
Abstract
Laccases are multicopper oxidases that are able to catalyze reactions involving a range of substrates, including phenols and amines, and this ability is related to the existence of different laccases. Basidiomycetes usually have more than one gene for laccase, but until now, this feature has not been demonstrated in a marine-derived fungus. Peniophora sp. CBMAI 1063 is a basidiomycete fungus isolated from a marine sponge that exhibits the ability to secrete significant amounts of laccase in saline conditions. In the present study, we identified laccase sequences from the transcriptome of Peniophora sp. CBMAI 1063 and used them to perform different molecular in silico analyses. The results revealed the presence of at least eight putative genes, which may encode ten different laccases with peptide lengths ranging from 482 to 588 aa and molecular weights ranging from 53.5 to 64.4 kDa. These laccases seem to perform extracellular activities, with the exception of one that may represent an intracellular laccase. The 10 predicted laccases expressed by Peniophora sp. CBMAI 1063 in laccase-induced media showed different patterns of N-glycosylation and isoelectric points and are divided into two classes based on the residue associated with the regulation of the redox potential of the enzyme. None of the predicted laccases showed more than 61% similarity to other fungal laccases. Based on the differences among the laccases expressed by Peniophora sp. CBMAI 1063, this marine-derived basidiomycete represents a valuable resource with strong potential for biotechnological exploitation.
Similar content being viewed by others

Introduction
Laccases (EC 1.10.3.2) are oxidoreductases that are widespread in nature and present in plants, insects, bacteria and fungi, though more expressly in the white rot fungal group (Giardina et al. 2010; Rivera-Hoyos et al. 2013). These enzymes seem to perform different physiological functions, such as lignin synthesis and degradation, spore pigmentation, cell wall elongation and stress defenses (Riva 2006; Giardina et al. 2010).
As a multicopper oxidase, the laccase has an active site with four copper ions. The copper ions are classified per Electron Paramagnetic Resonance (EPR) into three types: type 1—paramagnetic, “blue” ion; type 2—paramagnetic “non-blue” ion, and type 3—diamagnetic pair ion. In general, the type 1 copper ion are linked to two histidine residues, one cysteine residue, and one leucine or phenylalanine residue, while one type 2 and a pair of type 3 ions form a trinuclear cluster linked to eight histidine residues (Claus 2004; Giardina et al. 2010).
Sequence analyses have demonstrated that fungal laccases differ from other multicopper oxidases by a sequence signature corresponding to four conserved regions, namely, L1, L2, L3, and L4. These regions display not only the 12 residues that bind the copper ions but also non-ligand residues, which are involved in the three-dimensional structure of the active site (Kumar et al. 2003; Giardina et al. 2010).
Laccases are known to be capable of accepting a range of substrates such as phenols, amines, and diols, promoting the oxidation of these substrates while reducing molecular oxygen to water (Claus 2004; Riva 2006). Due to these features, laccases have been exploited for biotechnological applications, mainly in the pulp, paper and textile industries and biodegradation of a variety of xenobiotic compounds (Pezzella et al. 2015; Viswanath et al. 2014).
According to Bonugli-Santos et al. (2015), enzymes from marine-derived fungi may have different properties in comparison with that those produced by terrestrial relatives, due to different environmental conditions, such as salinity, temperature, and pressure. Considering the tolerance to saline conditions, these microorganisms are important microbial resources for biotechnological application in bioremediation, including degradation of polycyclic aromatic hydrocarbons (PAH) in ocean and marine sediments (Raghukumar et al. 2006; Passarini et al. 2011). Additionally, a large number of textile processes can generate effluents in saline and alkaline conditions, which can be efficiently decolorized/degraded by fungi from marine environments (Raghukumar et al. 2008; Verma et al. 2010; Chen et al. 2014).
Peniophora sp. CBMAI 1063 is a marine-derived basidiomycete that has the ability to express many laccases under saline and non-saline conditions (Bonugli-santos et al. 2010) and biodegrade 94% of the textile dye Reactive Black 5 (RB5) under saline conditions without the production of mutagenic products during the process (Bonugli-Santos et al. 2016). The culture conditions for laccase production by Peniophora sp. CBMAI 1063 have been optimized, and a patent have been requested (Bonugli-Santos et al. 2016).
