
Abstract
Background
The cholecystokinin receptor subtype 2 (CCK-2R) is an important target for diagnostic imaging and targeted radionuclide therapy (TRNT) due to its overexpression in certain cancers (e.g., medullary thyroid carcinoma (MTC)), thus matching with a theranostic principle. Several peptide conjugates suitable for the TRNT of MTC have been synthesized, including a very promising minigastrin analogue DOTA-(DGlu)6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 (CP04). In this contribution, we wanted to see whether CP04 binding affinity for CCK-2R is sensitive to the type of the complexed radiometal, as well as to get insights into the structure of CP04-CCK2R complex by molecular modeling.
Results
In vitro studies demonstrated that there is no significant difference in CCK-2R binding affinity and specific cellular uptake between the CP04 conjugates complexed with [68Ga]Ga3+ or [177Lu]Lu3+. In order to investigate the background of this observation, we proposed a binding model of CP04 with CCK-2R based on homology modeling and molecular docking. In this model, the C-terminal part of the molecule enters the cavity formed between the receptor helices, while the N-terminus (including DOTA and the metal) is located at the binding site outlet, exposed in large extent to the solvent. The radiometals do not influence the conformation of the molecule except for the direct neighborhood of the chelating moiety.
Conclusions
The model seems to be in agreement with much of structure-activity relationship (SAR) studies reported for cholecystokinin and for CCK-2R-targeting radiopharmaceuticals. It also explains relative insensitivity of CCK-2R affinity for the change of the metal. The proposed model partially fits the reported site-directed mutagenesis data.
Similar content being viewed by others

Background
With the enormous significance of cancer therapies for public health and the limited success of the currently available treatments (e.g., chemotherapy, external radiotherapy), there is an urgent need for development of novel—safer and more effective—strategies. One of the most intensively advancing and most promising directions in this realm is the targeted radionuclide therapy (TRNT). Its principle is to selectively accumulate the radionuclide-carrying pharmaceutical only in the tumor tissues so that the toxic effect of ionizing radiation may be restricted to the diseased cells with the negative impact on healthy ones being minimized. The same mechanism may be applied for the targeted diagnosis of cancers if the radionuclide used is a γ-emitter or positron emitter. In this case, a small dose of a radiopharmaceutical accumulated selectively in the cancer tissues precisely localizes the tumors. The combination of diagnostic and therapeutic properties renders radiopharmaceuticals ideal candidates for theranostics, i.e., allowing the early detection of the disease and patient stratification.
The radionuclide is (most often) brought to the site of action in the form of a complex with a chelator attached to a vector (small organic molecule, peptide, antibody, etc.) that is able to selectively target a specific type of cells. The selectivity here is possible thanks to the fact that many types of cancers overexpress particular proteins, especially receptors, on their surfaces. A properly chosen vector—with high affinity for an overexpressed receptor—enables concentration of the therapeutic radionuclide in the desired area and brings about the enhancement of its therapeutic action.
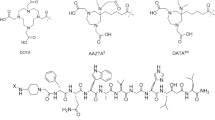
Among the cancer types which may benefit due to the treatment and diagnosis offered by targeted radionuclide delivery are medullary thyroid carcinomas (MTC). MTC belong still to one of the most challenging cancers. They are especially difficult to handle, especially in their advanced stages. Due to the high overexpression of the cholecystokinin receptor subtype 2 (CCK-2R) on MTC cells (incidence of > 90%) [1,2,3], it has been proposed that CCK-2R targeting peptides may serve as vectors for diagnostic imaging and TRNT of MTC. Several suitable peptide conjugates have been now reported with some of them showing high receptor affinity and ability for internalization [4]. One of these is a minigastrin (MG; H-Leu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) analogue CP04: DOTA-(DGlu)6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 (Fig. 1). Its radiolabeled complexes have been studied with respect to stability [5], receptor binding, internalization [4], in vivo tumor targeting in animals [6], and kidney retention [7]. These encouraging results prompted further clinical evaluation of [111In]In-CP04 within the ERA-NET project GRAN-T-MTC [8, 9].

