
Abstract
Bioethanol production using lignocellulosic biomass generates lignocellulosic bioethanol distillery wastewater (LBDW) that contains a large amount of xylose, making it a potential inexpensive source of xylose for biomaterials production. The main goal of this study was the production of useful enzymes from LBDW during treatment of this wastewater. In this study, we found that xylose strongly induced two yeast strains, Pseudozyma antarctica T-34 and GB-4(0), to produce novel xylanases, PaXynT and PaXynG, respectively. The nucleotide sequence of PaXynT [accession No. DF196774 (GAC73192.1)], obtained from the genome database of strain T-34 using its N-terminal amino acid sequence, was 91% identical to that of PaXynG (accession No. AB901085), and the deduced amino acid sequence is 98% identical. The specific activities of the purified PaXynT and PaXynG were about 52 U/mg. The optimal pH and temperature for both enzymes’ activities were 5.2 and 50°C, respectively. They hydrolyzed xylan to xylose and neither had β-xylosidase (EC 3.2.1.37) activity, indicating that they are endo-β-xylanases (EC 3.2.1.8). With these results, we expect that PaXyns can be employed in saccharizing lignocellulosic biomass materials for the production of useful products just like other endoxylanases. After 72 h of LBDW fed-batch cultivation using a jar-fermentor, strain GB-4(0) produced 17.3 U/ml (corresponding to about 0.3 g/l) of PaXynG and removed 63% of dissolved organic carbon and 87% of dissolved total phosphorus from LBDW. These results demonstrate the potential of P. antarctica for xylanase production during LBDW treatment.
Similar content being viewed by others


Introduction
Wastewater treatment system that uses yeasts could remove large amounts of organic compounds (10,000 COD mg/l/day), requires little space, and discharges little waste sludge (Yoshizawa 1978). This method is useful for treating food, brewing, and beverage industry wastewater (Yoshizawa 1978; Watanabe et al. 2009). Furthermore, the utilization of high strength wastewater is considered as an attractive way to produce useful materials, single cell protein, lactic acid, methane, and enzymes (lipase and protease) (Siso 1996; Angenent et al. 2004). Accordingly, using yeasts were suitable for high yield production of useful materials from high strength wastewater (Watanabe et al. 2013a, 2014a).
Producing bioethanol from lignocellulosic biomass generates lignocellulosic bioethanol distillery wastewater (LBDW) that contains high concentrations of organic compounds (30,000–60,000 COD mg/l). The cost of LBDW treatment directly increases the cost of bioethanol production. Since the ethanol-producing yeast Saccharomyces cerevisiae does not ferment xylose (Matsushika et al. 2008), the resulting LBDW is found to contain a large amount of xylose, making it a potential inexpensive source of fermentable xylose.
A non-pathogenic basidiomycetous yeast, Pseudozyma antarctica has an ability to assimilate various kinds of carbon sources (Boekhout 2011). Recently, we found that P. antarctica GB-4(0) efficiently removed organic compounds from shochu (a Japanese traditional distilled liquor) distillery wastewater (Watanabe et al. 2013b). The whole genome sequence of P. antarctica T-34 had already been determined and annotated (Morita et al. 2013). Moreover, we discovered that P. antarctica produces a cutinase-like enzyme (CLE), designated as PaE (22-kDa), which efficiently degrades various synthetic biodegradable polyesters (Kitamoto et al. 2011; Shinozaki et al. 2013), and that the PaE productivity was increased by xylose (Watanabe et al. 2014b). For scale-up production of PaE, under xylose feeding cultivation of P. antarctica using a jar fermentor (Watanabe et al. 2014b), we found a highly secreted 33-kDa unknown protein (unpublished data). If this 33-kDa unknown protein is a useful enzyme, it could be efficiently produced from large amounts of cheap xylose containing in LBDW during treatment.
A basidiomycetous yeast, Cryptococcus sp. strain S-2 (Masaki et al. 2012) and a yeast-like fungus, Aureobasidium pullulans (Dobberstein and Emeis 1989) secrete xylanases from xylose. Endo-β-xylanases (EC 3.2.1.8), which hydrolyze xylopyranosyl linkages of β-1,4-xylan, the principal type of hemicellulose, are useful for saccharizing lignocellulosic biomass materials for the production of bioethanol and other useful chemicals (Biely 1985). Xylanases are also utilized in paper and pulp industries (Pokhrel and Viraraghavan 2004) and pretreatment of animal feeds (Beg et al. 2001). The utilization of LBDW, a cheap substrate for xylanase production, would be an attractive way to reduce xylanase production costs, while treating LBDW.
In this study, we attempted to purify and identify the 33-kDa unknown proteins from P. antarctica T-34 and GB-4(0) using the genomic DNA sequence of T-34. After confirming that these proteins were xylanases, their biochemical properties were characterized. The xylanase of GB-4(0) was produced from LBDW derived from rice straw hydrolysate in large scale using a jar-fermentor.