In a previous study, two putative laccase genes from Peniophora sp. CBMAI 1063 were suggested based on fragments of approximately 150 bp (Bonugli-santos et al. 2010). However, complete laccase sequences were not available for this fungus. Therefore, the aims of the present study were to obtain the complete laccase sequences of the marine-derived fungus Peniophora sp. CBMAI 1063 (after being cultured under optimized conditions for laccase production) and to perform in silico analysis of all sequences in order to compare them with sequences from other basidiomycete fungi.
Materials and methods
Microorganism and culture conditions
Peniophora sp. CBMAI 1063 was isolated from the Brazilian sponge Amphimedon viridis collected in the town of São Sebastião, São Paulo, Brazil (Menezes et al. 2010) and taxonomically identified as reported by Bonugli-Santos et al. (2010). The strain is being maintained using different preservation methods at the Brazilian Collection of Environmental and Industrial Microorganisms—CBMAI (UNICAMP, SP, Brazil) and at the UNESP Central of Microbial Resources—CRM-UNESP (UNESP, SP, Brazil).
The fungus was cultivated for 7 days at 28 °C in a laccase expression-optimized medium (patent request deposited at Instituto Nacional de Propriedade Industrial—INPI under the number BR102014008502) composed of yeast extract (0.2%), bacteriological peptone (0.27%), malt extract (0.14%), d-glucose (0.27%), and artificial sea water adapted from Kester et al. (1967), ASW: 0.704% MgCl2, 0.098% CaCl2, 0.001% SrCl2, 1.555% NaCl, 0.261% Na2SO4, 0.044% KCL, 0.013% NaHCO3, 0.006% KBr and 0.002% H3BO3, supplemented with 2 mM CuSO4 as laccase inductor.
RNA extraction and sequencing
Total RNA from Peniophora sp. CBMAI 1063 was extracted using the RNeasyPlant Mini Kit (QIAGEN), according to manufacturer’s protocol. The integrity of the RNA was examined by 0.7% agarose gel electrophoresis, and the concentration was estimated using a NanoDrop 2000 spectrophotometer. The cDNA library construction and sequencing were performed in 1/3 lane using the Illumina Hiseq 2000 platform, paired-end 2 × 100 bp according to the manufacturer’s protocol from MACROGEN (Seoul, South Korea).
De novo assembly and functional annotation
The reads quality was assessed using the FastQC (Andrews 2010) program. Trimming of reads was performed with trimmomatic (Bolger et al. 2014) using the minimum quality filtering (Phred 20) functionality of this tool with a sliding window, which scans through reads from the 5′ end and removes subsequent bases from the 3′ end once the average quality score within the window drops below a user-specified value (minimum size 50 bp).
De novo assembly was performed using Trinity (Grabherr et al. 2011) with the parameter ‘min_kmer_cov 2’ following the method described by Haas et al. (2013). The use of this parameter increases the stringency for reads being assembled together (Chapman 2015). Thus, only the kmers that occur more than once are considered for the contigs, and the default is that all kmers are considered (Johnson 2015). We prepared a set of non-redundant contigs (unigenes) by selecting only the longest contigs among the isoforms.
The functional annotation was performed using the Blast2GO PRO version (Gotz et al. 2008) that describes the unigenes using the BLASTx algorithm (Altschul et al. 1990) with an E-value threshold of 1.0E−3 against the NCBI non-redundant (Nr) database to identify protein domains with the InterProScan (Zdobnov and Apweiler 2001) tool and assign the gene ontology (GO) and enzyme commission (EC) terms. Annotations using Blast2GO were conducted with 1.0E−6 as the E-value hit filter, 55 as the annotation cut-off and 5 as the GO weight.
Analysis of the laccase sequences
Sequences that returned from the Nr database as laccase were submitted to ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/). The ORFs with the largest lengths were selected, and the translated products were aligned using ClustalW (Bioedit 7.0). After the alignments, a search of the conserved regions L1, L2, L3, and L4 was performed according to Kumar et al. (2003), in order to obtain only true laccases.
