is the promoter P TEF which was amplified by REC-P-TEF1-F(GCS1) and gcs1-TEF1-R based on the yeast genome; The “DW(GCS1)” fragment is the downstream flanking region, which was amplified from the yeast genome by primers GCS1-F and GCS1-MID-R; then the four fragments “UP(GCS1)”, “amdSYM”, “TEF” and “DW(GCS1)” were jointed together by fusion PCR, resulting in the replacement cassette “UP(GCS1)-amdSYM(GCS1)-TEF(GCS1)-DW(GCS1)”. Similarly, the promoter replacement cassette “UP(GLO3)-amdSYM(GLO3)-TEF(GLO3)-DW(GLO3)” was constructed by using primer pairs “GLO3-up-120-F and GLO3-up-120-R-amds”, “amdS-F and REC-amdSYM-R(glo3)”, “REC-P-TEF1-F(glo3) and glo3-TEF1-R” and “GLO3-F and GLO3-MID-R” for amplification of fragments “UP(GLO3)”, “amdSYM(GLO3)”, “TEF(GLO3)” and “DW(GLO3)” respectively, and then jointing together by fusion PCR. The replacement of the GCS1 native promoter or the GLO3 native promoter by the TEF promoter was completed by transforming the replacement cassette “UP(GCS1)-amdSYM(GCS1)-TEF(GCS1)-DW(GCS1)” or “UP(GLO3)-amdSYM(GLO3)-TEF(GLO3)-DW(GLO3)” into the cell. Yeast strain AACK-GCS1 was constructed by transformation of the “UP(GCS1)-amdSYM(GCS1)-TEF(GCS1)-DW(GCS1)” cassette to strain AACK. The strains AACK-GLO3, YIGLO3, GLO3 + EG and GLO3 + AGL were constructed by transformation of the “UP(GLO3)-amdSYM(GLO3)-TEF(GLO3)-DW(GLO3)” cassette to strains AACK, YIGS16, CEN.PK530-6CK + EG and CEN.PK530-6CK + AGL, respectively. The transformants were selected on acetamide plate (Solis-Escalante et al. 2013). The acetamide medium contained 3 g/L KH2PO4, 0.5 g/L MgSO4·7H2O, 0.6 g/L acetamide, 6.6 g/L K2SO4, 1 mL/L of a trace element solution (Verduyn et al. 1992) and 1 mL/L a vitamin solution (Verduyn et al. 1992). The yeast strain YIGLO3GCS1 was constructed by the following steps: The amdSYM marker was removed from YIGLO3 genome in YPD medium, and the colonies were counter-selected on a fluoroacetamide plate as described (Solis-Escalante et al. 2013). Then the promoter replacement cassette “UP(GCS1)-amdSYM(GCS1)-TEF(GCS1)-DW(GCS1)” was transformed into the YIGLO3 amdSYM-removal strain, resulting in YIGLO3GCS1.
Under non-selective conditions, yeast strains were grown in YPD medium containing 10 g/L yeast extract, 20 g/L peptone and 20 g/L glucose. The yeast strains were cultured in SD-2xSCAA (Wittrup and Benig 1994) medium for recombinant protein production containing 20 g/L glucose, 6.9 g/L yeast nitrogen base without amino acids, 190 mg/L arginine, 400 mg/L aspartic acid, 1260 mg/L glutamic acid, 130 mg/L glycine, 140 mg/L histidine, 290 mg/L isoleucine, 400 mg/L leucine, 440 mg/L lysine, 108 mg/L methionine, 200 mg/L phenylalanine, 220 mg/L threonine, 40 mg/L tryptophan, 52 mg/L tyrosine, 380 mg/L valine, 1 g/L BSA, 5.4 g/L Na2HPO4, and 8.56 g/L NaH2PO4·H2O (pH = 6.0 by NaOH). The cultivation time for tube fermentation was 96 h. In the bioreactor batch fermentations, SD-2xSCAA medium was used. Strains were inoculated into 600 mL of SD-2xSCAA medium in a 1-L bioreactor (DasGip, Jülich, Germany) at 30 °C. The bioreactor system was run at 600 rpm, and 36 L/h air flow, and the pH value was maintained at 6 by the addition of NaOH.
