
Abstract
Iron is essential for most bacteria to survive, but excessive iron leads to damage by the Fenton reaction. Therefore, the concentration of intracellular free iron must be strictly controlled in bacteria. Riemerella anatipestifer (R. anatipestifer), a Gram-negative bacterium, encodes the iron uptake system. However, the iron homeostasis mechanism remains largely unknown. In this study, it was shown that compared with the wild type R. anatipestifer CH-1, R. anatipestifer CH-1Δfur was more sensitive to streptonigrin, and this effect was alleviated when the bacteria were cultured in iron-depleted medium, suggesting that the fur mutant led to excess iron accumulation inside cells. Similarly, compared with R. anatipestifer CH-1∆recA, R. anatipestifer CH-1∆recAΔfur was more sensitive to H2O2-induced oxidative stress when the bacteria were grown in iron-rich medium rather than iron-depleted medium. Accordingly, it was shown that R. anatipestifer CH-1∆recAΔfur produced more intracellular ROS than R. anatipestifer CH-1∆recA in iron-rich medium. Electrophoretic mobility shift assays showed that R. anatipestifer CH-1 Fur suppressed the transcription of putative iron uptake genes through binding to their promoter regions. Finally, it was shown that compared with the wild type, R. anatipestifer CH-1Δfur was significantly attenuated in ducklings and that the colonization ability of R. anatipestifer CH-1Δfur in various tissues or organs was decreased. All these results suggested that Fur is important for iron homeostasis in R. anatipestifer and its pathogenic mechanism.
Similar content being viewed by others


Introduction
Iron is an essential element for most organisms. In bacteria, iron is involved in several key metabolic processes, including respiration, tricarboxylic acid (TCA) cycling, oxygen transport, oxidative stress resistance, and DNA synthesis [1,2,3,4]. To survive, bacteria have evolved various systems to obtain iron from environment and/or host sources. However, excessive free iron in bacteria can produce toxic reactive oxygen species (ROS) and hydroxyl radicals through the Fenton reaction, which damage DNA, membranes and lipids [5, 6]. Therefore, bacteria developed scavenging systems (SOD isozymes, peroxidases, and catalases), and DNA repair systems (RecA, RecBCD, and RecF) to defend against oxidative stress [7,8,9,10]. RecA repairs oxidative DNA damage by combining with the RecF-like pathway or RecBCD pathway [8, 9]. In most Gram-negative bacteria, iron homeostasis is regulated by the ferric uptake regulator Fur [11,12,13,14,15]. When the concentration of intracellular free iron is high, the iron-associated Fur dimer binds to the promoter region of iron uptake genes to inhibit transcription, thus reducing iron intake. In contrast, when iron is deficient, iron dissociates from Fur, and Fur is released from the promoter region, leading to the increased transcription of iron uptake genes and thus increased iron intake [16].
Riemerella anatipestifer (R. anatipestifer), a Gram-negative bacterium belonging to the family Flavobacteriaceae, causes acute septicemia and infectious polyserositis in ducks, chickens, geese, and other avian species [17]. R. anatipestifer infection can give rise to high contagiousness and mortality in the duck industry [18], and at least 21 serotypes of R. anatipestifer without cross-protection have been identified [19, 20]. Besides, R. anatipestifer is resistant to multiple antibiotics [21,22,23,24], which is potentially related to its natural transformation ability [25]. Due to the presence of multiple serotypes and multidrug resistance, R. anatipestifer is hard to eradicate.
In previous studies, it has been shown that iron is essential for the survival of R. anatipestifer and that TonB plays an important role in iron and hemin uptake [26, 27]. Although genome analysis has shown that R. anatipestifer encodes putative iron uptake genes, the functions of most of them remain unclear [28]. It was shown that the putative iron-related TonB-dependent receptors B739_1208 and B739_1343 were important for pathogenesis, although they were not regulated by iron [29, 30]. Recently, it was found that some genes were significantly up-regulated under iron-limited conditions [31]. Although it has been shown that Fur of R. anatipestifer YM has a role in gene regulation, the role of Fur in iron homeostasis, oxidative stress resistance, and pathogenesis were not fully understood [32]. In this study, the role of R. anatipestifer CH-1 Fur in maintaining iron homeostasis, oxidative stress resistance, and pathogenesis were investigated.
Materials and methods
Bacterial strains, plasmids, and primers
The strains and plasmids used in this study are listed in Additional file 1. The primers used in this study are listed in Additional file 2.
Growth conditions
R. anatipestifer strains were grown routinely on LB agar supplemented with 5% sheep blood or in GCB liquid medium [25] at 37 °C with shaking. Iron-rich and iron-limited conditions were achieved by GCB medium and GCB medium supplemented with different concentrations of ethylenediamine-di-o-hydroxyphenylacetic acid (EDDHA) (Alfa chemistry, ACM1170021), respectively.
Construction of the R. anatipestifer CH-1ΔrecA, R. anatipestifer CH-1ΔrecAΔfur and R. anatipestifer CH-1ΔrecAΔfur pLMF03::fur complementation strains
Deletion of the genes was performed according to the natural transformation-based knockout method described in a previous study [25]. Briefly, the up- and downstream sequence of R. anatipestifer CH-1 recA were amplified by PCR using the primers listed in Additional file 2. The sequence containing the Cmp cassette was amplified from the R. anatipestifer CH-2 strain using primers CmpP1 and CmpP2 (Additional file 2). The PCR fragments (upstream, Cmp cassette, and downstream) were ligated using the overlap PCR method. The fused PCR fragments were purified and incubated with R. anatipestifer CH-1 for 1 h at 37 °C. Then, samples of the mixture were spread onto plates supplemented with chloramphenicol and incubated overnight at 37 °C. The correct clone was identified as described in a previous study [33]. The mutant R. anatipestifer CH-1ΔrecAΔfur was constructed by the same method on the basis of R. anatipestifer CH-1 Δfur, which was constructed in a previous study [33].
To construct the R. anatipestifer CH-1ΔrecAΔfurpLMF03::fur complementation strain, the plasmid pLMF03::fur was transformed into cells of the strain Escherichia coli S17-1, and the recombinant plasmid was introduced into the R. anatipestifer CH-1ΔrecAΔfur mutant strain via conjugation as described elsewhere [27]. The transconjugants were selected using blood agar plates supplemented with Cfx (1 μg/mL) and Kan (50 μg/mL) and identified by PCR amplification.
Streptonigrin sensitivity assay
For indirect quantification of the intracellular iron level, we performed a streptonigrin sensitivity assay as described previously [34]. Briefly, R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur were grown to OD600 = 1.0 in GCB medium, GCB medium supplemented with 100 μM EDDHA, and GCB medium supplemented with 100 μM EDDHA and 200 μM Fe(NO3)3 at 37 °C in a shaking incubator. Cells were harvested by centrifugation at 6000 rpm for 10 min, and pellets were diluted with fresh PBS up to OD600 = 0.5 and aliquoted at 1 mL/tube. Streptonigrin (Sigma-Aldrich, St. Louis, USA) was diluted to 1 µg/mL with sterile PBS, 0 µL, 50 µL, and 80 µL was added to each tube of bacterial solution, the final concentration of streptonigrin was 0 ng/mL, 50 ng/mL and 80 ng/mL, respectively. Then the samples were incubated in the static incubator at 37 °C for 30 min. After incubation, the bacterial solution was diluted and spread onto GCB plates for counting (T0, T50, and T80). After a 1-day incubation at 37 °C, the grown colonies were counted. The survival rate was calculated as (T50/T0) × 100% and (T80/T0) × 100%, and the experiments were performed in triplicate.