GeneRunner 5.0 was used to determine the size length of the coding sequence and the peptide chain. The peptide composition, molecular weight and isoelectric point (pI) were determined using ProtParam (http://web.expasy.org/protparam/) (Gasteiger et al. 2005). The similarity analysis with other fungal laccases and multicopper oxidases was performed using MegAlign (DNASTAR 14.1.0.115) (Eggert et al. 1998); the DNA and protein sequences from other organisms used in this analysis were obtained from the NCBI database. SignalP 4.1 (http://www.cbs.dtu.dk/services/SignalP/) was used, with SignalP 3.0 default, to recognize signal peptide for extracellular activity and predict cleavage sites for Peptidase I (Bendtsen et al. 2004; Petersen et al. 2011). The prediction of N-glycosylation sites was performed with the NetNGlyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/) (Vite-Vallejo et al. 2009), and the results were confirmed using GlicoEP (http://www.imtech.res.in/raghava/glycoep/submit.html) (Chauhan et al. 2013). The phylogenetic analysis was performed using MEGA 6.0 (Tamura et al. 2013). The distances were calculated using the neighbor-joining method and a bootstrap with 1000 pseudoreplications (Felsenstein 1985; Saitou and Nei 1987).
Accession numbers
The raw sequences data from the Peniophora sp. CBMAI 1063 transcriptome are available at Short Read Archives (SRA) GenBank database, deposited under the Accession Number No. SRR5799684 (BioProject: PRJNA392894). Putative laccase genes were also deposited in GenBank under the followed Accession Numbers: Lcc1 no. MF176136; Lcc2 no. MF176137; Lcc3 no. MF176138; Lcc3B no. MF176139; Lcc4 no. MF176140; Lcc5 no. MF176141; Lcc5B no. MF176142; Lcc6 no. MF176143; Lcc7 no. MF176144; Lcc8 no. MF176145.
Experimental in vitro validation
Two of the laccase sequences obtained from Peniophora sp. CBMAI was selected and cloned in Escherichia coli. The specific primers to each one of the sequences were designed using GeneRunner 5.0 (Additional file 1: Table S1). A first RT-PCR was performed according to the manufacture’s protocol (RevertAid H Minus Reverse Transcriptase—Thermo Scientific) with the oligo-dT primer to reverse transcribe the total mRNA of the fungus to cDNA. Afterward, laccase sequences amplification was performed by touchdown PCR using the designed primers. PCR conditions were as follows: 2 min of initial denaturation at 94 °C, followed by a touchdown step of 30 s from 74 °C to 62 °C (due to the difference of the forward and reverse annealing primers), 35 cycles of 30 s at 94 °C and 30 s at 62 °C and a final extension step of 5 min at 72 °C. PCR products were detected by 0.7% agarose gel electrophoresis, purified using the GeneJET gel Extraction Kit (Thermo Scientific) according to manufacturer’s protocol, and ligated into the pJET 1.2 cloning vector (Thermo Scientific). The E. coli DH10B strain was used as the cloning host, and six clones were selected to be sequenced using the Sanger method at MACROGEN (Seoul, South Korea).
Results
Transcriptome annotation
Sequencing generated 11,005,713,864 total bases and 108,967,464 reads. Trinity de novo assembly generated 36,981 contigs (including isoforms) with an average length of 1552 bp. A total of 16,663 non-redundant contigs (unigenes) were selected. The Blast2GO PRO results showed that 10,649 unigenes had significant similarity to known proteins in NCBI-Nr, 8367 had significant similarity with the InterPro domains and 3838 unigenes presented at least one GO term.
Among the unigenes submitted to the NR protein database (NCBI), 43% presented high similarity to other sequences, and all the top hits were related to terrestrial basidiomycetes. The Heterobasidion irregulare and Stereum hirsutum sequences presented the highest similarities to the Peniophora sp. CBMAI 1063 unigenes (Additional file 1: Figure S1).