Analytical methods
The cell dry weight and the concentration of glucose, ethanol and glycerol were detected as described previously (Hou et al. 2012a). Briefly, a high performance liquid chromatography (HPLC) (Dionex, Sunnyvale, CA, USA) was used with an Aminex HPX-87H column (Bio-Rad, Hercules, USA) at 65 °C. 5 mM H2SO4 was used as mobile phase with a flow rate of 0.6 mL/min.
Enzyme activity quantification
The amylase enzyme activity was quantified by the assay kit (Megazyme K-CERA, Wicklow, Ireland). α-Amylase from A. oryzae was used as a standard. The enzyme activity of endoglucanase was measured by a cellulase assay kit (Megazyme K-CELLG3) at 50 °C for 10 min. The enzyme activity of glucan 1,4-α-glucosidase was measured by an amyloglucosidase assay kit (Megazyme R-AMGR3, Wicklow, Ireland).
Intracellular α-amylase extraction
Yeast cells were harvested and washed by 1× phosphate buffer solution (pH 7.4) (PBS) twice. Then the cell pellets were resuspended in 1 mL 1× PBS with 10 μL of halt protease inhibitor cocktail (Thermo Fisher, Waltham, MA, USA). Next, 500 μL cell solution was transferred into 1.0 mm silica spheres lysing matrix tube (MP Biomedicals, Santa Ana, CA, USA). The cells were lysed using fastprep-24 tissue and cell homogenizer (MP Biomedicals, Santa Ana, CA, USA) at 6.5 m/s for 2 min. The cells were kept on ice during the 5 min interval between the two runs. The supernatant was collected by centrifugation for α-amylase quantification. The cell wall associated α-amylase was considered as intracellular α-amylase.
Reactive oxygen species measurement
The cells for reactive oxygen species (ROS) measurement were cultivated in tube at 30 °C and collected when OD600 reached 1. Then 1 OD600 of the cells of each samples were washed by 1× PBS twice and 50 mM sodium citrate buffer (pH 5) (SCB) once. The cell pellets were resuspended in 1 mL of SCB with 1 μL of 50 mM dihydrorhodamine 123 (Thermo Fisher, Waltham, MA, USA), and then were incubated in the dark at room temperature for 30 min. After the incubation, the cells were washed and resuspended in 1 mL of SCB. 200 μL of the cell samples were loaded into 96-well black plate for detection by fluorescence microplate reader (FLUOstar Omega, BMG LABTECH, Germany) with a 485-nm excitation filter and a 520-nm emission filter.
ER membrane staining
The cells for ER membrane staining were cultivated in tube at 30 °C and collected when OD600 reached 1. Then 1 OD600 of the cells were washed by 1× PBS twice and Hanks’ Balance Salt Solution without phenol red (HBSS) once. The cell pellets were resuspended in 1 mL of HBSS with 5 μL of ER-Tracker Blue-White DPX (Thermo Fisher, Waltham, MA, USA), and incubated for 30 min at 30 °C. After the incubation, the cells were washed and resuspended in 1 mL of HBSS. 200 μL of the cell samples were loaded into 96-well black plate for detection by fluorescence microplate reader with a 355-nm excitation filter and a 590-nm emission filter.
Statistical method
Student’s t test was used for statistical significance testing in this study.