Structure of CP04
Due to the properties of the macrocyclic chelator DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) N-terminally attached to the peptide sequence, CP04 can be radiolabeled with [111In]In3+ or [68Ga]Ga3+ for imaging or with [90Y]Y3+ and [177Lu]Lu3+ for therapy of MTC. However, another aspect of CP04 radiopharmaceutical properties that has remained only partially examined so far is whether their binding affinity to CCK-2R is influenced by the type of the incorporated radiometal. Such sensitivity has been demonstrated for, e.g., some radioconjugates targeting the bombesin [10] or somatostatin [11] receptors. In the case of CP04-based radiopharmaceuticals, Roosenburg et al. [12] have found apparent IC50 variations of rather moderate character in compounds labeled with [natIn], [natGa], and [natCu] or non-labeled. Our previous study showed different chromatographic behaviors of Ga-CP04 complex compared to Lu-CP04 (HPLC-relative retention to CP04: 1.08 and 0.85, respectively). A different coordination of these metal ions was also confirmed by LC-MS study [13]. In the present contribution, we supplement these findings by showing that no significant difference in competition binding and cellular uptake and internalization exists between Ga3+ and Lu3+ chelated CP04. Furthermore, we report that the metal exchange has no influence on the peptide conformation. We also propose the structural basis for the CP04-CCK2R binding based on molecular modeling of the peptide-ligand interactions.
Methods
Chemicals
CP04 (DOTA-(DGlu)6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) in GMP grade was purchased from piCHEM (Graz, Austria). The peptide was complexed with gallium and lutetium following the method described in our previous publication [13].
Cell culture
The A431 human epidermoid carcinoma cell line stably transfected with the human CCK2R (A431-CCK2R(+)) and mock transfected with an empty vector alone (A431-CCK2R(−)) were kindly provided by Prof. Luigi Aloj [14].
The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% of heat-inactivated fetal bovine serum and antibiotics (penicillin 100 U/mL and streptomycin 100 μg/mL) and cultivated at 37 °C in 5% CO2.
Competition binding assay
The binding affinity (the apparent 50% inhibitory concentration (IC50)) of [natGa]Ga-CP04 and [natLu]Lu-CP04 complexes for CCK2 receptor was determined in A431-CCK2R(+) cell line using [90Y]Y-CP04 as a competitive radioligand according to the method used by Roosenburg et al. [12]. [90Y]Y-CP04 was obtained with a molar activity of 2.05 GBq/μmol calculated at the time of experiment and 98% radiochemical purity as determined by radio HPLC method. Human epidermoid carcinoma (A431-CCK2(+)) cells were subcultured overnight in 12-well plates in concentration of 0.5 mln per well. Before the experiments, the cells were washed with DMEM medium and then [90Y]Y-labeled CP04 (2.05 GBq/μmol) was added to the cells in amount of 0.24 pmol per well (ca. 80,000 cpm) and incubated for 1 h at 37 °C with increasing concentrations (10−6–10−13 M) of the Ga and Lu complexes of CP04 peptide. After the incubation, the medium was removed and the cells were washed with phosphate-buffered saline (PBS). Subsequently, the cells were lysed with 1 M NaOH and collected and the cell-associated radioactivity was measured using a well-type gamma counter (1470 Wizard, Wallac). The IC50 value (half maximal inhibitory concentration) was calculated based on competition nonlinear binding curve using GraphPad Prism software Version 7.03. The results are expressed as a mean ± SD of three experiments performed in triplicate.
Cellular uptake and internalization
Cellular uptake/internalization of [68Ga]Ga- or [177Lu]Lu-labeled CP04 complexes in CCK2R-positive cells was carried out according to the method by Kaloudi et al. [15]. Approximately 100,000 cpm of [68Ga]Ga- or [177Lu]Lu-labeled CP04 (ca 1 ng of CP04/well) were added to the pre-incubated (24 h) cells (1 million cells in 2 mL of the medium) seeded in 6-well plates. The cells were incubated for 1 h at 37 °C. After incubation, the medium was removed and the cells were washed with cold 0.5% BSA-PBS. The binding to the cell surface receptors was determined by measurement of radioactivity of 1 mL of glycine buffer used for rinsing of cells. Then, 1 mL of 1 M NaOH was used for the lysis of the washed cells. Samples of the membrane-bound fractions and the internalized fractions (lysates) were measured for their radioactivity content using a well-type gamma counter (1470 Wizard, Wallac), and then, the percentage of membrane-bound and internalized fractions was calculated. The specific uptake to CCK2R receptors was calculated by subtracting the values obtained for A431-CCK2R(−) cells from the values obtained for A431-CCK2R(+) cells. At least four independent experiments were performed in triplicate.