The unigenes (3838) assigned to GO terms level 2 were classified into 39 functional groups belonging to three categories: molecular functions, biological process, and cellular process. Within molecular functions, “catalytic activity” and “binding” represented the most abundant subcategories with 1260 unigenes and 972 unigenes, respectively, while “metabolic processes”, “cellular processes”, and “single-organism processes” were the most representative subcategories in biological processes, with 1056, 956, and 757 unigenes, respectively. Finally, “cell”, with 471 unigenes, was the most representative functional group in cellular processes (Additional file 1: Figure S2).
Among the enzymes expressed by the fungus Peniophora sp. CBMAI 1063, transferases, with 180 unigenes, comprised the most representative group, followed by hydrolases with 169 unigenes and oxidoreductases with 111 unigenes.
Analysis and characterization of the laccase transcripts
Forty-seven sequences of laccase were found in the transcriptome. Among them, 13 presented all four conserved regions that are characteristic of known laccases. All putative laccases showed similarity to laccases from other basidiomycetes and multicopper oxidases from another Peniophora species. However, three putative laccase sequences were likely pseudogenes lacking a stop codon (comp15071_c0_seq1 and comp15071_c0_seq4) or presenting a stop codon interposed within the coding sequence (comp8257_c0_seq1).
Figure 1 shows the alignment of the 10 putative laccase sequences containing the four conserved regions and copper ligand sites. The sequences contained 1449–1767 bp, and all of them presented high GC contents, with the percentage ranging from 52.2 to 58.9%. The predicted polypeptide chain varied between 482 and 588 aa with peptide weights ranging from 53.5 to 64.4 kDa. The laccases found in the transcriptome represent extracellular laccases, with the exception of Lcc5B, which did not show a peptide cleavage site and seemed to be an intracellular enzyme. Table 1 shows the complete characterization with base pair length, peptide chain length, molecular weight, GC content, cleavage site for Peptidase I and theoretical pI, of all 10 putative laccases.

Predicted amino acid sequence alignments of all 10 putative laccases from Peniophora sp. CBMAI 1063. Amino acids with 100% matches are highlighted in black. Numbers above the amino acids indicate that they are copper ion-bound. Dots below the amino acids indicate conserved regions in the fungal laccases (L1, L2, L3 and L4)
Amino acid sequence analysis revealed that two types of laccases were expressed by Peniophora sp. CBMAI 1063 based on a variable copper type 1 ligand, which is related to the influence in the reduction–oxidation potential. At the variable position, six sequences contained leucine and four contained phenylalanine.
Except for Lcc5B, all laccases exhibited approximately four to ten sites that could be N-glycosylated; some sites were common to more than one sequence, and other sites were similar to those found in laccases from different fungi (Table 2).
The putative laccases of Peniophora sp. CBMAI 1063 showed high similarity (80–93%) to the multicopper oxidases found in the genome of Peniophora sp. (Nagy et al. 2015) but presented low similarity (below 60%) to other fungal laccases (Table 3).
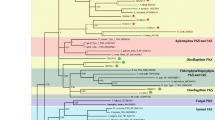
Data from phylogenetic analysis suggest a gene family with eight different genes, due to the formation of eight different clades involving all 10 putative laccases. Furthermore, according to the tree (Fig. 2) Lcc3 and Lcc3B should be considered identical laccases, as well as Lcc5 and Lcc5B. However, the amino acid analyses revealed that short insertions differentiated these laccases. This result leads to a conclusion that the enzymes Lcc3/Lcc3B and Lcc5/Lcc5B may arises from alternative splicing of the genes Lcc3 and Lcc5, respectively.

Phylogenetic tree based on the Lcc1 to Lcc8 sequences, other basidiomycete laccases and putative multicopper oxidases from Peniophora sp. (see Nagy et al. 2015). Two complete laccase families from Flammulina velutipes and Coprinopsis cinerea are presented in the tree. The scale bar indicates a distance equivalent to 0.1 amino acid substitutions per site
The gene family from Peniophora sp. CBMAI 1063 did not group with other fungal laccases and formed a separate cluster that included seven multicopper oxidases from Peniophora sp. However, Lcc8 grouped in a separated clade with only one other multicopper oxidase (Fig. 2).