In vitro growth rate determination
The in vitro growth rates of the test strains were determined by measuring the OD600 with a spectrophotometer (Eppendorf Biophotometer, Germany). Briefly, R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur were cultured overnight and inoculated into 20 mL of GCB liquid medium at an OD600 of 0.05, and growth rates at 37 °C were determined by measuring the OD600 every 2 h for 12 h. In parallel, R. anatipestifer CH-1 and R. anatipestifer CH-1Δfur were cultured overnight in iron-limited medium, then the overnight-cultured cells were subcultured into 20 mL of GCB or GCB supplemented with 50 μM EDDHA, 100 μM EDDHA or 200 μM EDDHA at an OD600 of 0.05, and growth rates were monitored by measuring OD600 as mentioned above. The data were analyzed using three independent experiments, with two replicate samples for each experiment.
H2O2 sensitivity assay
A hydrogen peroxide (H2O2) challenge assay was performed as described in a previous study, with slight modification [35]. The sensitivity of the fur mutant to H2O2 was determined using a strain lacking recA, which is defective in DNA repair and thus more sensitive to H2O2 than the parent strain [36]. The strains R. anatipestifer CH-1ΔrecA pLMF03, R. anatipestifer CH-1 ΔrecAΔfurpLMF03 and R. anatipestifer CH-1ΔrecAΔfurpLMF03::fur were grown in GCB liquid medium or GCB medium supplemented with EDDHA (25 μM or 50 μM) until the exponential phase (OD600 = 1.0–1.5). The cells were collected, washed and diluted in PBS to OD600 = 0.5, and aliquoted at 1 mL/tube. For the H2O2 challenge assay, each tube of the bacterial suspension was incubated with H2O2 (0, 5 or 10 mM) at 37 °C for 30 min. After exposure to H2O2, the bacteria were washed twice with PBS, and serial dilutions were spread onto GCB plates. After a 1-day incubation at 37 °C, the grown colonies were counted. The survival rate was calculated as described above, and the experiments were performed in triplicate.
Fluorescence dye-based intracellular ROS detection
To detect intracellular ROS levels, the fluorescent reporter dye 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA, Life Technologies) was used. Briefly, the strains R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur were grown in GCB liquid medium or GCB supplemented with EDDHA (25 μM or 50 μM) until the exponential phase (OD600 = 1.0–1.5). Cells were collected and washed and diluted in PBS to OD600 = 0.5, and 1 mL samples were collected. Then, the samples were resuspended in 1 mL of PBS containing 10 μM CM-H2DCFDA. Samples were incubated in the dark for 30 min at room temperature. The cultures were precipitated by centrifugation; the supernatants were removed and then the cells were resuspended in 1 mL of PBS containing 5 mM H2O2 or 10 mM H2O2. After 30 min of treatment in the dark at 37 °C, the cell suspensions (200 μL) were transferred to a dark 96-well plate. Fluorescence signals were measured using a Varioskan Flash (Thermo Scientific) with excitation/emission wavelengths of 495/520 nm. Bacterial cells resuspended in sterile PBS were used as a negative control, 1 mL of PBS containing 10 μM CM-H2DCFDA and supplemented with 100 μM H2O2 was used as a positive control, and 1 mL of sterile PBS containing 10 μM CM-H2DCFDA was used as a black control. The incubation conditions were the same as those of the experimental groups, and the experiments were performed in triplicate.
qRT-PCR
Real-time PCR was performed as described in a previous study [27]. Briefly, R. anatipestifer CH-1, R. anatipestifer CH-1Δfur and R. anatipestifer CH-1ΔfurpLMF03::fur were grown in GCB or GCB supplemented with 100 μM EDDHA to exponential phase (OD600 = 1.0–1.5), and RNA was extracted by the RNeasy Minikit procedure (Qiagen). cDNA synthesis was performed with reverse transcriptase (HiScript Q RT SuperMix for qPCR gDNA wiper, R223-01, Vazyme, Nanjing, China). Real-time PCR was performed with SYBR Green master mix (Q111-03, Vazyme) using a CFX Connect real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA). Then, the transcription levels of TonB-dependent receptor genes B739_0103 and B739_0173 were detected in each sample using specific primers [33]. Relative fold changes were calculated as described previously with the threshold cycle (ΔΔCT) method, considering the efficiency of the PCR for each target [37]. Quantitative measurements were performed on biological samples in triplicate, and the results were normalized to findings with the R. anatipestifer housekeeping gene recA [27].
Electrophoretic mobility shift assays (EMSAs)
DNA mobility shift assays were performed using the method described in a previous study, with minor modifications [38]. The promoter regions of the B739_0173 and B739_0173-coding regions (204 bp and 214 bp, respectively) were amplified by PCR with the primers B739_0173 promoter P1/P2 and B739_0173 coding region P1/P2 (Additional file 2). Fifty to two hundred fifty nanograms of B739_0173 promoter DNA or 250 ng of B739_0173-coding region DNA was mixed with 4 µg of Fur6His protein in binding buffer (40 mM Tris–HCl, pH 8.0, 50 mM KCl, 2 mM DTT, 6% glycerol, 0.2 mM MnCl2 or 0.2 mM EDTA) in a 20 µL (final volume) mixture and incubated at 37 °C for 30 min. A 6% nondenaturing polyacrylamide gel in 0.5 × TBE running buffer was prerun for 30 min at 100 V and loaded with 20 µL of the binding reaction mixture. After being run for 2 h at 100 V, the gel was stained with Goldview and Coomassie Brilliant Blue.
LD50 determination
The median lethal dose (LD50) was measured to evaluate virulence as previously described [30]. Briefly, R. anatipestifer CH-1, R. anatipestifer CH-1Δfur and R. anatipestifer CH-1ΔfurpLMF03::fur were cultured in TSB medium at 37 °C with shaking until the exponential growth phase (OD600 = 1.0–1.5), and the bacteria were collected and washed and diluted in PBS. Each strain was prepared at the following doses: 5 × 1010 CFU/mL, 5 × 109 CFU/mL, 5 × 108 CFU/mL, and 5 × 107 CFU/mL. Subsequently, the above doses of the bacteria were injected intramuscularly into the ducklings (10 ducklings/group), with each duckling receiving 0.2 mL. Once the ducklings exhibited signs of moribundity, they were euthanized via forced CO2 inhalation, and dead ducklings were subjected to R. anatipestifer identification by PCR and Gram staining. The mortality of the ducklings was recorded daily for 7 days post-challenge. The LD50 was calculated by using the Reed-Muench method [39].
Colonization assays
To assess bacterial colonization ability in ducklings, 3-day-old ducklings were infected intramuscularly with R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur (109 CFU/duckling). The initial bacterial number was estimated by OD600 and counted by spreading on blood plates. At 24 h and 48 h post-infection, six surviving ducklings in each test group were randomly selected and euthanized by forced CO2 inhalation. Liver, spleen, brain and blood from the heart were collected and weighed. The samples were homogenized in PBS (0.1 g of sample/0.9 mL of PBS) using a Nasco WHIRL–PAK (B01245WA, USA) as described previously [30]. The homogenized contents were serially diluted in PBS buffer and spread on blood agar plates supplemented with 50 μg/mL kanamycin to determine the bacterial CFU since R. anatipestifer is naturally resistant to kanamycin [26]. The plates were incubated at 37 °C overnight for counting and calculating the loads per gram of tissue [40].