NMR measurements
For the purpose of NMR measurements, samples of the CP04 complexes were dissolved in 90% H2O/10% D2O (total volume of about 0.6 ml). The NMR spectra with WATERGATE solvent suppression were recorded at 400.13 MHz proton frequency and at 303 K on a Bruker DRX 400 spectrometer. The 1D 1H-NMR spectra were recorded according to standard procedures with a time domain of 32-k data points and a spectral width of 6410.27 Hz [16,17,18,19,20,21]. The free-induction decay was acquired for 2.556 s with a dwell time set to 78.0 μsec. The relaxation delay was set to 1 s. The sweep width of the 2D homonuclear TOCSY and ROESY spectra was 5592.84 Hz in the direct and indirect 1H-dimension, respectively. The free-induction decay was acquired for 183.1 msec with a dwell time set to 89.4 μsec. The relaxation delay was also set to 1 s. All spectra were zerofilled prior to Fourier transformation, and sine apodisation functions were applied in both dimensions using MestReNova [22].
Homology modeling
The CCK2 receptor structure was obtained via homology modeling by using the SWISS-MODEL Homology Modeling server [23,24,25]. The template was the structure of δ-opioid receptor bound to naltrindole (PDB accession code: 4EJ4) which was selected based on sequence identity, similarity, and coverage as suggested by the server (no user intervention). All the default parameters were retained.
Molecular docking
Several conformations of the unlabeled CP04 were generated and then docked to the CCK-2R homology model by using AutoDock Vina [26]. The binding box was selected to encompass the intrahelical cavity as well as extracellular loops and then extended to free volume around the receptor outlet; exhaustiveness was set to 20. The best scored binding poses were manually inspected for compatibility with mutagenesis data reported for CCK derivatives binding to CCK-2R [27].
Results and discussion
In vitro binding properties
With the purpose of checking the sensitivity of CCK-2R/CP04 interaction to the change of metal coupled to the peptide, [177Lu]Lu- and [68Ga]Ga-CP04 complexes were prepared and assayed for affinity to human CCK-2R by competition binding experiments in A431-CCK2R(+) cells. In both cases, the apparent affinity for hCCK-2R was in the low nanomolar range and was found comparable for [natGa]Ga-CP04 and [natLu]Lu-CP04 (IC50= 1.15 ± 0.39 nM, n = 3, and 1.02 ± 0.28 nM, n = 3, respectively).
Both complexes efficiently internalized in A431-CCK-2R(+) cells after 1-h incubation at 37 °C, showing a minor, 4% portion of radioactivity bound on the cell membrane, as expected for receptor agonists, with high internalization (over 70%) (Table 1). There was no statistically significant difference between the [177Lu]Lu- and [68Ga]Ga-CP04 complexes with regard to cellular uptake (according to the Student t test).
These observations prompted us to investigate their structural basis by NMR and molecular modeling.
NMR studies
The complexes were subject to NMR TOCSY and ROESY experiments so to see whether the type of metal influences the structure of the compound. The signal dispersion in the 1D 1H NMR spectra (Fig. 2) suggests a rather dynamic conformational ensemble of molecules in solution. The respective 1H NMR signals were assigned on the basis of their characteristic chemical shifts, as well as TOCSY and ROESY correlations (Table 2). Most of the chemical shifts originating from amino acids in both complexes are almost exactly the same, except for those very close to the site of complexation, i.e., originating from the two last glutamic acid moieties. As a result, it can be concluded that no conformational difference between the two complexes is observed. The differences in the chemical shifts of the D-Glu1–2 can be ascribed to different coordination structures. In the crystal structure of [Ga-DOTA-D-Phe-NH2], the ion is coordinated by only two carboxylate groups, with the remaining carboxylate arm pendant [28]. Contrarily, no free carboxylate is found in DOTA (or DOTA derivatives) crystal structures with lanthanides (like Lu(III)) [28, 29]. The presence or lack of the free carboxylate group in the chelator moiety, as well as different geometries of the macrocycle bringing about local perturbation in a structure, is reflected by different chemical shifts corresponding to the amino acids neighboring the DOTA moiety.

The overlay of the 1H-1H TOCSY (blue) and ROESY (red) spectra of Ga3+-CP04 complex (upper spectra) and Lu3+-CP04 complex (lower spectra). The red spots with no blue ones below indicate ROESY correlations between distinct amino acids
A close inspection of the ROESY correlations indicates that only intraresidual and (a few) sequential cross correlations between amino acids could be detected (please refer to the overlay of ROESY and TOCSY spectra, Fig. 2). Therefore, no secondary structure can be identified, which further confirms that both complexes form a dynamic ensemble of conformations rather than an organized rigid structure.