Materials and methods
Strains
Pseudozyma antarctica GB-4(0) was isolated from rice husks (Kitamoto et al. 2011) whereas P. antarctica T-34 was isolated from Mt. Tsukuba soil sample (Kitamoto et al. 1990). These strains were deposited in the Genebank at the National Institute of Agrobiological Sciences (NIAS), Japan [accession numbers: T-34, MAFF 306900; GB-4(0), MAFF 306999].
Cultivation conditions
The two yeast strains were pre-cultivated in test tubes containing 5 ml of YM medium (0.3% yeast extract, 0.3% malt extract, 0.5% peptone, 1% dextrose) at 30°C with reciprocal shaking at 160 rpm for 24 h. The pre-cultures (100 μl) were added to 100 ml flasks containing 20 ml of modified fungal minimum medium (FMM) (0.3% yeast extract, 0.2% NaNO3, 0.06% KH2PO4, 0.06% MgSO4·7H2O) with 8% xylose or beechwood xylan. Xylose and other nutrients were separately autoclaved (121°C, 20 min). The cultures were cultivated at 30°C with rotary shaking at 200 rpm for 96 h.
Every 24 h, 1-ml aliquots of the culture were harvested and centrifuged at 15,000 rpm for 5 min. The pellets were dried at 105°C for 2 h and their dry cell weights were measured to investigate cell growth. At the same time, the xylanase activities of the supernatant were measured as described below.
Enzyme activity
Xylanase activity was determined using xylan as the substrate at 30°C for 30 min. The reaction mixture contained 15 mM sodium acetate buffer (pH 5.2), 1.0% (w/v) beechwood xylan (Sigma-Aldrich Japan), and 100 μl of culture supernatant in a total volume of 1 ml. After the reaction, the amount of reducing sugar was analyzed by the modified Somogyi–Nelson method (Hatanaka and Kobara 1980) with d-xylose as the standard. One unit (U) of xylanase activity was defined as 1 μmol of d-xylose liberated per min in the reaction mixture.
β-Xylosidase activity was also determined using the synthetic substrate p-nitrophenyl-β-d-xylopyranoside (pNP-X) (Iefuji et al. 1996). The reaction mixture contained 200 μl of 4 mM pNP-X solution, 600 μl of 30 mM sodium acetate buffer (pH 5.2) and 200 μl of enzyme solution in a total volume of 1 ml. After 30 min, 2 ml of 0.2 M Tris–HCl buffer (pH 8.5) was added to stop the enzyme reaction. One unit of β-xylosidase activity was defined as 1 μmol of p-nitrophenol released per min.
Electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to the method of Laemmli (1970) using a 14.1% polyacrylamide slab gel. Proteins were visualized by Coomassie brilliant blue (CBB) staining using Phast Gel Blue R (GE Healthcare) and by silver staining (2D-Silver stain II “Daiichi”; Daiichi Pure Chemicals Co., Ltd. Tokyo, Japan).
The proteins were transferred to PVDF membranes (Pall, Port Washington, NY, USA) by semidry blotting. The 33-kDa band corresponding to P. antarctica T-34 protein, which was designated as PaXynT was excised and its N-terminal amino acid sequence was determined using ABI Procise 491HT (Applied Biosystems, Foster City, CA, USA).
Genomic DNA isolation
The two yeast strains were cultivated in 5 ml of YM medium overnight. The cultures were centrifuged, and genomic DNA was isolated from the cell pellets using Dr. GenTLE for Yeast (Takara, Kyoto, Japan).