Duck serum bactericidal assay
Duck serum was obtained from the whole blood of 7-day-old ducklings via jugular vein bleeding. Blood samples were centrifuged twice (3500 rpm for 5 min) to obtain non-inactivated serum, and the serum was heat-inactivated at 55 °C for 1 h to obtain inactivated serum, which was stored at −20 °C before use. Bacterial survival in serum was determined as described in a previous study, with minor modifications [41]. Briefly, R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur were grown in GCB liquid medium to the exponential phase (OD600 = 1.0–1.5), the viable bacteria were washed twice with PBS, and the concentration of bacteria was adjusted to 109 CFU/mL. Then, the mixture containing the cell suspension and 50% non-inactivated duck serum or 50% inactivated serum were incubated at 37 °C for 0.5 h and 1 h. The number of surviving bacteria was then determined by GCB plate counting. The survival rate was calculated as follows: the number of viable bacteria treated with non-inactivated duck serum or inactivated serum compared to the number of viable bacteria without treatment. The experiments were performed in triplicate.
Statistical analysis
All experimental data are expressed as the mean ± 1 standard deviation (SD). Statistical analysis was performed using GraphPad Prism 7.00 (GraphPad Software, CA, USA) and SPSS Statistics 20 for Windows. The independent Student’s t-test was utilized to compare two groups, and one-way analysis of variance (ANOVA) or two-way ANOVA was used to compare multiple groups. P < 0.05 was considered significant.
Results
The R. anatipestifer CH-1 fur mutant was more sensitive to streptonigrin
To identify whether the fur mutation led to an increased intracellular free iron concentration in R. anatipestifer CH-1, we checked the sensitivity of the bacteria to streptonigrin since it is bactericidal in the presence of iron [34, 42]. Firstly, the bacteria were grown in iron rich medium, and the collected bacteria were used to measure the sensitivity to streptonigrin as described in “Materials and methods”. As shown in Figure 1A, the survival rate of R. anatipestifer CH-1ΔfurpLMF03 was approximately sevenfold lower than that of R. anatipestifer CH-1pLMF03 after treatment with 50 ng/mL streptonigrin. The survival rate of R. anatipestifer CH-1ΔfurpLMF03 was ~20-fold lower than that of R. anatipestifer CH-1pLMF03 when the concentration of streptonigrin was increased to 80 ng/mL. Moreover, the survival rate of the fur mutant strain was restored by the expression of Fur in trans (Figure 1A). To further verify that this effect was caused by iron, 100 μM the iron chelator EDDHA was added to the medium when the bacteria were cultured. Under this condition, the survival rates of all the strains were enhanced when treated with 50 ng/mL or 80 ng/mL streptonigrin. The survival rates of R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1Δfur pLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur were ~80%, ~75%, and ~80%, respectively, when treated with 50 ng/mL streptonigrin under iron-limited conditions (Figure 1B). In parallel, the survival rates of R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur were ~40%, ~35%, and ~40%, respectively, when treated with 80 ng/mL streptonigrin under iron-limited conditions (Figure 1B). Moreover, the sensitivity of all the strains to streptomycin was restored when iron(III) nitrate was added to the iron-limited medium (Figure 1C). These results suggest that Fur-deficient cells were strongly sensitive to streptonigrin, potentially due to excess iron inside the cells.

The sensitivity of R. anatipestifer CH-1 and its fur mutant to streptonigrin. The R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur strains were grown in GCB medium with no addition (A), with 100 μM EDDHA (B) or with 100 μM EDDHA and 200 μM Fe(NO3)3 (C) at 37 °C in a shaking incubator to the exponential phase (OD = 1.0–1.5). The bacteria were collected and suspended in PBS at 0.5 OD/mL, and were exposed to streptonigrin (50 ng/mL and 80 ng/mL) for 30 min. The survival rate of each culture was determined as described in the “Materials and methods”. The error bars represent the standard deviations of three independent experiments and three replicate samples for each experiment. Statistical significance was determined using two-way ANOVA (****P < 0.0001, ** P < 0.01, * P < 0.05).
The effect of the fur mutation on growth was not caused by excess iron in cells
As Fur plays an important role in the global gene regulation, the deletion of fur may diminish bacterial growth, therefore, we first tested whether the absence of fur influences R. anatipestifer CH-1 growth. The results showed that the growth ability of the fur mutant was significantly decreased in GCB medium compared to that of the wild type (Figure 2A) and that it was restored by the expression of Fur in the mutant strain in trans (Figure 2A). To investigate whether the growth defects of Δfur were caused by potential intracellular increased iron concentration, we measured the growth curve of R. anatipestifer CH-1 and R. anatipestifer CH-1Δfur in GCB supplemented with the different concentration of iron chelator EDDHA. As shown in Figure 2B, the growth of R. anatipestifer CH-1 became slower when 50 μM, 100 μM or 200 μM was added to the medium. At the same time, the results showed that the addition of 50 μM, 100 μM or 200 μM EDDHA to the GCB medium did not benefit the growth of the fur mutant (Figure 2C). This result indicates that the effect of Fur mutation on growth is not caused by excess iron in cells.

The growth curves of R. anatipestifer CH-1 and its fur mutant. A R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur cells were cultured overnight and then inoculated into 20 mL of GCB liquid medium at an OD600 of 0.05, the growth rates were determined at 37 °C with shaking cultivation (180 rpm) by measuring the OD600 every 2 h for 12 h. R. anatipestifer CH-1 (B) or R. anatipestifer CH-1Δfur (C) was precultured in iron-limited medium overnight and then inoculated into 20 mL of GCB or GCB supplemented with 50 μM EDDHA, 100 μM EDDHA and 200 μM EDDHA at an OD600 of 0.05, and the growth rates were monitored as above. The error bars represent the standard deviations of three independent experiments and three replicate samples for each experiment. Statistical significance was determined using two-way ANOVA (****P < 0.0001, ** P < 0.01).
The fur mutant is more sensitive to H2O2-induced oxidative stress
Increased intracellular iron levels promote the decomposition of H2O2 and formation of hydroxyl radicals through the Fenton reaction, which damage cellular components [43]. The fur mutant was more sensitive to streptonigrin, which could be caused by increased intracellular iron concentrations. Thus, it was hypothesized that the fur mutant is more sensitive to H2O2. Compared with the wild type, the fur mutant did not display significantly increased sensitivity to H2O2 (data not shown). We then detected the sensitivity of the fur mutant strain to H2O2 in the R. anatipestifer CH-1 strain lacking recA, which is defective in DNA repair [36]. After exposure to 5 mM H2O2 and 10 mM H2O2, the survival rate of R. anatipestifer CH-1ΔrecAΔfur pLMF03 decreased significantly compared with that of R. anatipestifer CH-1ΔrecApLMF03 when cultured in GCB medium (Figure 3A). In parallel, we added different concentrations of EDDHA to GCB medium and then checked the sensitivity of these strains to H2O2. As Figure 3B shows, when the bacteria were cultured in GCB containing 25 μM EDDHA, the survival rate of R. anatipestifer CH-1ΔrecAΔfurpLMF03 was also decreased significantly compared with that of R. anatipestifer CH-1ΔrecApLMF03 when treated with 5 mM or 10 mM H2O2. However, when the bacteria were cultured in GCB containing 50 μM EDDHA, the survival rate of R. anatipestifer CH-1ΔrecAΔfurpLMF03 did not significantly decrease compared to that of R. anatipestifer CH-1ΔrecApLMF03 when treated with 5 mM and 10 mM H2O2 (Figure 3C). These results suggested that the deletion of fur in R. anatipestifer CH-1 significantly increased the sensitivity of this strain to H2O2 in iron-sufficient medium due to excess iron inside the cells.