CP04 binding mode from molecular docking
In order to elucidate the molecular basis of the CP04/CCK-2R interactions, the conjugate was docked to the receptor homology model (no CCK-2R crystal structure has been reported so far). The molecular docking predicts that CP04 enters the CCK2R binding pocket with the C-terminus directed towards the intracellular side (Fig. 3). Numerous contacts between the peptide and the receptor are created. Discussing these (as well as in further parts of the manuscript), for the sake of convenience, the CP04 amino acid residues are counted back starting from the C-terminus and the number denoted with the minus sign in the superscript. The aromatic side chain of the C-terminal Phe−1 is located in the “aromatic box” formed by Trp2185.39, His207ECL2, Tyr1894.61, and Trp209ECL2. The phenyl ring lies parallel to the rings of Trp2185.39 and His207ECL2, while perpendicular to the ring of Tyr1894.61. The charged side chain of the penultimate Asp−2 points to His207ECL2. The positioning of Met−3 side chain is less tight. The side chain is oriented towards Thr1112.60, but as there is a lot of free volume around it, it is probable that multiple orientations co-exist. In the case of Trp−4, the aromatic indole forms hydrophobic interactions with Trp2185.39, Leu2225.43 and Asn3536.55. A cation-π interaction is formed between Arg3566.58 and the aromatic ring of Tyr−6. Additional stabilization of the bound conformation may come here from a hydrogen bond between the Tyr−6 phenol function and the carbonyl of Ala3576.59.

CP04 (green) docked to the homology model of the CCK2 receptor (gray): (a) general view and (b, c) focus on the interactions of the four C-terminal residues with the receptor presented from two perspectives
The further (counting from the C-terminus) amino acid residues (including the D-Glu’s of the “spacer”) and DOTA are located at the binding site outlet where the receptor helices are located quite far from each other. Large free volume in this vicinity, solvent exposition, and a simultaneous lack of strong binding partners for flexible residues of the spacer arm make it likely that this part of CP04 assumes many conformations without sharp energetic minima.
The proposed binding mode fits the SAR of CCK/gastrin derivatives and MG-based radiopharmaceuticals
Endogenous CCK-2R ligands include several cholecystokinin (CCK) and gastrin peptides of varying length (Fig. 4). In the past, their sequences were starting points for development of many peptide CCK-2R agonists. As a consequence, rich structure-activity relationship (SAR) data is available.

Sequences of the endogenous CCK-2R ligands. C-terminal WMDF-NH2 sequence is pharmacophoric for this receptor
The presented binding model of CP04 to CCK-2R fits the structure-activity relationships of the peptide agonists of this receptor (on the assumption that CP04 binding mode overlaps with that of other derivatives). Especially, the positioning of the pharmacophoric C-terminal WMDF-NH2 seems to correspond well to the observations gathered in SAR studies (Table 3).
The herein reported binding mode can be also used to rationalize much of the binding data reported so far for minigastrin-based vectors and radiopharmaceuticals. In the light of our model, it is well understood why the chelating moiety has to be attached to the N-terminus while the derivatives with the ligand coupled via the C-terminal amide or in the para position of the Phe−1 exhibit a significant decrease in affinity of even several orders of magnitude [30]. A voluminous moiety at the C-terminus cannot be fit in the binding pocket together with the Trp-Met-Asp-Phe-NH2 and requires a complete rearrangement of the binding mode.