Sequencing of the PaXyn genes
The N-terminal peptide sequence of the PaXynT was searched in the annotated whole genome sequences of P. antarctica T-34 (Morita et al. 2013), and was used to obtain its genomic sequence. To determine the nucleotide sequence of the unknown protein produced by P. antarctica GB-4(0), designated as PaXynG, forward primers (PaXynF1, PaXynF2, PaXynF3, and PaXynF4) and reverse primers (PaXynR1, PaXynR2, PaXynR3, and PaXynR4) were designed based on the genomic sequence of PaXynT. The primer sequences and their positions in the T-34 gene are shown in Table 1. The entire gene for PaXynG was obtained by PCR amplification using the above primers and genomic DNA of P. antarctica GB-4(0) as template. The reaction was performed using TaqEX (Takara, Kyoto, Japan) according to the manufacturer’s instructions. Amplified DNA fragments were subjected to 1% agarose gel electrophoresis and were purified using QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). The sequences of the PCR products were determined using BigDye Terminator Cycle Sequence V3.1 Kit with the same primers and Applied Biosystems 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Purification
After 96 h flask cultivation of both strains, the culture supernatants obtained after centrifugation (7,000×g, 10 min, 4°C) were filtered using a membrane filter (ADVANTEC® C045A090C, 0.45 μm, Toyo Roshi Kaisha, Ltd. Japan). One-ml aliquots of filtrates were applied to a TSK-GEL3000SWXL column (Tosoh) in 50 mM acetate sodium buffer (pH 5.2) containing 0.3 M NaCl at a flow rate of 0.5 ml/min. The absorbance was measured at 280 nm. Fractions containing enzyme activity were concentrated, desalted using Amicon Ultra-15 10000 MWCO (MILLIPORE), and filtered using a syringe filter (ADVANTEC® DISMIC®-25cs, 0.2 μm, Toyo Roshi Kaisha, Ltd.). The filtrate (100 μl) was mixed with 900 μl of 50 mM Na-phosphate buffer (pH 7.1) containing 1.3 M ammonium sulfate. The mixture (1 ml) was applied to a hydrophobic interaction column (Phenyl Superose HR 5/5; GE Healthcare UK Ltd.) with a linear gradient (1.2–0 M) of ammonium sulfate in 50 mM Na-phosphate buffer (pH 7.1) at a flow rate of 0.5 ml/min. Fractions containing enzyme activity were concentrated, desalted, and filtered as described above. Protein concentration was measured with a protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA).
Enzyme pH and temperature properties
Optimal pH for enzyme activity was determined at 30°C for 30 min with beechwood xylan as substrate using several 15 mM buffers: sodium acetate-HCl (pH 2.0–4.0), sodium acetate (pH 4.0–5.2), sodium phosphate (pH 5.2–6.8), and Tris–HCl (pH 6.8–8.0). To estimate pH stability, the enzyme solutions were incubated at pH ranging from 2.0 to 8.0 using the above buffers for 1 h at 30°C. The residual xylanase activity was then determined at pH 5.2. Optimal temperature for activity was determined at pH 5.2 for 30 min at different temperatures (30–70°C) with beechwood xylan as substrate. Thermostability was estimated by incubating the enzyme sample in water bath at 30–70°C for 30 min. After heat treatment, samples were chilled on ice and residual xylanase activity was determined as described above.
Xylan hydrolysis and end product analysis
Purified PaXynG was used to determine the end products of beechwood xylan hydrolysis. The reactions were conducted at pH 5.2 and 30°C. After incubations at 10, 20, and 30 min, the reactions were stopped by boiling. The hydrolysis products were separated by thin-layer chromatography (TLC) according to the method of La Grange et al. (2001). Samples of the reaction mixtures, containing 20 μg of xylose-equivalent sugars, were applied to a TLC sheet (Silica gel 60 F254, Merck, Darmstadt, Germany) that was placed in a chamber containing 7:1:2 solvent mixture of n-propanol:ethanol:H2O. A xylooligosaccharides standard mixture (20 μg/μl) containing xylose (X1), xylobiose (X2), xylotriose (X3), and xylotetraose (X4) was also used. The sugar spots were identified by spraying 1% anthrone dissolved in 75% sulfuric acid, followed by heating (Morita et al. 2010). Hydrolyzed xylose was measured using a d-xylose assay kit (Megazyme, Ireland) according to the manufacturer’s manual.
Jar-fermentor cultivation
Pseudozyma antarctica GB-4(0) was used for the jar-fermentor cultivation experiments. A 30-ml pre-culture was grown in a 300-ml flask at 30°C with rotary shaking at 200 rpm for 24 h in YM medium. This culture was then used to inoculate the 5-l jar fermentor containing 3 l of PaXyn production medium [0.2% yeast extract, 0.2% NaNO3, 0.2% (NH4)2SO4, 0.04% KH2PO4, 0.04% MgSO4·7H2O, and 2% xylose]. Batch cultivation was performed until all the xylose was depleted (around 24 h). Then, xylose-fed-batch cultivation was performed to induce PaXynG production by adding fed medium [0.2% yeast extract, 0.085% YNB w/o amino acid and ammonium sulfate (Difco) and 20% xylose] at a feeding rate of 500 ml/day using a peristaltic pump. Xylose and other nutrients were separately autoclaved (121°C, 20 min).
LBDW derived from rice straw hydrolysate was kindly provided by Biomaterial in Tokyo Co., Ltd. The concentration of xylose (6.7%) was measured with a d-xylose assay kit (Megazyme, Ireland). Analysis of LBDW using standard methods (APHA et al. 1998) showed the following components: dissolved organic carbon (DOC), 52,000 mg/l, dissolved total nitrogen (DTN), 550 mg/l, and dissolved total phosphorus (DTP), 600 mg/l. It has a pH of 4.2. The pre-culture (30 ml) prepared as described above was inoculated into 2–l of 4-times-diluted LBDW. After 24 h cultivation, LBDW was continuously added at a rate of 1 l/day using a peristaltic pump.