The sensitivity of R. anatipestifer CH-1ΔrecA and its derived strains to H2O2-induced oxidative stress. R. anatipestifer CH-1ΔrecApLMF03, R. anatipestifer CH-1ΔrecAΔfurpLMF03 and R. anatipestifer CH-1ΔrecAΔfurpLMF03::fur cells were cultured in GCB medium with no addition (A), with 25 μM EDDHA (B) or with 50 μM EDDHA (C) at 37 °C with shaking cultivation (180 rpm) to the exponential growth phase (OD600 = 1.0–1.5), and the cells were collected and diluted in PBS to 0.5 OD/mL. Then, the cell suspensions were incubated with 0 mM, 5 mM or 10 mM H2O2 at 37 °C for 30 min. The survival rate of each culture was determined as described in the “Materials and methods”. The error bars represent the standard deviations of three independent experiments and three replicate samples for each experiment. Statistical significance was determined using two-way ANOVA (****P < 0.0001, *** P < 0.001, ** P < 0.01).
Deletion of fur causes increased intracellular ROS when treated with H2O2
Next, we determined if the absence of fur resulted in increased ROS in R. anatipestifer CH-1 in iron-rich medium after treatment with H2O2. The intracellular total ROS activity was measured by using CM-H2DCFDA, a permeability indicator of ROS [44]. Notably, after treatment with 5 mM H2O2, the fluorescence intensity of R. anatipestifer CH-1ΔfurpLMF03 was approximately 90 AU (absorbance unit), which was approximately twofold higher than that of R. anatipestifer CH-1pLMF03 in iron-rich medium, suggesting an increase in ROS in the fur deletion strain (Figure 4A). In parallel, we added different concentrations of EDDHA to GCB medium and checked the fluorescence intensity of these strains. When 25 µM EDDHA was added to the GCB medium, the fluorescence intensity of R. anatipestifer CH-1ΔfurpLMF03 was approximately 1.5-fold higher than that of R. anatipestifer CH-1pLMF03 (Figure 4B). When 50 µM EDDHA was added to the GCB medium, there was no difference in the fluorescence intensity between R. anatipestifer CH-1pLMF03 and R. anatipestifer CH-1ΔfurpLMF03 (Figure 4C). In addition, the fluorescence intensity of the fur-deficient strain could be restored by the expression of fur in trans. Overall, compared with that of the wild-type strain, the ROS content of the fur mutant strain increased in iron-sufficient medium when treated with H2O2.

Fluorescence-based ROS detection using the CM-H2DCFDA probe. R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur cells were grown in GCB liquid medium with no addition (A), with 25 μM EDDHA (B) or with 50 μM EDDHA (C) at 37 °C in a shaking incubator until the exponential growth phase (OD600 = 1.0–1.5). Then, the cells were collected and diluted in PBS to 0.5 OD/mL, and the bacterial suspensions were treated with 10 μM CM-H2DCFDA for 30 min before treatment with 5 mM H2O2 or 10 mM H2O2 for 30 min in the dark. After exposure to H2O2, the cells from each culture were added to a dark 96-well plate, and the fluorescence signals were measured as described in the “Materials and methods”. The error bars represent the standard deviations of three independent experiments and three replicate samples for each experiment. Statistical significance was determined using two-way ANOVA (****P < 0.0001, *** P < 0.001).
Fur binds to the promoters of putative iron uptake genes
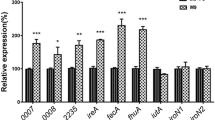
The above results showed that the absence of fur led to a potentially increase in intracellular iron content, indicating that Fur is involved in the regulation of iron transport. Therefore, to verify whether R. anatipestifer CH-1 Fur can regulate the transcription of putative iron uptake-related genes, we detected the mRNA levels of TonB-dependent receptor genes B739_0103 and B739_0173, which were up-regulated in an iron-limited environment and considered iron uptake-related genes [31, 33]. The results showed that the transcription of B739_0103 and B739_0173 was markedly increased in R. anatipestifer CH-1∆fur compared to that in the wild-type strain, and their transcript levels were not affected when 100 µM EDDHA was added to the GCB medium (Figure 5). Moreover, the increased transcription was fully restored to the wild-type level by the complementation of fur (Figure 5). These results indicated that Fur inhibits the transcription of the iron uptake genes B739_0103 and B739_0173 in R. anatipestifer CH-1.

Relative mRNA levels of B739_0103 and B739_0173 in R. anatipestifer CH-1 and its derived strains. R. anatipestifer CH-1, R. anatipestifer CH-1Δfur and R. anatipestifer CH-1ΔfurpLMF03::fur were grown in GCB or GCB supplemented with 100 μM EDDHA at 37 °C in a shaking incubator to the exponential growth phase (OD600 = 1.0–1.5). Total RNA and cDNA of these strains were prepared as described in the “Materials and methods”, and then the transcription of B739_0103 and B739_0173 was measured by qRT-PCR. Relative fold changes are reported in comparison with the parent strain. “c” means the complementary strain R. anatipestifer CH-1ΔfurpLMF03::fur. Statistical significance was determined using two-way ANOVA (****P < 0.0001).
To explore how Fur regulates iron uptake genes, EMSAs were performed as described in the “Materials and methods”. Since Mn2+ has more stable chemical properties than Fe2+, it is a typical surrogate for iron to maintain the regulatory activity of Fur [45]. As shown in Figure 6, only in the reaction buffer containing 200 µM MnCl2, the incubation of the promoter region of the putative iron uptake gene B739_0173 with purified Fur6His led to the formation of DNA–protein complexes, which showed clearly retarded migration in the gels, and the complex formation was increased with higher DNA concentrations. The results suggest that Fur binds to the promoter region of target genes and that binding occurs only in the presence of Mn2+. As a negative control, the DNA fragment of the coding region did not form a complex with Fur6His (Figure 6). Taken together, these results provide evidence that Fur inhibits the transcription of iron uptake-related genes by binding to the promoter region of these genes in R. anatipestifer CH-1.

Electrophoretic mobility shift assay (EMSA) for recombinant Fur6His binding with the promoter of B739_0173. The DNA of the B739_0173 promoter region and B739_0173-coding region were amplified by PCR, and 50–250 ng of B739_0173 promoter DNA or 250 ng of B739_0173-coding region DNA was mixed with 4 µg of Fur6His protein in binding buffer at 37 °C for 30 min. The samples were electrophoresed on a gel as described in the “Materials and methods”. (A) The gel was stained with Goldview. (B) The gel was stained with Coomassie Brilliant Blue. The experiment was repeated three times, the B739_0103 promoter region with Fur6His showed similar results, and a representative image is shown.
Fur contributes to the virulence and colonization ability of R. anatipestifer CH-1
In our previous study, we demonstrated that a fur deletion strain of R. anatipestifer showed reduced virulence in the Galleria mellonella model [33]. To investigate if Fur plays a role in the pathogenesis of R. anatipestifer CH-1 in poultry, we used a duckling model [29, 30] to determine the LD50 of the fur mutant. The calculated LD50 value of R. anatipestifer CH-1Δfur pLMF03 was greater than 1012 CFU, whereas the LD50 values of R. anatipestifer CH-1pLMF03 and the complementation strain were 108 CFU and 109 CFU, respectively. These results showed that the fur mutation led to reduced virulence of R. anatipestifer CH-1 in ducklings.
To test whether the reduced virulence is due to a decrease in bacterial colonization ability, groups of ducklings were inoculated with 109 CFU of R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 or R. anatipestifer CH-1ΔfurpLMF03::fur in the leg. Twenty-four hours and 48 h post-infection, the bacterial loads in the liver, spleen, brain, and the blood from the heart of the ducklings were determined. As shown in Figure 7A, at 24 h post-inoculation, the number of recovered colonies from various tissues and organs for the R. anatipestifer CH-1 fur mutant was significantly reduced compared to that of the parent strain (P < 0.0001) (Figure 7A). Similarly, at 48 h post-inoculation, the amount of colonized R. anatipestifer CH-1Δfur in various tissues and organs was also significantly decreased compared to that of the parent strain (Figure 7B). Moreover, compared to 24 h post-infection, the gap between the fur mutant and the parent strain was increased (Figure 7B). In addition, the bacterial loads in each tissue of the complementation strain R. anatipestifer CH-1ΔfurpLMF03::fur at 24 h and 48 h were comparable to those of the wild-type strain (Figure 7). These results indicated that Fur not only contributes to the colonization of R. anatipestifer CH-1 in duckling tissues, such as the liver, spleen, brain, and the blood from the heart, but also protected R. anatipestifer CH-1 from host clearance.