Results
Overexpression of GCS1 or GLO3 improves the titer of α-amylase in the SEC16-overexpression strain
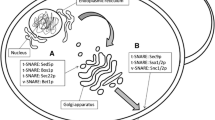
To increase the protein secretory capacity in a SEC16 overexpression strain by enlarging the retrograde trafficking pathway, two ARF GAPs mediating COPI vesicle formation, Gcs1p and Glo3p were overexpressed (Fig. 1a). Overexpression of GCS1 or GLO3 enhanced the titer of α-amylase 19.7 and 25.1%, respectively, when the cells were grown in tube cultures for 96 h (Fig. 1b). The final biomass yield of the GCS1 or GLO3 overexpression strains, YIGCS1 and YIGLO3, were slightly higher than the reference strain YIGS16 (Fig. 1b). Interestingly, we found that overexpression of GCS1 or GLO3 in the reference strain AACK (yeast strain without overexpression of SEC16) significantly decreased the titer of α-amylase secretion (Fig. 1c). We also tried to overexpress GCS1 and GLO3 simultaneously in YIGS16, yet the α-amylase titer did not further increase compared with that of single overexpression of GLO3 in YIGS16 (resulting in strain YIGLO3) (Additional file 1: Figure S1). This may be due to a redundant function provided by GCS1 and GLO3, each one ensuring sufficient vesicular transport from the Golgi apparatus to the ER (Poon et al. 1999). Hence, only overexpression of either GCS1 or GLO3 was considered in subsequent experiments.

Overexpression of GCS1 or GLO3 by promoter replacement in the SEC16-overexpression strain (YIGS16) has a positive effect on α-amylase secretion. a Constructions of the vesicle trafficking engineering strains. SEC16 is involved in COPII vesicle formation; GCS1 and GLO3 are involved in COPI vesicle formation. b The titer and biomass of YIGS16, YIGCS1 and YIGLO3. c The titer and biomass of AACK, AACK-GCS1 and AACK-GLO3. *P < 0.05; **P < 0.01; ***P < 0.001. Measurements are reported as the average value ± standard deviation from independent triplicates
Overexpression of GCS1 or GLO3 does not increase ROS
Recombinant protein production may cause ROS accumulation in cells due to the requirement for additional capacity requirement for oxidative protein folding in the ER (Tu and Weissman 2002; Tyo et al. 2012). In this study, we aimed to return more necessary components for the anterograde trafficking process in YIGS16 to strengthen the recombinant protein transportation by amplifying the retrograde trafficking, and we therefore evaluated how the ROS level was altered in the strains with a more balanced vesicle trafficking. From this analysis we found that even though the GCS1 and GLO3 overexpression strains had increased α-amylase secretion this was not associated with additional ROS accumulation (Additional file 1: Figure S2). This implied that overexpression of GCS1 or GLO3 did not cause increased cellular stress.
Engineering the retrograde trafficking increases the surface of the ER membrane
As one of the functions of the retrograde transportation is retrieving the necessary proteins with the lipids for the continuous anterograde trafficking (Poon et al. 1999), we were interested to test if there was any change in ER membrane surface in the engineered strains. We have shown previously that moderate expression of SEC16 reduced ER membrane surface (AACK) so we therefore measured the ER membrane surfaces in YIGS16, YIGCS1 and YIGLO3 by ER labeling with the ER-Tracker Blue-white DPX (Echevarria et al. 2003), and found that the surfaces of ER membranes in YIGCS1 and YIGLO3 were slightly higher than in YIGS16 (Fig. 2a). Meanwhile, we also stained the ER membranes of AACK, AACK-GCS1 and AACK-GLO3, and GCS1 and GLO3 overexpression in AACK also resulted in an increase in ER surface area (Fig. 2b). This suggested that the ER membrane was enlarged when GCS1 or GLO3 were overexpressed. Enlarged ER membrane was likely a result of more lipids returned to the ER by enhanced retrograde trafficking.

The amounts of ER membrane of strains a YIGS16, YIGCS1 and YIGLO3. b AACK, AACK-GCS1 and AACK-GLO3 using ER-Tracker Blue-White DPX. The ER membrane changes were quantified by their fluorescence intensity. *P < 0.05; ***P < 0.001. Measurements are reported as the average value ± standard deviation from independent triplicates
Strain YIGLO3 has higher α-amylase yield throughout the cultivation
As both strains YIGCS1 and YIGLO3 showed increased final α-amylase titer in tube fermentations (Fig. 1b), we investigated α-amylase secretion throughout a fermentation process. Strains were therefore cultured in batch bioreactors, and samples were taken for analysis throughout the fermentation (Fig. 3a, b). The secreted α-amylase yield (titer/biomass ratio) was calculated at five stages of the process, which included the exponential growth phase (OD600nm ≈ 1), the end of the glucose growth phase, the middle of the ethanol growth phase, the end of the ethanol growth phase and at the end of the fermentation. The α-amylase yield was higher for YIGLO3 in all stages compared with the reference strain YIGS16, while YIGCS1 only showed a higher α-amylase yield in the end (Fig. 3c). There was no significant difference in the percentage of intracellular α-amylase and intracellular α-amylase per cell for the three strains (Fig. 3d and Additional file 1: Figure S3).