In vitro validation
The most expressed laccase, according with FPKM factor (data not shown), did not present stop codon in its sequence and was considered as pseudogene thus two other laccases were selected based on high similarity with the most expressed laccase also using FPKM factor (data not shown): Lcc3 and Lcc3B. Although amplifications showed sequences with the expected size, it was not possible to clone and sequence fragments from Lcc3. Six clones from Lcc3B were sequenced and compared with the sequence obtained in the transcriptome. After amplification, the Lcc3B sequence showed approximately 1500-bp band in the agarose gel (Fig. 3). The sequence of the cloned fragment was 100% identical to the sequence of Comp15071_c0_seq5 from transcriptome (Table 1).

PCR of comp15071_c0_seq5 (Lcc5B) with three different polymerases. Bands with the length of approximately 1.500 bp correspond to the predicted size of the sequence. a amplification with Pfu platinum DNA polymerase (Thermo Scientific), b 1-kb ladder (Promega), c amplification with Taq DNA polymerase (Promega), d amplification with Phusion DNA polymerase (Thermo Scientific)
Discussion
According to Giardina et al. (2010), most of the fungal laccases are glycoproteins with extracellular activity and molecular weights ranging from 60 to 70 kDa. The majority of putative laccases expressed by Peniophora sp. CBMAI 1063 had molecular weights near or higher than 60 kDa, corresponding to extracellular enzymes. However, Lcc5B seems to play an intracellular role. The existence of an intracellular laccase has already been reported in Trametes versicolor (Schlosser et al. 1997), Pleurotus ostreatus (Palmieri et al. 2000), and Flammulina velutipes (Wang et al. 2015) and may be related in these organisms to the low molecular weight phenol oxidation, cell division and elongation processes (Baldrian 2006; Wang et al. 2015).
Eggert et al. (1998), suggested three classes of laccases based on the variable residues that bind the copper type 1 ion (molecular analysis). Class 1 has methionine, class 2 has leucine, and class 3 has phenylalanine at this position. According to this classification, six putative laccases from Peniophora sp. CBMAI 1063 belong to class 2, while four laccases belong to class 3. Site-directed mutagenesis of the residues that occupy this position seems to interfere with the redox potential due to the alteration in the coordination of the T1 copper ion (Xu et al. 1996, 1999). The theoretical pI prediction ranged from 4.21 to 6.12, based on differences found in the amino acid compositions of the putative laccases. These results were expected, and together with other results, these data reinforce the idea that the laccases from Peniophora sp. CBMAI 1063 may act on different substrates under acidic conditions.
Laccases generally have an expressive glycosidic portion, which may represent approximately 10–45% of the total mass (Claus 2004). Mannose seems to be the most representative carbohydrate in fungal laccases, and in association with other sugars, mannose constitutes the glycosidic moiety. The glycosidic portion guarantee the stability in the enzyme, minimize protease susceptibility, signal extracellular activity, and influence redox potential (Dwivedi et al. 2011; Vite-Vallejo et al. 2009). In the present study, different N-glycosylation sites were predicted for nine putative laccases, which presented among 4–10 possible sites. However, some sites were too close to each other to allow simultaneous glycosylation. In this sense, sites that were homologous to those found in other fungal laccases could in fact be glycosylated.
The occurrence of multiple laccase genes seems to be recurrent in many basidiomycete genomes. The first laccase gene family was reported in Agaricus bisporus, which exhibited two different laccase genes in the same chromosome (Giardina et al. 2010). Afterward, other gene families were reported in Trametes villosa, and F. velutipes with 13 and 11 genes (Wang et al. 2015), respectively, and Coprinopsis cinerea with 17 genes (Kilaru et al. 2006). Representatives of the genus Peniophora were also reported as laccase producers with at least five different laccase isoenzymes (Niku-Paavola et al. 2004).
However, there were no data in the consulted literature related to the presence of a multiple-laccase gene family from a marine-derived basidiomycete. In the present study, 8 putative laccase genes with 10 possible enzyme products were found in the transcriptome of Peniophora sp. CBMAI 1063.