Colonization of R. anatipestifer CH-1 and its fur mutant in ducklings at 24 h and 48 h post-infection. Doses (200 μL) of 109 CFU of R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1 ΔfurpLMF03::fur were prepared and injected intramuscularly into 3-day-old ducklings (20 ducklings/group). At 24 h (A) and 48 h (B) post-infection, bacteria were isolated from the livers, spleens, brains, and the blood from the heart, as described in the “Materials and methods”. The data points represent the CFU/g values of the indicated organs in individual ducklings; the bars show the mean values (n = 6). Statistical significance was determined using two-way ANOVA (****P < 0.0001, ** P < 0.01).
The fur mutant is susceptible to non-inactivated duck serum
Compared to that of wild type, R. anatipestifer CH-1Δfur had a decreased colonization ability in blood. Therefore, it can be hypothesized that the fur mutant is susceptible to duck serum. To further investigate this hypothesis, the survival rates of R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur in 50% non-inactivated duck serum were measured. As shown in Figure 8A, after exposure to this serum for 0.5 h, the survival rate of R. anatipestifer CH-1 pLMF03 was ~70%, while the survival rate of R. anatipestifer CH-1Δfur pLMF03 was significantly decreased compared to the parent strain. After incubation with 50% non-inactivated duck serum for 1 h, the survival rates of R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur were approximately 30%, 5% and 30%, respectively (Figure 8A). As a control, 50% inactivated duck serum had neither an effect on the survival of R. anatipestifer CH-1pLMF03 nor on the survival of R. anatipestifer CH-1ΔfurpLMF03, and there is no difference in survival rates among all strains when treated with 50% inactivated duck serum for 0.5 h or 1 h (Figure 8B). These results demonstrated that the lack of Fur has a detrimental effect on serum resistance, which may also lead to a decrease in the virulence of R. anatipestifer CH-1.

The sensitivity of R. anatipestifer CH-1 and its derived strains to non-inactivated duck serum. The non-inactivated duck serum and inactivated duck serum were obtained by centrifuging from the jugular vein-collected blood of 7-day-old ducklings as described in the “Materials and methods”. R. anatipestifer CH-1pLMF03, R. anatipestifer CH-1ΔfurpLMF03 and R. anatipestifer CH-1ΔfurpLMF03::fur were grown in GCB liquid medium at 37 °C in a shaking incubator to exponential phase (OD600 = 1.0–1.5). Cells were collected and diluted in PBS to 109 CFU/mL, and the bacterial suspensions were incubated with 50% non-inactivated duck serum (A) or 50% inactivated duck serum (B) for 0.5 h or 1 h at 37 °C. Bacterial survival was enumerated by plating and counting colonies the following day as described in the “Materials and methods”. The error bars represent the standard deviations of three independent experiments and three replicate samples for each experiment. Statistical significance was determined using two-way ANOVA (****P < 0.0001).
Discussion
Iron is an essential element for the survival and growth of most bacteria; however, it can be toxic when present in excess [5, 6]. In some bacteria, iron inside bacterial cells is tightly regulated by the ferric uptake regulator Fur [46,47,48]. R. anatipestifer, an iron-dependent bacterium, has unclear mechanisms to regulate iron transport [32]. In the R. anatipestifer CH-1 genome, B739_0252 was annotated as a Fur family transcriptional regulator since it contains a Fur_like domain at amino acids 28–151. A protein BLAST analysis indicated that the Fur of R. anatipestifer CH-1 had low identity compared with well-characterized Fur proteins of other bacteria, such as E. coli (25% identity and 40% similarity), Campylobacter jejuni (25% identity and 39% similarity), and Pseudomonas aeruginosa (24% identity and 39% similarity). In this study, we determined the role of Fur in the physiology and virulence of R. anatipestifer CH-1.
Many studies have led to a classic model of Fur regulation in response to different iron conditions [46, 49, 50]. When the intracellular iron concentration is high, Fur-Fe2+ represses the expression of iron acquisition genes by binding upstream of these genes. When the intracellular iron concentration is low, Fur-Fe2+ dimers dissociate, which relieves the inhibition of iron acquisition genes, leading to an increased intracellular iron concentration. In this study, we found that the sensitivity of R. anatipestifer CH-1Δfur to streptonigrin was significantly higher than that of R. anatipestifer CH-1, and the sensitivity was affected by the external iron concentrations. The antibiotic streptonigrin is bactericidal in the presence of iron, indicating that the lack of fur may cause an increase in free intracellular iron concentration in R. anatipestifer CH-1. Besides, in an iron-rich environment, the deletion of the fur gene could affect the growth of R. anatipestifer CH-1. Since the lack of Fur may increase the free intracellular iron concentration, it was hypothesized that the growth defect of the mutant strain is due to dysregulated iron acquisition. However, supplementation with different concentration of EDDHA in iron-rich medium did not improve the growth ability of the fur mutant, indicating that the growth defect of the fur mutant strain is not caused by an imbalance in iron uptake. This was not surprising, since in addition to iron metabolism regulation, Fur was also shown to be involved in other cellular processes as a global regulator [13, 48].
The inactivation of fur may lead to unrestrained iron uptake, thus leading to the accumulation of free iron in the cytoplasm when the bacteria are grown in iron-rich conditions. Finally, it will result in excessive iron-catalyzed production of ROS [51]. In this study, we also found that after fur deletion, the strain was more sensitive to H2O2 and increased levels of intracellular ROS could be detected. In summary, the higher susceptibility to streptonigrin and H2O2 and the accumulation of ROS in the Fur-deficient strain suggest that a key role of Fur in R. anatipestifer is to avoid iron intoxication and oxidative stress.
In our previous studies, it was shown that the putative TonB-dependent receptor genes B739_0103 and B739_0173 were up-regulated under iron-limited conditions [31], and this phenomenon prompted us to check whether this regulation relies on Fur in R. anatipestifer CH-1. Fur plays a role through binding to the promoter region of its target gene, and the putative Fur-box sequence (5′-GATAATGATAATCATTATC-3′) has been found in R. anatipestifer YM [1, 32, 52]. Sequence comparison showed that the sequence of the Fur box was also present in the promoter regions of B739_0103 and B739_0173. As expected, it was shown that the transcription of B739_0103 and B739_0173 was significantly up-regulated in the fur mutant, suggesting that Fur may inhibit the transcription of iron uptake genes in R. anatipestifer CH-1. Moreover, it was shown that Fur was able to bind to the promoter region of B739_0173 rather than the coding sequence in the presence of Mn2+. From these results, it can be concluded that R. anatipestifer CH-1 Fur is involved in regulating the transcription of iron uptake genes by binding to their promoters and that this process requires the participation of metal ions, which is different from the function of Fur in Helicobacter pylori and C. jejuni. In H. pylori and C. jejuni, Fur can form a dimer even without iron as a cofactor and directly bind to the promoter region of the target gene, which is called apo-Fur regulation [53,54,55].