Batch fermentation of the strain YIGS16, YIGCS1 and YIGLO3. The time course of a the biomass and b the α-amylase titer. c The titer/biomass ratio and d the percentage of intracellular α-amylase of the three strains at five different time points. Measurements are reported as the average value ± standard deviation from independent triplicates
Besides increased α-amylase secretion, physiological changes were also found for the engineered strains, especially for YIGLO3. Both YIGCS1 and YIGLO3 had higher maximum specific growth rates and higher final biomass titers compared with strain YIGS16 (Table 2, Fig. 3a). YIGLO3 had a significantly higher specific α-amylase production rate and yield of α-amylase in the glucose phase (Table 2). Additionally, YIGLO3 had a higher specific ethanol production rate, higher ethanol production peak, lower specific glycerol production rate and lower glycerol production peak compared with YIGS16 (Table 2, Additional file 1: Figures S4b, c). In contrast, no significant difference was found in these parameters between YIGCS1 and YIGS16. Furthermore, the glucose consumption profile was similar for all three strains (Additional file 1: Figure S4a), and there were no significant changes in the specific glucose uptake rates among the three strains (Table 2).
Overexpression of GLO3 improves secretion of two other recombinant proteins
To investigate whether overexpression of GLO3 can also increase the production of other recombinant proteins, two heterologous proteins, Trichoderma reesei endoglucanase I and Rhizopus oryzae glucan-1,4-α-glucosidase were evaluated. The production of both proteins significantly increased, about 30%, in the GLO3 overexpression strain, and just as for YIGLO3 the final biomass titer of both strains increased about 5% compared to that of the corresponding reference strain (Fig. 4).

Secretion of two recombinant proteins, endoglucanase and glucan 1,4-α-glucosidase, in the reference strain and GLO3-overexpression strain. ***P < 0.001 Measurements are reported as the average value ± standard deviation from independent quadruplicates
Discussion
ER-to-Golgi translocation of recombinant proteins is mediated by COPII-coated vesicles (Jensen and Schekman 2011). Previously, we showed that moderate overexpression (but not high-level overexpression) of SEC16 in yeast improved protein secretion by enhancing the anterograde transport from the ER to the Golgi apparatus (Bao et al. 2017). Meanwhile, the enhanced ER-to-Golgi flux caused by expressing SEC16 resulted in more ER membranes and ER membrane proteins to be directed to the anterograde vesicles to the Golgi, and this resulted in reduced ER membranes (Bao et al. 2017). COPI-coated vesicles are responsible for retrieving these essential components from the Golgi apparatus to the ER for continuous anterograde trafficking (Poon et al. 1999). Here by overexpression of GCS1 or GLO3 we increased the retrograde trafficking from the Golgi to the ER in the YIGS16 background strain expressing SEC16 and we also detected increased amounts of ER membranes in YIGCS1 and YIGLO3. Hereby more essential components were recycled to the ER and could be reused for anterograde trafficking, resulting in increased protein secretion.