According to Valderrama et al. (2003), most of the fungal laccase multigene families arise from duplication events. If the duplication occurs after the last speciation, laccase genes from the same family groups will be in the same clade in a neighbor-joining analysis. On the other hand, if the duplication event occurs before the last speciation, these genes may assemble with other laccase families. These evolutionary relationships lead to a conclusion that the majority of the laccase genes in Peniophora sp. CBMAI 1063 arose from the last speciation, except for Lcc8, which may have arisen from an earlier duplication event. Although all laccases from Peniophora sp. CBMAI 1063 grouped with the multicopper oxidases from Peniophora sp., the sequence analysis revealed that these multicopper oxidases also exhibited the laccase signature (data not shown).
Different laccase genes in a single genome suggest that the enzymes play different physiological functions in the organism. Laccases have been associated with fruiting body development, spore pigmentation, pathogenesis, cell elongation, the duplication process, the stress response, and lignin bioconversion (Giardina et al. 2010; Rivera-Hoyos et al. 2013). Neighbor-joining analysis allowed a prediction laccase function using its similarity to other identified genes. However, none of the putative genes grouped with a well-identified gene, so further studies are needed to unveil all of the functions of the laccase isoenzymes in the Peniophora sp. CBMAI 1063 physiology.
In optimized conditions, Peniophora sp. CBMAI 1063 was able to express at least 10 different laccases based on peptide chain length, peptide composition, molecular weight, glycosylation pattern, and cellular activity site. It is important to highlight that in a previous study carried out by our research group, the marine-derived fungus Peniophora sp. CBMAI 1063, after has being cultured in the optimized conditions for laccase production (the same conditions used in the present study), was able to produce great amounts of laccase only in the presence of artificial seawater (saline condition) and copper sulfate (data not published yet).
Considering the marine origins of the new putative laccases, it is expected a high-salt tolerance from these enzymes, which represents a great potential to apply them in industrial and/or environmental processes performed under saline conditions. To this end, studies related to the expression and characterization of these enzymes, involving genetic improvement and heterologous expression, should be performed.
Abbreviations
- aa:
-
amino acid
- bp:
-
base pair
- CBMAI:
-
Coleção Brasileira de Micro-organismos de Ambiente e Indústria
- cDNA:
-
complementary deoxyribonucleic acid
- DNA:
-
deoxyribonucleic acid
- Lcc:
-
laccase
- MCO:
-
multi-copper oxidase
- mRNA:
-
messenger ribonucleic acid
- NCBI:
-
National Center for Biotechnological Information
- NR:
-
non-redundant
- ORF:
-
open reading frame
- PCR:
-
polymerase chain reaction
- pI:
-
isoelectric point
- RNA:
-
ribonucleic acid
- RT-PCR:
-
reverse transcriptase-polymerase chain reaction
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Baldrian P (2006) Fungal laccases-occurrence and properties. FEMS Microbiol Rev 30:215–242
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Bonugli-santos RC, Durrant LR, Sette LD (2010) Laccase activity and putative laccase genes in marine-derived basidiomycetes. Fungal Biol 114:863–872
Bonugli-Santos RC, dos Santos Vasconcelos MR, Passarini MRZ, Vieira GAL, Lopes VCP, Mainardi PH, dos Santos JA, dos Azevedo Duarte L, Otero IVR, da Silva Yoshida AM, Feitosa VA, Pessoa A, Sette LD (2015) Marine-derived fungi: diversity of enzymes and biotechnological applications. Front Microbiol 6:1–15
Bonugli-Santos RC, Vieira GAL, Collins C, Fernandes TCC, Marin-Morales MA, Murray P, Sette LD (2016) Enhanced textile dye decolorization by marine-derived basidiomycete Peniophora sp. CBMAI 1063 using integrated statistical design. Environ Sci Pollut Res 23:8659–8668