The Fur protein contributes to virulence in animal models for numerous bacterial pathogens [32, 48, 56,57,58,59,60], but the precise mechanism of the attenuation of fur mutants is not completely clear. In R. anatipestifer, previous works identified that the absence of Fur could reduce virulence in ducklings and in Galleria mellonella larvae [32, 33]. In agreement with these studies, it was shown that the LD50 of the fur-deficient strain in ducklings was significantly higher (more than 104 times) than that of the wild-type strain. The colonization ability of the fur mutant in ducklings was greatly diminished. Moreover, compared to the wild type, the R. anatipestifer CH-1∆fur mutant was more easily eliminated by the host.
As a mechanism of host defense against bacterial pathogen invasion, host innate immune cells, such as macrophages and neutrophils, produce superoxide radicals and hydrogen peroxide to kill invading bacteria [61]. Recent studies have shown that the host also uses iron or other metal toxicity at the site of infection to kill and control bacterial infection [62,63,64]. As antagonistic strategies, bacterial pathogens have evolved systems such as ROS detoxification, macromolecule damage repair, and metal efflux systems to survive in the host. Here, we can conclude that the decreased virulence of R. anatipestifer CH-1∆fur in ducks is partly due to its reduced resistance to oxidative stress. Moreover, we found that compared to the parent strain, the fur mutant was more easily killed by the non-inactivated duck serum. This supports the fact that fur deletion might lead to a decreased virulence of R. anatipestifer in ducks. It has been reported that a decrease in virulence of the fur mutant may be related to a reduction in the activity of enzymes required for protection against ROS, and changes in the expression of virulence factors in the fur mutant [12, 65]. Whether R. anatipestifer Fur regulates the expression of oxidative stress response enzymes and virulence genes needs to be investigated further. Regardless, the attenuated R. anatipestifer CH-1 fur mutant may provide the basis for future investigations of an attenuated vaccine. Overall, this study provides evidence of the essentiality of Fur in maintaining iron homeostasis, oxidative stress resistance and pathogenesis in R. anatipestifer CH-1.
Availability of data and materials
The nucleotide sequences of R. anatipestifer CH-1 were deposited in GenBank under accession number CP003787. The accession number of ferric uptake regulator Fur is following: Fur of Riemerella anatipestifer CH-1 (GenBank: AFR34859.1), Fur of Escherichia coli (GenBank: EFJ3478230.1), Fur of Campylobacter jejuni (GenBank: VTQ54023.1), Fur of Pseudomonas aeruginosa (GenBank: MXH36461.1). The datasets generated and/or analysed during the current study are available from the corresponding authors on reasonable request.
Abbreviations
- TCA:
-
Tricarboxylic acid
- ROS:
-
Reactive oxygen species
- GCB:
-
Gonorrhoeae-culture broth
- EDDHA:
-
Ethylenediamine-N,N′-bis ((2-hydroxyphenyl) acetic acid)
- H2O2 :
-
Hydrogen peroxide
- OD600 :
-
Optical density at 600 nm
- CM-H2DCFDA:
-
5-(And-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester
- ΔΔCT:
-
Threshold cycle
- EMSA:
-
Electrophoretic mobility shift assay
- LD50 :
-
Median lethal dose
- ANOVA:
-
Analysis of variance
- SD:
-
Standard deviation
- Amp:
-
Ampicillin
- Kan:
-
Kanamycin
- Cfx:
-
Cefoxitin
References
Andrews SC, Robinson AK, Rodriguez-Quinones F (2003) Bacterial iron homeostasis. FEMS Microbiol Rev 27:215–237. https://doi.org/10.1016/S0168-6445(03)00055-X
Huynh C, Andrews NW (2008) Iron acquisition within host cells and the pathogenicity of Leishmania. Cell Microbiol 10:293–300. https://doi.org/10.1111/j.1462-5822.2007.01095.x
Nairz M, Schroll A, Sonnweber T, Weiss G (2010) The struggle for iron - a metal at the host-pathogen interface. Cell Microbiol 12:1691–1702. https://doi.org/10.1111/j.1462-5822.2010.01529.x
Chasteen ND, Harrison PM (1999) Mineralization in ferritin: an efficient means of iron storage. J Struct Biol 126:182–194. https://doi.org/10.1006/jsbi.1999.4118
Fenton HJ (1894) Oxidation of tartaric acid in presence of iron. J Chem Soc 65:899–910
Imlay JA, Chin SM, Linn S (1988) Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240:640–642. https://doi.org/10.1126/science.2834821
Imlay JA (2008) Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem 77:755–776. https://doi.org/10.1146/annurev.biochem.77.061606.161055
Aranda J, Bardina C, Beceiro A, Rumbo S, Cabral MP, Barbe J, Bou G (2011) Acinetobacter baumannii RecA protein in repair of DNA damage, antimicrobial resistance, general stress response, and virulence. J Bacteriol 193:3740–3747. https://doi.org/10.1128/JB.00389-11
Stohl EA, Seifert HS (2006) Neisseria gonorrhoeae DNA recombination and repair enzymes protect against oxidative damage caused by hydrogen peroxide. J Bacteriol 188:7645–7651. https://doi.org/10.1128/JB.00801-06
Lopez E, Elez M, Matic I, Blazquez J (2007) Antibiotic-mediated recombination: ciprofloxacin stimulates SOS-independent recombination of divergent sequences in Escherichia coli. Mol Microbiol 64:83–93. https://doi.org/10.1111/j.1365-2958.2007.05642.x
Stojiljkovic I, Baumler AJ, Hantke K (1994) Fur regulon in gram-negative bacteria. Identification and characterization of new iron-regulated Escherichia coli genes by a fur titration assay. J Mol Biol 236:531–545. https://doi.org/10.1006/jmbi.1994.1163
Kurabayashi K, Agata T, Asano H, Tomita H, Hirakawa H (2016) Fur represses adhesion to, invasion of, and intracellular bacterial community formation within bladder epithelial cells and motility in uropathogenic Escherichia coli. Infect Immun 84:3220–3231. https://doi.org/10.1128/IAI.00369-16
Hu YH, Sun L (2016) The global regulatory effect of Edwardsiella tarda Fur on iron acquisition, stress resistance, and host infection: a proteomics-based interpretation. J Proteomics 140:100–110. https://doi.org/10.1016/j.jprot.2016.04.005
Choi J, Ryu S (2019) Regulation of iron uptake by fine-tuning the iron responsiveness of the iron sensor Fur. Appl Environ Microbiol 85:e03026-e3118. https://doi.org/10.1128/AEM.03026-18
Santos R, Batista BB, da Silva Neto JF (2020) Ferric uptake regulator Fur coordinates siderophore production and defense against iron toxicity and oxidative stress and contributes to virulence in Chromobacterium violaceum. Appl Environ Microbiol 86:e01620-e1720. https://doi.org/10.1128/AEM.01620-20
Beauchene NA, Mettert EL, Moore LJ, Keleş S, Willey ER, Kiley PJ (2017) O2 availability impacts iron homeostasis in Escherichia coli. Proc Natl Acad Sci U S A 114:12261–12266