In the mammalian cells, interference of COPI vesicle formation by the addition of ArfGEF inhibitor brefeldin A shows a negative effect on ER export (Ward et al. 2001), which may result from the shortage of the protein components required for the COPII vesicle assembly that occurs since the retrograde trafficking from the Golgi to the ER was inadequate (Brandizzi and Barlowe 2013). A similar phenomenon was observed that the secretory pathway was impaired when the anterograde trafficking was disturbed by the disruption of COPII vesicle formation using Sar1p [H79G], a GTP locked version (Aridor et al. 1995; Pepperkok et al. 1998; Ward et al. 2001). These results imply that the balance between anterograde and retrograde trafficking plays an important role in the secretory pathway. Wildtype S. cerevisiae has a limited protein secretory capacity and secretes naturally only a few proteins such as α-factor, invertase, and aspartyl protease (Carlson et al. 1983; Idiris et al. 2010; Komano et al. 1999). In our previous study, we found that increasing the anterograde trafficking by moderate expression of SEC16 improves the yeast secretory pathway (Bao et al. 2017), which implies the natural anterograde trafficking flow in S. cerevisiae is inadequate. When only GLO3 or GCS1 was overexpressed in the wildtype strain AACK, the titer of α-amylase was decreased. This may be due to the inadequacy of the anterograde trafficking as the retrograde trafficking was overflown. However, when GLO3 or GCS1 were overexpressed in the anterograde trafficking amplifying strain YIGS16, the secretory capacity could be further increased. This might be the trafficking flow between the ER and the Golgi apparatus was improved by engineering the retrograde trafficking, reaching a better balance. In the controlled condition, the effect of GCS1 overexpression on protein secretion was inconspicuous compared with GLO3. This is due to that Glo3p plays a major role in the COPI vesicle formation (Poon et al. 1999), which could also explain AACK-GLO3 has an even lower α-amylase titer than AACK-GCS1 (Fig. 1c).
ER expansion by the deletion of the lipid-regulator OPI1 alleviates ER stress and increases the secretion of IgG in S. cerevisiae (de Ruijter et al. 2016; Schuck et al. 2009). Interestingly, the overexpression of GCS1 or GLO3 increases the amounts of ER membranes in AACK and YIGS16, but has different effects on the secretion, which might be due to the two different manners of ER expansion. In our case, the ER was enlarged by amplifying the retrograde trafficking, while it was expanded through inducing the lipid biosynthesis genes by the deletion of OPI1 (Schuck et al. 2009).
In our previous study, we found that moderate overexpression of SEC16 causes a lower specific growth rate and final biomass yield compared with the reference strain (Bao et al. 2017). Here, the retrograde trafficking engineered strains, YIGCS1 and YIGLO3, showed higher specific growth rate and higher final biomass yield, which indicated that cellular stress caused by SEC16 overexpression was reduced by anaplerosis of the retrograde trafficking. This could be the result that the trafficking flows were balanced between the ER and the Golgi apparatus through engineering the retrograde trafficking in the anterograde trafficking amplifying strain. Accordingly, there was no indication of increased oxidative stress in strains YIGCS1 and YIGLO3, as measured by ROS, despite a higher flux through the secretory pathway. In addition, we also observed that the titers were increased and the biomass was slightly, but significantly, in the strains with a balanced trafficking compared with the only anterograde trafficking amplifying strain, when overexpressing two other recombinant proteins. This implied that this vesicle trafficking balanced system could be used a general strategy for designing improved recombinant protein producing strains.
In conclusion, our strategy may be generally applicable for improving recombinant protein production in yeast, as we have shown that the strategy resulted in improved secretion of three different recombinant proteins when GLO3 was overexpressed. In conclusion, we report the positive effect of engineering the retrograde trafficking in a SEC16 moderate overexpression yeast strain on the secretion of three different recombinant proteins. We detected increased ER membrane surface and an unchanged ROS accumulation in the engineered strains, which suggest that increased trafficking turnover benefits recombinant protein secretion as well as the cellular stress level.