Chapman MA (2015) Transcriptome sequencing and marker development for four underutilized legumes. Appl Plant Sci 3:1400111
Chauhan JS, Rao A, Raghava GPS (2013) In silico platform for prediction of N-, O- and C-glycosites in eukaryotic protein sequences. PLoS ONE 8:e67008
Chen H, Wang M, Shen Y, Yao S (2014) Optimization of two-species whole-cell immobilization system constructed with marine-derived fungi and its biological degradation ability. Chin J Chem Eng 22:187–192
Claus H (2004) Laccases: structure, reactions, distribution. Micron 35:93–96
Dwivedi UN, Singh P, Pandey VP, Kumar A (2011) Structure–function relationship among bacterial, fungal and plant laccases. J Mol Catal B Enzym 68:117–128
Eggert C, Lafayette PR, Temp U, Eriksson KEL, Dean JFD (1998) Molecular analysis of a laccase gene from the white rot fungus Pycnoporus cinnabarinus. Appl Environ Microbiol 64:1766–1772
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution (NY) 39:783–791
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press, Totowa, pp 571–607
Giardina P, Faraco V, Pezzella C, Piscitelli A, Vanhulle S, Sannia G (2010) Laccases: a never-ending story. Cell Mol Life Sci 67:369–385
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512
Johnson M (2015) De novo transcriptome assembly using Trinity. http://blog.mossmatters.net/wp-content/uploads/2015/07/using_trinity.pdf
Kester DR, Duedall IW, Connors DN, Pytkowicz RM (1967) Preparation of artificial seawater. Limnol Oceanogr 12:176–179
Kilaru S, Hoegger PJ, Kues U (2006) The laccase multi-gene family in Coprinopsis cinerea has seventeen different members that divide into two distinct subfamilies. Curr Genet 50:45–60
Kumar SVS, Phale PS, Durani S, Wangikar PP (2003) Combined sequence and structure analysis of the fungal laccase family. Biotechnol Bioeng 83:386–394
Menezes CBA, Bonugli-Santos RC, Miqueletto PB, Passarini MRZ, Silva CHD, Justo MR, Leal RR, Fantinatti-Garboggini F, Oliveira VM, Berlinck RGS, Sette LD (2010) Microbial diversity associated with algae, ascidians and sponges from the north coast of São Paulo state, Brazil. Microbiol Res 165:466–482
Nagy G, Riley R, Tritt A, Adam C, Daum C, Floudas D, Sun H, Yadav JS, Pangilinan J, Larsson K, Matsuura K, Barry K, Labutti K, Kuo R, Ohm RA, Bhattacharya SS, Shirouzu T, Yoshinaga Y, Martin FM, Grigoriev IV, Hibbett DS (2015) Comparative genomics of early-diverging mushroom-forming fungi provides insights into the origins of lignocellulose decay capabilities. Mol Biol Evol 33:959–970
Niku-Paavola ML, Fagerström R, Kruus K, Viikari L (2004) Thermostable laccases produced by a white-rot fungus from Peniophora species. Enzym Microb Technol 35:100–102
Palmieri G, Giardina P, Bianco C, Fontanella B, Sannia G (2000) Copper induction of laccase isoenzymes in the ligninolytic fungus Pleurotus ostreatus. Appl Environ Microbiol 66:920–924
Passarini MRZ, Rodrigues MVN, Da Silva M, Sette LD (2011) Marine-derived filamentous fungi and their potential application for polycyclic aromatic hydrocarbon bioremediation. Mar Pollut Bull 62:364–370
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Pezzella C, Guarino L, Piscitelli A (2015) How to enjoy laccases. Cell Mol Life Sci 72:923–940
Raghukumar C, Shailaja MS, Parameswaran PS, Singh SK (2006) Removal of polycyclic aromatic hydrocarbons from aqueous media by the marine fungus NIOCC#312: involvement of lignin-degrading enzymes and exopolysaccharides. Indian J Mar Sci 35:373–379
Raghukumar C, D’souza-Ticlo D, Verma AK (2008) Treatment of colored effluents with lignin-degrading enzymes: an emerging role of marine-derived fungi. Crit Rev Microbiol 34:189–206
Riva S (2006) Laccases: blue enzymes for green chemistry. Trends Biotechnol 24:219–226
Rivera-Hoyos CM, Morales-Álvarez ED, Poutou-Piñales RA, Pedroza-Rodríguez AM, Rodríguez-Vázquez R, Delgado-Boada JM (2013) Fungal laccases. Fungal Biol Rev 27:67–82
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schlosser D, Grey R, Fritsche W (1997) Patterns of ligninolytic enzymes in Trametes versicolor. Distribution of extra- and intracellular enzyme activities during cultivation on glucose, wheat straw and beech wood. Appl Microbiol Biotechnol 47:412–418