The cultivation conditions were as follows: aeration rate was 2 LPM; agitation value was 500 rpm; dissolved oxygen (DO) was maintained at around 25% of saturated value; pH was controlled at 6.0 with 14% ammonia solution, which also provided a nitrogen source for the culture; temperature was 30°C.
Accession number
The accession number of the genomic sequence encoding PaXynG of P. antarctica GB-4(0) registered in DBBJ is AB901085.
Results
Production of xylanase from P. antarctica GB-4(0)
When cultivated in flask with modified FMM with 8% xylose, P antarctica GB-4(0) produces a 33-kDa unknown protein (Figure 1b, indicated by the arrow). Productions of some xylanases are reported to have been induced by xylose (Furukawa et al. 2009; Gielkens et al. 1999; Masaki et al. 2012; Dobberstein and Emeis 1989). Among the Pseudozyma species found to have the ability to secrete xylanases are P. hubiensis NCIM3574 (Adsul et al. 2009) and P. brasiliensis sp. nov. strain GHG001 (Borges et al. 2014). These findings led us to speculate that the 33-kDa unknown protein produced by P. antarctica strains could also be xylanases. With this assumption, we investigated the time course of xylanase activity of P. antarctica GB-4(0) culture.

Time course of xylanase (PaXynG) production by P. antarctica GB-4(0) with modified FMM containing 8% xylose in flask cultivation. a Cell growth (closed triangles) and xylanase (open squares) production. Each result is the average of three different experiments. Error bars show standard deviations. b SDS-PAGE of supernatants (5 μl) periodically sampled from the flask cultivation. The arrow at 33 kDa indicates PaXyn bands.
Xylanase activity was detected in the culture and was found to have gradually increased with time (Figure 1a). At 96 h, 49.9 U/ml of xylanase activity and cell growth with an OD660 of 106.7 were obtained (Figure 1a). Simultaneously, the intensity of the 33-kDa band of GB-4(0) on the SDS-PAGE gel increased with time (Figure 1b). Xylan, being the substrate of xylanases, generally induces xylanase production by microorganisms. However, with modified FMM using 8% xylan instead of xylose under the same cultivation conditions described previously, P. antarctica GB-4(0) showed less xylanase activity (14.0 U/ml). The 33-kDa band was also detected in this culture by SDS-PAGE analysis (data not shown).
Nucleic acid and amino acid analysis of the two xylanases
Another P. antarctica strain, T-34, also produced xylanase (18.5 U/ml) under the same conditions with xylose, and the 33-kDa unknown protein band was also detected on SDS-PAGE gel (data not shown). In order to identify the xylanase of strain GB-4(0), we referred to the DNA sequence of the gene that encodes the 33-kDa unknown protein of strain T-34 based on the database for its whole genome sequence (Morita et al. 2013).
The N-terminal sequence (amino acids 35–44 after cleavage of a signal peptide) of the 33-kDa unknown protein from P. antarctica T-34 indicated that it corresponded to a gene with accession No. DF196774 (GAC73192.1) in the genome of P. antarctica T-34. This gene contains the putative promoter region (600 bp), 1,026-bp ORF, the putative terminator region (300 bp), and one putative intron (624–688) comprised of 64 nucleotides.
The deduced amino acid sequence contains 341 amino acid residues (accession No. M9ME65) and the predicted size of the mature protein (307 amino acid residues, 32.9 kDa) is almost the same as that estimated by SDS-PAGE electrophoresis. A search of DDBJ (http://www.ddbj.nig.ac.jp/) yielded a number of xylanases belonging to glycosyl hydrolases (GH) 10 family, whose amino acid sequences are 57–77% identical to the 33-kDa unknown protein of P. antarctica T-34 (Table 2). These results lend support to our speculation that the 33-kDa unknown protein is also a xylanase (PaXyn).
The genomic sequence encoding PaXynT of P. antarctica T-34, was used to design primers for the amplification of the genomic sequence encoding PaXynG of P. antarctica GB-4(0). A single PCR product was obtained and sequenced (1,954 bp, accession No. AB901085). The nucleotide sequence encoding PaXynG was found to be 91% identical to that encoding PaXynT over the range −600 to 1,390 of T-34. The deduced amino acid sequence (341 amino acid residues) of PaXynG is 98% identical to that of PaXynT.
Purification and characterization of PaXynG
The PaXynG was purified by gel-chromatography and hydrophobic interaction-chromatography as described in Table 3. Purification to homogeneity was confirmed by observation of a single 33-kDa band on a silver stained SDS-PAGE gel (Figure 2). Its specific activity was about 52 U/mg.

SDS-PAGE of PaXynG of P. antarctica GB-4(0). Lane M molecular standard, lane 1 culture medium, lane 2 TSK-Gel G3000SWXL-purified PaXynG, lane 3 Phenyl Superose HR 5/5-purified PaXynG.