Abbreviations
- ER:
-
endoplasmic reticulum
- UPR:
-
unfolded protein response
- ERES:
-
endoplasmic reticulum exit site
- COPII:
-
coat protein complex II
- COPI:
-
coat protein complex I
- ArfGEF:
-
Arf1p guanine exchange factor
- GAP:
-
GTP activating protein
- ROS:
-
reactive oxygen species
References
Aridor M, Bannykh SI, Rowe T, Balch WE (1995) Sequential coupling between COPII and COPI vesicle coats in endoplasmic-reticulum to Golgi transport. J Cell Biol 131(4):875–893. https://doi.org/10.1083/jcb.131.4.875
Bao J, Huang M, Petranovic D, Nielsen J (2017) Moderate expression of SEC16 increases protein secretion by Saccharomyces cerevisiae. Appl Environ Microbiol 83(14):e03400–e03416
Brandizzi F, Barlowe C (2013) Organization of the ER–Golgi interface for membrane traffic control. Nat Rev Mol Cell Biol 14(6):11
Carlson M, Taussig R, Kustu S, Botstein D (1983) The secreted form of invertase in Saccharomyces cerevisiae is synthesized from mRNA encoding a signal sequence. Mol Cell Biol 3:439–447
Cox J, Mann M (2011) Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem 80:273–299
de Ruijter JC, Koskela EV, Frey AD (2016) Enhancing antibody folding and secretion by tailoring the Saccharomyces cerevisiae endoplasmic reticulum. Microb Cell Fact 15:87. https://doi.org/10.1186/s12934-016-0488-5
Echevarria W, Leite MF, Guerra MT, Zipfel WR, Nathanson MH (2003) Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat Cell Biol 5(5):440–446
Feizi A, Österlund T, Petranovic D, Bordel S, Nielsen J (2013) Genome-scale modeling of the protein secretory machinery in yeast. PLoS ONE 8(5):e63284. https://doi.org/10.1371/journal.pone.0063284
Hawkins RD, Hon GC, Ren B (2010) Next-generation genomics: an integrative approach. Nat Rev Genet 11(7):476–486
Hong KK, Nielsen J (2012) Metabolic engineering of Saccharomyces cerevisiae: a key cell factory platform for future biorefineries. Cell Mol Life Sci CMLS 69(16):2671–2690
Hou J, Tyo K, Liu Z, Petranovic D, Nielsen J (2012a) Engineering of vesicle trafficking improves heterologous protein secretion in Saccharomyces cerevisiae. Metab Eng 14(2):120–127
Hou J, Tyo KE, Liu Z, Petranovic D, Nielsen J (2012b) Metabolic engineering of recombinant protein secretion by Saccharomyces cerevisiae. FEMS Yeast Res 12(5):491–510
Huang M, Bao J, Nielsen J (2014) Biopharmaceutical protein production by Saccharomyces cerevisiae: current state and future prospects. Pharm Bioprocess 2(2):167–182
Huang M, Bai Y, Sjostrom SL, Hallstrom BM, Liu Z, Petranovic D, Uhlen M, Joensson HN, Andersson-Svahn H, Nielsen J (2015) Microfluidic screening and whole-genome sequencing identifies mutations associated with improved protein secretion by yeast. Proc Natl Acad Sci USA 112(34):E4689–E4696
Idiris A, Tohda H, Kumagai H, Takegawa K (2010) Engineering of protein secretion in yeast: strategies and impact on protein production. Appl Microbiol Biotechnol 86(2):403–417. https://doi.org/10.1007/s00253-010-2447-0
Jensen D, Schekman R (2011) COPII-mediated vesicle formation at a glance. J Cell Sci 124:1–4
Komano H, Rockwell N, Wang GT, Krafft GA, Fuller RS (1999) Purification and characterization of the yeast glycosylphosphatidylinositol-anchored, monobasic-specific aspartyl protease yapsin 2 (Mkc7p). J Biol Chem 274(34):24431–24437
Liu Z, Tyo KE, Martinez JL, Petranovic D, Nielsen J (2012) Different expression systems for production of recombinant proteins in Saccharomyces cerevisiae. Biotechnol Bioeng 109(5):1259–1268
Martínez JL, Meza E, Petranovic D, Nielsen J (2016) The impact of respiration and oxidative stress response on recombinant α-amylase production by Saccharomyces cerevisiae. Metab Eng Commun 3:205–210
Nielsen J (2013) Production of biopharmaceutical proteins by yeast: advances through metabolic engineering. Bioengineered 4(4):207–211
Pelham HRB (1995) Sorting and retrieval between the endoplasmic-reticulum and Golgi-apparatus. Curr Opin Cell Biol 7(4):530–535