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Valderrama B, Oliver P, Medrano-soto A, Vazquez-duhalt R (2003) Evolutionary and structural diversity of fungal laccases. Antonie Van Leeuwenhoek 89:289–299
Verma AK, Raghukumar C, Verma P, Shouche YS, Naik CG (2010) Four marine-derived fungi for bioremediation of raw textile mill effluents. Biodegradation 21:217–233
Viswanath B, Rajesh B, Janardhan A, Kumar AP, Narasimha G (2014) Fungal laccases and their applications in bioremediation. Enzym Res 2014:163242
Vite-Vallejo O, Palomares LA, Dantán-González E, Ayala-Castro HG, Martínez-Anaya C, Valderrama B, Folch-Mallol J (2009) The role of N-glycosylation on the enzymatic activity of a Pycnoporus sanguineus laccase. Enzym Microb Technol 45:233–239
Wang W, Liu F, Jiang Y, Wu G, Guo L, Chen R, Chen B, Lu Y, Dai Y, Xie B (2015) The multigene family of fungal laccases and their expression in the white rot basidiomycete Flammulina velutipes. Gene 563:142–149
Xu F, Shin W, Brown SH, Wahleithner JA, Sundaram UM, Solomon EI (1996) A study of a series of recombinant fungal laccases and bilirubin oxidase that exhibit significant differences in redox potential, substrate specificity, and stability. Biochim Biophys Acta 1292:303–311
Xu F, Palmer AE, Yaver DS, Berka RM, Gambetta GA, Brown SH, Solomon EI (1999) Targeted mutations in a Trametes villosa laccase axial perturbations of the T1 copper. J Biol Chem 274:12372–12375
Zdobnov EM, Apweiler R (2001) InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17:847–848
Authors’ contributions
All authors contributed to the design of the experiments. IVRO performed the experiments and drafted the manuscript. MF contributed with transcriptome data analysis. All authors read and approved the final manuscript.
Authors information
IVRO obtained his B.Sc in Biological Sciences at Federal University of Mato Grosso do Sul and his M.Sc. in Applied Microbiology at São Paulo State University, Brazil. Now he is a Ph.D. studying in Applied Microbiology at São Paulo State University.
MF obtained her Ph.D. in Cellular and Molecular Biology at São Paulo State University, Brazil. Now she is a Postdoctoral researcher working with genomic, transcriptomic and metagenomics.
MBJ obtained his Ph.D. in Sciences (Biochemistry) at University of São Paulo. He is a senior professor attached to the Department of Biochemistry and Microbiology of São Paulo State University, Brazil.
HF obtained his Ph.D in Biochemistry at University of Oxford, England. He is a professor attached to the Department of Biochemistry and Microbiology of São Paulo State University, Brazil.
LDS obtained his Ph.D. in Food Science (Applied Microbiology) at University of Campinas. She is a professor attached to the Department of Biochemistry and Microbiology of São Paulo State University, Brazil.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This study was supported by Grants financed by FAPESP (Reference Numbers: #2013/19486-0 and #2016/07957-7) and scholarships (IVRO) funded by CNPq, Conselho Nacional de Desenvolvimento Científico e Tecnológico—Brazil (#159488/2014-1) and (MF) by FAPESP (Reference Number: #2014/17950-4).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
13568_2017_526_MOESM1_ESM.pdf
Additional file 1. Table S1. Specific primers designed for Comp15071_c0_seq5 with tails to bind amplification products in the cloning vector. Figure S1. Species distribution of all homologous unigenes. Figure S2. Gene ontology (GO) classification of assembled unigenes (Level 2). GO terms were distributed into three ontologies: molecular functions—blue bars; biological process—red bars; and cellular component—green bars.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Otero, I.V.R., Ferro, M., Bacci, M. et al. De novo transcriptome assembly: a new laccase multigene family from the marine-derived basidiomycete Peniophora sp. CBMAI 1063. AMB Expr 7, 222 (2017). https://doi.org/10.1186/s13568-017-0526-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-017-0526-7