Hess C, Enichlmayr H, Jandreski-Cvetkovic D, Liebhart D, Bilic I, Hess M (2013) Riemerella anatipestifer outbreaks in commercial goose flocks and identification of isolates by MALDI-TOF mass spectrometry. Avian Pathol 42:151–156. https://doi.org/10.1080/03079457.2013.775401
Ruiz J, Sandhu T (2013) Riemerella anatipestifer infection. In: Swayne DE (ed) Disease of Poultry, 13th edn. Wiley Blackwell publishing, Ames, pp 823–828
Pathanasophon P, Sawada T, Tanticharoenyos T (1995) New serotypes of Riemerella anatipestifer isolated from ducks in Thailand. Avian Pathol 24:195–199. https://doi.org/10.1080/03079459508419059
Pathanasophon P, Phuektes P, Tanticharoenyos T, Narongsak W, Sawada T (2002) A potential new serotype of Riemerella anatipestifer isolated from ducks in Thailand. Avian Pathol 31:267–270. https://doi.org/10.1080/03079450220136576
Zhong CY, Cheng AC, Wang MS, Zhu DK, Luo QH, Zhong CD, Li L, Duan Z (2009) Antibiotic susceptibility of Riemerella anatipestifer field isolates. Avian Dis 53:601–607. https://doi.org/10.1637/8552-120408-ResNote.1
Zhang X, Wang MS, Liu MF, Zhu DK, Biville F, Jia RY, Chen S, Sun KF, Yang Q, Wu Y, Zhao XX, Chen XY, Cheng AC (2017) Contribution of RaeB, a putative RND-type transporter to aminoglycoside and detergent resistance in Riemerella anatipestifer. Front Microbiol 8:2435. https://doi.org/10.3389/fmicb.2017.02435
Luo HY, Liu MF, Wang MS, Zhao XX, Jia RY, Chen S, Sun KF, Yang Q, Wu Y, Chen XY, Biville F, Zou YF, Jing B, Cheng AC, Zhu DK (2018) A novel resistance gene, lnu(H), conferring resistance to lincosamides in Riemerella anatipestifer CH-2. Int J Antimicrob Agents 51:136–139. https://doi.org/10.1016/j.ijantimicag.2017.08.022
Huang L, Yuan H, Liu MF, Zhao XX, Wang MS, Jia RY, Chen S, Sun KF, Yang Q, Wu Y, Chen XY, Cheng AC, Zhu DK (2017) Type B chloramphenicol acetyltransferases are responsible for chloramphenicol resistance in Riemerella anatipestifer. China Front Microbiol 8:297. https://doi.org/10.3389/fmicb.2017.00297
Liu M, Zhang L, Huang L, Biville F, Zhu D, Wang M, Jia R, Chen S, Sun K, Yang Q, Wu Y, Chen X, Cheng A (2017) Use of natural transformation to establish an easy knockout method in Riemerella anatipestifer. Appl Environ Microbiol 83:e00127-e217. https://doi.org/10.1128/AEM.00127-17
Liao H, Cheng X, Zhu D, Wang M, Jia R, Chen S, Chen X, Biville F, Liu M, Cheng A (2015) TonB Energy transduction systems of Riemerella anatipestifer are required for iron and hemin utilization. PLoS One 10:e0127506. https://doi.org/10.1371/journal.pone.0127506
Liu M, Wang M, Zhu D, Wang M, Jia R, Chen S, Sun K, Yang Q, Wu Y, Chen X, Biville F, Cheng A (2016) Investigation of TbfA in Riemerella anatipestifer using plasmid-based methods for gene over-expression and knockdown. Sci Rep 6:37159. https://doi.org/10.1038/srep37159
Wang X, Liu W, Zhu D, Yang L, Liu M, Yin S, Wang M, Jia R, Chen S, Sun K, Cheng A, Chen X (2014) Comparative genomics of Riemerella anatipestifer reveals genetic diversity. BMC Genomics 15:479. https://doi.org/10.1186/1471-2164-15-479
Wang M, Zhang P, Zhu D, Wang M, Jia R, Chen S, Sun K, Yang Q, Wu Y, Chen X, Biville F, Cheng A, Liu M (2017) Identification of the ferric iron utilization gene B739_1208 and its role in the virulence of R. anatipestifer CH-1. Vet Microbiol 201:162–169. https://doi.org/10.1016/j.vetmic.2017.01.027
Liu M, Huang M, Shui Y, Biville F, Zhu D, Wang M, Jia R, Chen S, Sun K, Zhao X, Yang Q, Wu Y, Chen X, Cheng A (2018) Roles of B739_1343 in iron acquisition and pathogenesis in Riemerella anatipestifer CH-1 and evaluation of the RA-CH-1DeltaB739_1343 mutant as an attenuated vaccine. PLoS One 13:e0197310. https://doi.org/10.1371/journal.pone.0197310
Liu M, Huang M, Zhu D, Wang M, Jia R, Chen S, Sun K, Yang Q, Wu Y, Biville F, Cheng A (2017) Identifying the genes responsible for iron-limited condition in Riemerella anatipestifer CH-1 through RNA-Seq-Based analysis. Biomed Res Int 2017:8682057. https://doi.org/10.1155/2017/8682057
Guo Y, Hu D, Guo J, Li X, Guo J, Wang X, Xiao Y, Jin H, Liu M, Li Z, Bi D, Zhou Z (2017) The role of the regulator Fur in gene regulation and virulence of Riemerella anatipestifer assessed using an unmarked gene deletion system. Front Cell Infect Microbiol 7:382. https://doi.org/10.3389/fcimb.2017.00382
Liu M, Huang M, Huang L, Biville F, Zhu D, Wang M, Jia R, Chen S, Zhao X, Yang Q, Wu Y, Zhang S, Huang J, Tian B, Chen X, Liu Y, Zhang L, Yu Y, Pan L, Ur Rehman M, Cheng A (2019) New perspectives on Galleria mellonella larvae as a host model using Riemerella anatipestifer as a proof of concept. Infect Immun 87:00072–00119. https://doi.org/10.1128/iai.00072-19
Crosa LM, Crosa JH, Heffron F (2009) Iron transport in Francisella in the absence of a recognizable TonB protein still requires energy generated by the proton motive force. Biometals 22:337–344. https://doi.org/10.1007/s10534-008-9170-7
Liu M, Tian X, Wang M, Zhu D, Wang M, Jia R, Chen S, Zhao X, Yang Q, Wu Y, Zhang S, Huang J, Tian B, Chen X, Liu Y, Zhang L, Yu Y, Biville F, Pan L, Rehman MU, Cheng A (2019) Development of a markerless gene deletion strategy using rpsL as a counterselectable marker and characterization of the function of RA0C_1534 in Riemerella anatipestifer ATCC11845 using this strategy. PLoS One 14:e0218241. https://doi.org/10.1371/journal.pone.0218241
Imlay JA, Linn S (1986) Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide. J Bacteriol 166:519–527. https://doi.org/10.1128/jb.166.2.519-527.1986
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. https://doi.org/10.1093/nar/29.9.e45
Huang L, Tian X, Liu M, Wang M, Biville F, Cheng A, Zhu D, Jia R, Chen S, Zhao X, Yang Q, Wu Y, Zhang S, Huang J, Tian B, Yu Y, Liu Y, Zhang L, Pan L, Rehman MU, Chen X (2019) DprA is essential for natural competence in Riemerella anatipestifer and has a conserved evolutionary mechanism. Front Genet 10:429. https://doi.org/10.3389/fgene.2019.00429
Reed LJ, Muench H (1938) A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497
Chareyre S, Mandin P (2018) Bacterial iron homeostasis regulation by sRNAs. Microbiol Spectr 6:10–17. https://doi.org/10.1128/microbiolspec.RWR-0010-2017
Huang SH, Wang CK, Peng HL, Wu CC, Chen YT, Hong YM, Lin CT (2012) Role of the small RNA RyhB in the Fur regulon in mediating the capsular polysaccharide biosynthesis and iron acquisition systems in Klebsiella pneumoniae. BMC Microbiol 12:148. https://doi.org/10.1186/1471-2180-12-148
Yeowell HN, White JR (1982) Iron requirement in the bactericidal mechanism of streptonigrin. Antimicrob Agents Chemother 22:961–968
Zheng M, Doan B, Schneider TD, Storz G (1999) OxyR and SoxRS regulation of fur. J Bacteriol 181:4639–4643