Analogously to the CCK and MG derivatives prepared for general medicinal chemistry, the MG-based vectors are most sensitive to the changes in the final C-terminal tetrapeptide sequence and the similar exchanges are tolerated (e.g., Met−3 to norleucine [31] or methoxinine [32]). Variations in the spacer residues between the chelator and the Trp-Met-Asp-Phe-NH2 influence the affinity only to a minor extent [4]. In the past, the derivatives with spacers of different lengths were reported (1–6 residues [31, 33, 34]) and with different residues (His [31], Gln, Ser, PEG [34]) instead of the hexa-Glu sequence. All these variations had only little impact on binding. The same applies to the stereochemistry of the spacer as the hexa-L-Glu derivative has similar affinity as the hexa-D-Glu analogue [7]. The derivatives with cyclized (rigidified) elements in the spacer do not have a much improved affinity [35]. Furthermore, little impact on affinity is seen also with different chelating agents at the N-terminus (DOTA, DOTAGA, DTPA, HYNIC) [30, 33, 35,36,37]. The presence and the character of metal give none but small changes in the binding strength. Some minigastrin analogues complexed with natIn have been reported to have IC50 identical (within error) to the one of metal-free conjugates [7, 34]. Roosenburg and colleagues prepared CP04 analogues with three different chelators unmetaled or in complex with three different metals (12 variants in total), and the IC50 values varied between 0.8 and 1.5 nM [12]. In the contribution herein presented we see no significant difference in binding of compounds with Ga and Lu even though Ga3+-DOTA and Lu3+-DOTA have different coordination structures (one free carboxylate arm in the chelate with Ga3+ [28, 29]). The affinity of shorter minigastrin analogue with AAZTA as a chelator (AAZTA-MG) also did not exhibit sensitivity with regard to the incorporated metal [38].
The relative insensitivity of the binding affinity to the changes in the spacer, chelating moiety, and the presence/absence of metal as well as its exact type is again understandable within our binding model. The N-terminal part of CP04 (and by analogy: N-terminal part of the discussed radiopharmaceuticals) is predicted to be located at the receptor binding site outlet. Due to a large free volume in this vicinity, lack of strong interaction partners, as well as the solvent exposure of the site, accompanied by significant flexibility of the peptide chain, it is not unreasonable to suppose that this substructure may take several energetically similar conformations. With a few poses in equilibrium and no tight arrangement of the N-terminus, different modifications (spacer length, exchange of amino acids in the spacer, cyclization, and various chelators and metals) do not disturb the binding to a significant degree. For example, if any electrostatic mismatch lowering affinity might occur upon a particular structural change in the conjugate, the molecule should be easily able to accommodate a different binding pose avoiding unfavorable contacts. And contrarily, a possible additional drug-receptor contact in this vicinity should not improve the binding a lot since in the solvent-exposed environment, the strength of a single intermolecular electrostatic interaction is reduced. The experimentally observed small variations are too subtle to be explained or predicted by the used modeling methods. An attempt of such prediction would require also incorporation of the amino terminal domain of the receptor (absent in this model and in many crystallographic structures of GPCRs; CCK-2R N-terminus shows no significant homology to any protein found in the PDB database [39]).
The proposed binding mode in the light of earlier receptor studies
Extensive experimental studies had been performed on CCK2 receptor in order to determine the structural basis of the interactions with ligands. Unfortunately, not all of them coincide with our predictions. Recently, based on photoaffinity labeling studies and hints from mutagenesis, it has been proposed that Phe120ECL1 interacts with Phe−1 of a CCK-like probe compound [40]. Contrarily, according to our binding pattern, this residue does not have any direct interaction with CP04. In the past, photoaffinity labeling studies suggested also Phe122ECL1 and Thr119ECL1 to be involved in interactions with CCK [41]; however, in our model, no contact with CP04 is predicted. On the other hand, in agreement with the results presented here, fluorescence spectroscopy studies supported the entrance of the CCK C-terminus into the intramembranous helical bundle [42].
Regarding the site-directed mutagenesis, some of the reported data do not seem to fit our model. The mutation of residues such as Arg571.35, Tyr611.39, Phe120ECL1, Thr1934.65, Trp3466.48, and Val3496.51 has been shown to influence more or less the binding affinity of cholecystokinin peptide [27]. In the binding mode presented herein, these residues are rather distant from any of the CP04 residues. Still, several other results from mutagenesis are easily reconcilable with our model (Table 4).
Here let us note that all comparisons between the experimental data and our model are burdened with several issues. First of all, most of the experimental studies were performed for cholecystokinin or variants thereof. While the C-terminal residues are identical in those and in CP04, the remaining parts of the molecules may influence the binding mode of the common sequences. Secondly, the loss of affinity upon a mutation could mean that a particular residue is involved in interactions with the ligand in the final binding pose, but it cannot be excluded that such a residue only takes part in the process of ligand entrance or induced-fit. The same applies to the results coming from the photoaffinity labeling or fluorescence spectroscopy.