Its optimal pH was 5.2, and the optimal temperature was 50°C (Table 4). It was stable over a wide pH range (3.0–8.0), retaining 97% of their original activities after 1 h incubation (data not shown). The temperature at which the enzyme lost half of their activity (a measure of thermostability) was 57°C. It had no β-xylosidase activity. The same results were obtained with purified PaXynT (Table 4).
A TLC analysis of products revealed that purified PaXynG hydrolyzed beechwood xylan to xylose (X1), xylobiose (X2), xylotriose (X3), xylotetraose (X4), and other xylooligosaccharides (Figure 3). The released xylose was confirmed by detection with d-xylose assay kit (data not shown).

TLC analysis of products resulting from the hydrolysis of beechwood xylan by purified PaXynG for the indicated incubation times. Sugar standards (M) correspond to xylose (X1), xylobiose (X2), and xylotriose (X3). The spot indicated by the arrow is xylose (X1).
Production of highly concentrated PaXynG by xylose-fed-batch cultivation using a jar-fermentor
Because P. antarctica GB-4(0) produced larger amounts of PaXyn than T-34 in flask cultivation, GB-4(0) was used in jar fermentor cultivation. After the initial low concentration of xylose (2%) was consumed (24 h), xylose was fed into the PaXyn production medium. This resulted in a DO value around 50% of saturation (data not shown); and 232.4 U/ml (corresponding to 4.5 g/l) of PaXynG was obtained after 72 h (Figure 4).

PaXynG production by P. antarctica GB-4(0) with xylose feeding using a jar fermentor. Cell growth (closed triangles) and PaXynG production (open squares) with 2% xylose. Xylose feeding began after 24 h. Each result is the average of two different experiments. Error bars show standard deviations.
Simultaneous production of PaXynG from LBDW during its treatment using a jar-fermentor
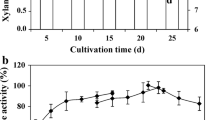
In a preliminary examination, we found that P. antarctica GB-4(0) and T-34 could not grow on LBDW unless it was diluted 4–5 times with water (data not shown). Thus, LBDW-fed-batch cultivation was initially carried out with 4-times-diluted LBDW. After the initial carbon sources, that could be utilized by P. antarctica GB-4(0), was consumed (24 h), non-diluted LBDW was fed into the culture using a peristaltic pump. This resulted in a DO value around 60% of saturation (data not shown); 17.3 U/ml (0.3 g/l) of PaXynG was obtained after 72 h (Figure 5a). PaXynG was detected as a 33-kDa band on a CBB stained SDS-PAGE gel (Figure 5b).

LBDW-fed cultivation of P. antarctica GB-4(0) using a jar fermentor. a Cell growth (closed triangles) and PaXynG production (open squares) with 4-times-diluted LBDW. LBDW feeding began after 24 h. Each result is the average of two different experiments. Error bars show standard deviations. b SDS-PAGE of supernatants (5 μl) periodically sampled from the jar fermentor cultivation. The arrow at 33 kDa indicates PaXynG bands.
In terms of wastewater treatment, over 99% of the xylose content of the LBDW was consumed after 72-h cultivation of P. antarctica GB-4(0). The resulting effluent DOC and DTP were 12,000 and 48 mg/l, respectively. The total amount of influent DOC ((26,000 mg + 104,000 mg)/4 l) and DTP ((300 mg + 1,200 mg)/4 l) were calculated as 32,500 and 375 mg/l, respectively. Thus, considering these values, the removal ratios were calculated to be 63 and 87%, respectively. Day removal ratio of DOC (6.8 kg/m3/day) was much higher compared to that removed by activated sludge (0.5–1.0 kg/m3/day) (Watanabe et al. 2013a).
Discussion
In this study, we attempted to produce useful materials from LBDW by cultivating P. antarctica during treatment of this wastewater. We first identified the highly-secreted 33-kDa unknown proteins in the culture of P. antarctica GB-4(0) and T-34 cultivated with xylose to be novel xylanases (PaXyns). Next, we determined the genomic sequence for PaXyn of P. antarctica T-34 (PaXynT) using the annotated whole genome sequence (Morita et al. 2013). P. antarctica GB-4(0) produced higher PaXyn than P. antarctica T-34 (Figure 1). Using the sequence encoding PaXynT, we successfully identified the genomic sequence for PaXyn of P. antarctica GB-4(0) (PaXynG). Xylanases are classified into two substantial groups, GH 10 and 11 family, on the basis of their structures (Kimura et al. 2010). The sequence similarity of these PaXyns indicated that they belong to GH10 family xylanases which exhibit higher affinity for shorter liner β-1,4-xylooligosaccharides than GH11 family xylanases (Biely et al. 1997).