Le SB, Hailer MK, Buhrow S, Wang Q, Flatten K, Pediaditakis P, Bible KC, Lewis LD, Sausville EA, Pang YP, Ames MM, Lemasters JJ, Holmuhamedov EL, Kaufmann SH (2007) Inhibition of mitochondrial respiration as a source of adaphostin-induced reactive oxygen species and cytotoxicity. J Biol Chem 282:8860–8872. https://doi.org/10.1074/jbc.M611777200
Sarvan S, Yeung A, Charih F, Stintzi A, Couture JF (2019) Purification and characterization of Campylobacter jejuni ferric uptake regulator. Biometals 32:491–500. https://doi.org/10.1007/s10534-019-00177-5
Lee JW, Helmann JD (2007) Functional specialization within the Fur family of metalloregulators. Biometals 20:485–499. https://doi.org/10.1007/s10534-006-9070-7
Fillat MF (2014) The FUR (ferric uptake regulator) superfamily: diversity and versatility of key transcriptional regulators. Arch Biochem Biophys 546:41–52. https://doi.org/10.1016/j.abb.2014.01.029
Troxell B, Hassan HM (2013) Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front Cell Infect Microbiol 3:59. https://doi.org/10.3389/fcimb.2013.00059
Bagg A, Neilands JB (1987) Ferric uptake regulation protein acts as a repressor, employing iron (II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry 26:5471–5477. https://doi.org/10.1021/bi00391a039
Hantke K (2001) Iron and metal regulation in bacteria. Curr Opin Microbiol 4:172–177. https://doi.org/10.1016/s1369-5274(00)00184-3
Cornelis P, Wei Q, Andrews SC, Vinckx T (2011) Iron homeostasis and management of oxidative stress response in bacteria. Metallomics 3:540–549. https://doi.org/10.1039/c1mt00022e
Yu C, Genco CA (2012) Fur-mediated global regulatory circuits in pathogenic Neisseria species. J Bacteriol 194:6372–6381. https://doi.org/10.1128/JB.00262-12
Carpenter BM, Gilbreath JJ, Pich OQ, McKelvey AM, Maynard EL, Li ZZ, Merrell DS (2013) Identification and characterization of novel Helicobacter pylori apo-fur-regulated target genes. J Bacteriol 195:5526–5539. https://doi.org/10.1128/JB.01026-13
Ernst FD, Bereswill S, Waidner B, Stoof J, Mader U, Kusters JG, Kuipers EJ, Kist M, van Vliet AHM, Homuth G (2005) Transcriptional profiling of Helicobacter pylori Fur- and iron-regulated gene expression. Microbiology 151:533–546. https://doi.org/10.1099/mic.0.27404-0
Butcher J, Sarvan S, Brunzelle JS, Couture JF, Stintzi A (2012) Structure and regulon of Campylobacter jejuni ferric uptake regulator Fur define apo-Fur regulation. Proc Natl Acad Sci U S A 109:10047–10052. https://doi.org/10.1073/pnas.1118321109
Gancz H, Censini S, Merrell DS (2006) Iron and pH homeostasis intersect at the level of Fur regulation in the gastric pathogen Helicobacter pylori. Infect Immun 74:602–614. https://doi.org/10.1128/IAI.74.1.602-614.2006
Pich OQ, Merrell DS (2013) The ferric uptake regulator of Helicobacter pylori: a critical player in the battle for iron and colonization of the stomach. Future Microbiol 8:725–738. https://doi.org/10.2217/fmb.13.43
Ebanks RO, Goguen M, Knickle L, Dacanay A, Leslie A, Ross NW, Pinto DM (2013) Analysis of a ferric uptake regulator (Fur) knockout mutant in Aeromonas salmonicida subsp. salmonicida. Vet Microbiol 162:831–841. https://doi.org/10.1016/j.vetmic.2012.10.038
Tanui CK, Shyntum DY, Priem SL, Theron J, Moleleki LN (2017) Influence of the ferric uptake regulator (Fur) protein on pathogenicity in Pectobacterium carotovorum subsp. brasiliense. PLoS One 12:e0177647. https://doi.org/10.1371/journal.pone.0177647
Perard J, Nader S, Levert M, Arnaud L, Carpentier P, Siebert C, Blanquet F, Cavazza C, Renesto P, Schneider D, Maurin M, Coves J, Crouzy S, Michaud-Soret I (2018) Structural and functional studies of the metalloregulator Fur identify a promoter-binding mechanism and its role in Francisella tularensis virulence. Commun Biol 1:93. https://doi.org/10.1038/s42003-018-0095-6
Bogomolnaya LM, Tilvawala R, Elfenbein JR, Cirillo JD, Andrews-Polymenis HL (2020) Linearized siderophore products secreted via MacAB efflux pump protect salmonella enterica serovar typhimurium from oxidative stress. mBio 11:3. https://doi.org/10.1128/mBio.00528-20
VanderWal AR, Makthal N, Pinochet-Barros A, Helmann JD, Olsen RJ, Kumaraswami M (2017) Iron efflux by PmtA is critical for oxidative stress resistance and contributes significantly to group A Streptococcus virulence. Infect Immun 85:00091–00117. https://doi.org/10.1128/IAI.00091-17
Turner AG, Djoko KY, Ong CY, Barnett TC, Walker MJ, McEwan AG (2019) Group A Streptococcus co-ordinates manganese import and iron efflux in response to hydrogen peroxide stress. Biochem J 476:595–611. https://doi.org/10.1042/BCJ20180902
Johnson MD, Kehl-Fie TE, Klein R, Kelly J, Burnham C, Mann B, Rosch JW (2015) Role of copper efflux in pneumococcal pathogenesis and resistance to macrophage-mediated immune clearance. Infect Immun 83:1684–1694. https://doi.org/10.1128/IAI.03015-14
Pajuelo D, Hernandez-Cabanyero C, Sanjuan E, Lee CT, Silva-Hernandez FX, Hor LI, MacKenzie S, Amaro C (2016) Iron and Fur in the life cycle of the zoonotic pathogen Vibrio vulnificus. Environ Microbiol 18:4005–4022. https://doi.org/10.1111/1462-2920.13424
Acknowledgements
We thank Dr Francis Biville (Institute Pasteur) and Philippe Delepelaire (Institut de Biologie Physico-Chimique, CNRS Université Paris Diderot) for valuable discussions.
Funding
This work was supported by the National Natural Science Foundation of China (grant No. 32072825, http://www.nsfc.gov.cn/), Sichuan Science and Technology Program (2020YJ0344), China Agricultural Research System (CARS-42–17), and Sichuan Veterinary Medicine and Drug Innovation Group of the China Agricultural Research System (SCCXTD-2020–18), as well as National Undergraduates Innovating Experimentation Project (201910626019).
Author information
Authors and Affiliations
Contributions
ML, MW and AC conceived and designed the research. ML, MH, JL, and MW performed experiments and wrote the manuscript. MH, JL, DZ, RJ, SC, QT, and XZ participated in the experiments. QY, YW, SZ, and JH contributed analysis tools. XO, SM, DS, and QG supervised the studies and corrected the manuscripts. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
One-day-old Pekin ducklings were purchased from Grimaud Farms in Chengdu (Sichuan, China) and housed at our animal facilities with free access to food and water. This study was carried out in accordance with the recommendations of the local animal welfare bodies and the Sichuan Agricultural University ethics committee (SYXK2014-187).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, M., Liu, M., Liu, J. et al. Functional characterization of Fur in iron metabolism, oxidative stress resistance and virulence of Riemerella anatipestifer. Vet Res 52, 48 (2021). https://doi.org/10.1186/s13567-021-00919-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-021-00919-9