Pepperkok R, Lowe M, Burke B, Kreis TE (1998) Three distinct steps in transport of vesicular stomatitis virus glycoprotein from the ER to the cell surface in vivo with differential sensitivities to GTP gamma S. J Cell Sci 111(Pt 13):1877–1888
Plath K, Mothes W, Wilkinson BM, Stirling CJ, Rapoport TA (1998) Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell 94(6):795–807
Poon PP, Cassel D, Spang A, Rotman M, Pick E, Singer RA, Johnston GC (1999) Retrograde transport from the yeast Golgi is mediated by two ARF GAP proteins with overlapping function. EMBO J 18:555–564
Reaves ML, Rabinowitz JD (2011) Metabolomics in systems microbiology. Curr Opin Biotechnol 22(1):17–25
Robinson AS, Hines V, Wittrup KD (1994) Protein disulfide isomerase overexpression increases secretion of foreign proteins in Saccharomyces cerevisiae. Bio/Technology 12(4):381–384
Schröder M (2008) Engineering eukaryotic protein factories. Biotechnol Lett 30(2):187–196
Schuck S, Prinz WA, Thorn KS, Voss C, Walter P (2009) Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol 187(4):525–536
Shusta EV, Raines RT, Pluckthun A, Wittrup KD (1998) Increasing the secretory capacity of Saccharomyces cerevisiae for production of single-chain antibody fragments. Nat Biotechnol 16(8):773–777
Snyder M, Gallagher JE (2009) Systems biology from a yeast omics perspective. FEBS Lett 583(24):3895–3899
Solis-Escalante D, Kuijpers NG, Bongaerts N, Bolat I, Bosman L, Pronk JT, Daran JM, Daran-Lapujade P (2013) amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res 13(1):126–139
Supek F, Madden DT, Hamamoto S, Orci L, Schekman R (2002) Sec16p potentiates the action of COPII proteins to bud transport vesicles. J Cell Biol 158(6):1029–1038. https://doi.org/10.1083/jcb.200207053
Szul T, Sztul E (2011) COPII and COPI traffic at the ER–Golgi interface. Physiology 26(5):348–364. https://doi.org/10.1152/physiol.00017.2011
Tu BP, Weissman JS (2002) The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol Cell 10(5):983–994
Tyo KE, Liu Z, Petranovic D, Nielsen J (2012) Imbalance of heterologous protein folding and disulfide bond formation rates yields runaway oxidative stress. BMC Biol 10:16
Valkonen M, Penttila M, Saloheimo M (2003) Effects of inactivation and constitutive expression of the unfolded-protein response pathway on protein production in the yeast Saccharomyces cerevisiae. Appl Environ Microbiol 69:2065–2072
Verduyn C, Postma E, Scheffers WA, Van Dijken JP (1992) Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8(7):501–517. https://doi.org/10.1002/yea.320080703
Ward TH, Polishchuk RS, Caplan S, Hirschberg K, Lippincott-Schwartz J (2001) Maintenance of Golgi structure and function depends on the integrity of ER export. J Cell Biol 155(4):557–570. https://doi.org/10.1083/jcb.200107045
Wittrup KD, Benig V (1994) Optimization of amino-acid supplements for heterologous protein secretion in Saccharomyces cerevisiae. Biotechnol Tech 8(3):161–166. https://doi.org/10.1007/Bf00161582
Authors’ contributions
JB and JN conceived the study. JB and MH designed and performed the experiments. DP and JN supervised the project. JB, MH, DP and JN analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was financially supported by the Novo Nordisk Foundation, the Knut and Alice Wallenberg Foundation and Swedish Strategic Research Foundation. We thank Dr. Verena Siewers, Dr. José L. Martínez, Dr. Guokun Wang and Dr. Xin Chen for useful comments and discussions.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article and its additional files.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was funded by the Novo Nordisk Foundation, the Knut and Alice Wallenberg Foundation and Swedish Strategic Research Foundation.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1.
Additional table and figures.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bao, J., Huang, M., Petranovic, D. et al. Balanced trafficking between the ER and the Golgi apparatus increases protein secretion in yeast. AMB Expr 8, 37 (2018). https://doi.org/10.1186/s13568-018-0571-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-018-0571-x