The optimal pH, optimal temperature, pH stability, and thermostability of the both PaXyns are almost the same (Table 4). They produced xylose (X1), xylobiose (X2), xylotriose (X3), and other xylooligosaccharides from xylan (Figure 3) and did not have any β-xylosidase activity (Table 4). These data indicate that they are endoxylanases (1,4-β-d-xylan xylanohydrolases (EC 3.2.1.8)). Since these PaXyns did not have any β-xylosidase activity, they could not hydrolyze xylobiose to xylose. Thus, the detected xylose might have resulted from the hydrolysis of xylotriose and other xylooligosaccharides by PaXyns. With this result, it is possible that just like the other endoxylanases (Biely 1985), PaXyns could be employed in saccharizing lignocellulosic biomass materials for the production of useful products.
The production of PaXyns was strongly induced by xylose; a yield of 4.5 g/l of PaXynG was produced by P. antarctica GB-4(0) in 72 h (0.0625 g/l/h) with xylose fed-batch cultivation using a jar fermentor (Figure 4). This yield is comparable with 8.1 g/l of recombinant xylanase (PtxynA) of Paecilomyces thermorphila produced by a methylotrophic yeast Pichia pastoris in 228 h (0.0355 g/l/h) with methanol fed-batch cultivation using a jar fermentor (Fan et al. 2012). The results show that P. antarctica is an attractive producer of native xylanase. After centrifugation, about 3 l of supernatant was obtained from 3.5 l of jar culture. The rate of xylanase production (produced PaXynG, 4.5 g/l × 3 l = 13.5 g)/(consumed xylose, 60 g + 200 g = 260 g) by this strain was calculated to be 0.052 g/g.
The hydrolysate of lignocelluloses contains not only fermentable sugars but also toxic compounds that inhibit cell growth, such as furans and organic acids (Okuda et al. 2008). Since these compounds are also generated during fermentation and distillation processes, they are expected to be present in LBDW. It is probably the reason why P. antarctica GB-4(0) could not grow in non-diluted LBDW. However, with LBDW fed-batch cultivation, about 0.3 g/l of PaXynG was produced by P. antarctica GB-4(0) in 72 h. After centrifugation, about 3.5 l of supernatant was obtained from 4.0 l of jar culture. This is equivalent to the xylanase production rate of about 0.006 g/g (produced PaXynG, 0.3 g/l × 3.5 l = 1.05 g/consumed xylose of LBDW, 34.5 g + 134 g = 168.5 g).
PaXynG production by P. antarctica GB-4(0) from LBDW, resulted in the removal of 63% of DOC and 87% of DTP. Because ammonia solution was added for pH control and nitrogen source, DTN removal ratio could not be accurately estimated. However, since the initial DTN was low, the residual DTN is expected to consist mainly of PaXynG which could be easily recovered by ultrafiltration. In the case of shochu distillery wastewater, we previously confirmed that the yeast-treated wastewater was efficiently treated by a combination of nitrification/denitrification cycle treatment and activated sludge process (Watanabe et al. 2009, 2013c). In a laboratory-scale demonstration, 50 cycles (25 days) removed 98.9% of DOC, 95.7 of DTN, and 94.1% of DTP from barley shochu distillery wastewater (Watanabe et al. 2013c). Thus, it can be expected that the remaining DOC, DTN, and DTP of P. antarctica GB-4(0)-treated LBDW could also be easily removed by additional conventional wastewater treatment methods. These results indicated that xylanase production by P. antarctica GB-4(0) from LBDW could also contribute to the cutting of LBDW treatment cost.
Abbreviations
- COD:
-
chemical oxygen demand
- DOC:
-
dissolved organic carbon
- DO:
-
dissolved oxygen
- DTN:
-
dissolved total nitrogen
- DTP:
-
dissolved total phosphorus
- FMM:
-
fungal minimum medium
- LBDW:
-
lignocellulosic bioethanol distillery wastewater
- PaXynG:
-
xylanase of P. antarctica GB-4(0)
- PaXynT:
-
xylanase of P. antarctica T-34
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TLC:
-
thin-layer chromatography
References
Adsul MG, Bastwade KB, Gokhale DV (2009) Biochemical characterization of two xylanases from yeast Pseudozyma hubiensis producing only xyloologosaccharides. Bioresour Technol 100:6488–6495. doi:10.1016/j.biortech.2009.07.064
Angenent LT, Karim K, Al-Dahhan MH, Wrenn BA, Domíguez-Espinosa R (2004) Production of bioenergy and biochemical from industrial and agricultural wastewater. Trends Biotechnol 22:477–485. doi:10.1016/j.tibtech.2004.07.001
APHA, AWWA, WEF (1998) Standard methods for the examination of water and wastewater, 20th edn. American Public Health Association, Washington DC
Beg QK, Kapoor M, Mahajan L, Hoondal GS (2001) Microbial xylanase and their industrial applications: a review. Appl Microbiol Biotechnol 56:326–338. doi:10.1007/s002530100704
Biely P (1985) Microbial xylanolytic systems. Trends Biotechnol 3:286–290. doi:10.1016/0167-7799(85)90004-6
Biely P, Vršanská M, Tenkanen M, Kluepfel D (1997) Endo-β-1,4-xylanase families: differences in catalytic properties. J Biotechnol 57:151–166. doi:10.1016/S0168-1656(97)00096-5
Boekhout T (2011) Pseudozyma Bandoni emend. Boekhout (1985) and a comparison with the yeast state of Ustilago maydis (De Candolle) Corda (1842). The Yeasts, a Taxonomic study, vol 3, Fifth edn, pp 1857–1868
Borges TA, de Souza AT, Squina FM, Riaño-Pachón DM, dos Santos RAC, Machado E et al (2014) Biochemical characterization of an endoxylanase from Pseudozyma brasiliensis sp. nov. strain GHG001 isolated from the intestinal tract of Chrysomelidae larvae associated to sugarcane roots. Process Biochem 49:77–83. doi:10.1016/j.procbio.2013.10.004
Dobberstein J, Emeis CC (1989) β-Xylanase produced by Aureobasidium pullulans CBS 58475. Appl Microbiol Biotechnol 32:262–268. doi:10.1007/BF00184971
Fan G, Katrolia P, Jia H, Yang S, Yan Q, Jiang Z (2012) High-level expression of a xylanase gene from thermophilic fungus Paecilomyces thermophila in Pichia pastoris. Biotechnol Lett 34:2043–2048. doi:10.1007/s10529-012-0995-3
Furukawa T, Shida Y, Kitagami N, Mori K, Kato M, Kobayashi T et al (2009) Identification of specific binding sites for XYR1, a transcriptional activator of cellulolytic and xylanolytic genes in Trichoderma reesei. Fungal Genet Biol 46:564–574. doi:10.1016/j.fgb.2009.04.001
Gielkens MMC, Dekkers E, Visser J, de Graaff LH (1999) Two cellobiohydrolase-encoding genes from Aspergillus niger require d-xylose and the xylanolytic transcriptional activator XlnR for their expression. Appl Environ Microbiol 65:4340–4345
Hatanaka C, Kobara Y (1980) Determination of glucose by a modification of Somogyi–Nelson method. Agric Biol Chem 44:2943–2949
Iefuji H, Chino M, Kato M, Iimura Y (1996) Acid xylanase from yeast Cryptococcus sp. S-2: purification, characterization, cloning, and sequencing. Biosci Biotechnol Biochem 60:1331–1338. doi:10.1271/bbb.60.1331
Kimura T, Mizutani T, Sun J, Kawazu T, Karita S, Sakka M et al (2010) Stable production of thermotolerant xylanase B of Clostridium stercorarium in transgenic tobacco and rice. Biosci Biotechnol Biochem 74:954–960. doi:10.1271/bbb.90774
Kitamoto D, Akiba S, Hioki C, Tabuchi T (1990) Extracellular accumulation of mannosylerythritol lipids by a strain of Candida antarctica. Agric Biol Chem 54:31–36
Kitamoto HK, Shinozaki Y, Cao X, Konishi M, Morita T, Tago K et al (2011) Phyllosphere yeasts rapidly break down biodegradable plastics. AMB Express 1:44. doi:10.1186/2191-0855-1-44
La Grange DC, Pretorius IS, Claeyseens M, van Zyl WH (2001) Degradation of xylan to d-xylose by recombinant Saccharomyces cerevisiae coexpressing the Aspergillus niger β-xylosidase (xlnD) and the Trichoderma reesei xylanase II (xyl2) genes. Appl Environ Microbiol 67:5512–5519. doi:10.1128/AEM.67.12.5512-5519.2001
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 277:680–685
Masaki K, Tsuchioka H, Hirano T, Kato M, Ikeda H, Iefuji H (2012) Construction of a new recombinant protein expression system in the basidiomycetous yeast Cryptococcus sp. strain S-2 and enhancement of the production of a cutinase-like enzyme. Appl Microbiol Biotechnol 93:1627–1636. doi:10.1007/s00253-011-3680-x
Matsushika A, Watanabe S, Kodakari T, Makino K, Sawayama S (2008) Bioethanol production from xylose by recombinant Saccharomyces cerevisiae expression xylose reductase, NADP+-dependent xylitol dehydrogenase, and xylulokinase. J Biosci Bioeng 105:296–299. doi:10.1263/jbb.105.296
Morita T, Ito E, Fukuoka T, Imura T, Kitamoto D (2010) The role of PaAAC1 encoding a mitochondrial ADP/ATP carrier in the biosynthesis of extracellular glycolipids, mannosylerythritol lipids, in the basidiomycetous yeast Pseudozyma antarctica. Yeast 27:379–388. doi:10.1002/yea.1761
Morita T, Koike H, Koyama Y, Hagiwara H, Ito E, Fukuoka T et al (2013) Genome sequence of the basidiomycetous yeast Pseudozyma antarctica T-34, a producer of the glycolipid biosurfactants mannosylerythritol lipids. Genome Announc 1:e00064-13. doi:10.1128/genomeA.00064-13
Okuda N, Ninomiya K, Katakura Y, Shioya S (2008) Strategies for reducing supplemental medium cost in bioethanol production from waste house wood hydrolysate by ethanologenic Escherichia coli: inoculation size increase and coculture with Saccharomyces cerevisiae. J Biosci Bioeng 105:90–96. doi:10.1263/jbb.105.90
Pokhrel D, Viraraghavan T (2004) Treatment of pulp and paper mill wastewater—a review. Sci Total Environ 333:37–58. doi:10.1016/j.scitotenv.2004.05.017
Shinozaki Y, Morita T, Cao X, Yoshida S, Koitabashi M, Watanabe T et al (2013) Biodegradable plastic-degrading enzyme from Pseudozyma antarctica: cloning, sequencing, and characterization. Appl Microbiol Biotechnol 97:2951–2959. doi:10.1007/s00253-012-4188-8
Siso MIG (1996) The biotechnological of utilization of cheese whey: a review. Bioresour Technol 57:1–11. doi:10.1016/0960-8524(96)00036-3
Watanabe T, Masaki K, Iwashita K, Fujii T, Iefuji H (2009) Treatment and phosphorus removal from high-concentration organic wastewater by the yeast Hansenula anomala J224 PAWA. Bioresour Technol 100:1781–1785. doi:10.1016/j.biortech.2008.10.006
Watanabe T, Iefuji H, Kitamoto HK (2013a) Treatment of, and Candida utilis biomass production from shochu wastewater; the effects of maintaining a low pH on DOC removal and feeding cultivation on biomass production. Springerplus 2:514. doi:10.1186/2193-1801-2-514
Watanabe T, Iefuji H, Kitamoto HK (2013b) Breeding of wastewater treatment yeasts having high nitrogen removal ability. J Brew Soc Jpn 108:823–829 (In Japanese)
Watanabe T, Fujii T, Iefuji H, Kitamoto HK (2013c) Treatment and reutilization of Shochu wastewater using yeasts. Yousui Haisui 55:605–612 (In Japanese)
Watanabe T, Shinozaki Y, Suzuki K, Koitabashi M, Yoshida S, Sameshima-Yamashita Y et al (2014a) Production of a biodegradable plastic-degrading enzyme from cheese whey by the phyllosphere yeast Pseudozyma antarctica GB-4(1)W. J Biosci Bioeng 118:183–187. doi:10.1016/j.jbiosc.2014.01.007
Watanabe T, Shinozaki Y, Yoshida S, Koitabashi M, Sameshima-Yamashita Y, Fujii T et al (2014b) Xylose induces the phyllosphere yeast Pseudozyma antarctica to produce a cutinase-like enzyme which efficiently degrades biodegradable plastics. J Biosci Bioeng 117:325–329. doi:10.1016/j.jbiosc.2013.09.002
Yoshizawa K (1978) Treatment of waste-water discharged from sake brewery using yeast. J Ferment Technol 56:389–395
Authors’ contributions
TW designed the study, carried out most of the biological studies and statistical analyses, and drafted the manuscript. KS carried out the purification of PaXyns. IS analyzed xylanase activity. TM identified genomic sequence of PaXyn of P. antarctica T-34. HK helped in the genomic sequence analysis of PaXyns. YS helped in the purification of PaXyns. HU helped in the characterization of PaXyns. MK interpreted the data and revised the manuscript. HKK conceived and planned the study, and helped in drafting the manuscript. All authors read and approved the final manuscript.
Acknowledgments
We thank Prof. Haruyuki Iefuji, Ehime University, Japan, for his helpful discussions and Mrs. Xiao-hong Cao, for technical assistance. LBDW derived from hydrolysate of rice straw was kindly provided by Biomaterial in Tokyo Co., Ltd. This work was supported by a grant of science and technology research promotion program for agriculture, forestry, fisheries and food industry; by a Research Fellowship of the Japan Society for the Promotion of Science (JSPS) for Young Scientists, and by a JSPS KAKENHI Grant (No. 23 10107).
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Watanabe, T., Suzuki, K., Sato, I. et al. Simultaneous bioethanol distillery wastewater treatment and xylanase production by the phyllosphere yeast Pseudozyma antarctica GB-4(0). AMB Expr 5, 36 (2015). https://doi.org/10.1186/s13568-015-0121-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-015-0121-8