In our binding mode, the positioning of the C-terminal tetrapeptide sequence differs considerably from the one proposed for cholecystokinin in earlier studies [43, 44]. These differences seem inevitable as the earlier homology models had been built on rhodopsin or β-adrenergic receptor as templates. Our model uses the inactive crystal of delta opioid receptor. This structure and those ones differ as to the relative positions of the helices and the geometries of the loops, and consequently, the binding sites do differ too. However, our prediction agrees with those earlier propositions in that the terminal tetrapeptide sequence enters the cavity formed between the helices. For the sake of completeness, let us mention here that other homology models have been reported too [45, 46], but as they were used for modeling of small molecular binders, no comparisons can be made here.
Conclusions
Although usually the modeling and docking studies are employed in the design of new molecules, in this work, we referred to them from the radiopharmacy routine perspective in order to better understand the tracer’s biological behavior. The structure of DOTA complex is not the same when either Ga3+ or Lu3+ is incorporated; however, our competition binding assay as well as cellular uptake and internalization studies demonstrated that there is no significant difference in affinity for CCK-2R between the Ga- and Lu-bearing CP04 complexes. NMR spectroscopy shows that the metal influences only the conformation of the direct neighborhood of the chelating site. We also propose a binding mode for CP04 and CCK-2R based on homology modeling of the receptor and molecular docking. In this model, the conjugate dips down between the helices with the C-terminal end, while the N-terminal part including DOTA and the metal is at the receptor outlet. The residues of the carboxy terminus form many well-defined interactions with the receptor binding site. Much of these seem consistent with experimental SAR data for cholecystokinin and derivatives. Our in silico model allows also the rationalization of the so far reported binding data for minigastrin-based radiopharmaceuticals. It also partially fits the experimental receptor studies (mainly mutagenesis).
References
Reubi JC, Waser B. Unexpected high incidence of cholecystokinin-B/gastrin receptors in human medullary thyroid carcinomas. Int J Cancer. 1996;67:644–7.
Reubi JC, Schaer JC, Waser B. Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997;57:1377–86.
Reubi JC. CCK receptors in human neuroendocrine tumors: clinical implications. Scand J Clin Lab Invest. 2001;61:101–4.
Aloj L, Aurilio M, Rinaldi V, D’ambrosio L, Tesauro D, Peitl PK, et al. Comparison of the binding and internalization properties of 12 DOTA-coupled and 111In-labelled CCK2/gastrin receptor binding peptides: a collaborative project under COST action BM0607. Eur J Nucl Med Mol Imaging Springer-Verlag. 2011;38:1417–25.
Ocak M, Helbok A, Rangger C, Peitl PK, Nock BA, Morelli G, et al. Comparison of biological stability and metabolism of CCK2 receptor targeting peptides, a collaborative project under COST BM0607. Eur J Nucl Med Mol Imaging. 2011;38:1426–35.
Laverman P, Joosten L, Eek A, Roosenburg S, Peitl PK, Maina T, et al. Comparative biodistribution of 12 111In-labelled gastrin/CCK2 receptor-targeting peptides. Eur J Nucl Med Mol Imaging. 2011;38:1410–6.
Kolenc Peitl P, Tamma M, Kroselj M, Braun F, Waser B, Reubi JC, et al. Stereochemistry of amino acid spacers determines the pharmacokinetics of 111In-DOTA-Minigastrin analogues for targeting the CCK2/gastrin receptor. Bioconjug Chem American Chemical Society. 2015;26:1113–9.
Pawlak D, Rangger C, Kolenc Peitl P, Garnuszek P, Maurin M, Ihli L, et al. From preclinical development to clinical application: kit formulation for radiolabelling the minigastrin analogue CP04 with In-111 for a first-in-human clinical trial. Eur J Pharm Sci. 2016;85:1–9.
Maina T, Konijnenberg MW, Kolenc Peitl P, Garnuszek P, Nock BA, Kaloudi A, et al. Preclinical pharmacokinetics, biodistribution, radiation dosimetry and toxicity studies required for regulatory approval of a phase I clinical trial with 111In-CP04 in medullary thyroid carcinoma patients. Eur J Pharm Sci. 2016;91:236–42.
Koumarianou E, Loktionova NS, Fellner M, Roesch F, Thews O, Pawlak D, et al. 44Sc-DOTA-BN[2-14]NH 2 in comparison to 68Ga-DOTA-BN[2-14]NH 2 in pre-clinical investigation. Is 44Sc a potential radionuclide for PET. Appl. Radiat. Isot. Elsevier. 2012;70:2669–76.
Fani M, Braun F, Waser B, Beetschen K, Cescato R, Erchegyi J, et al. Unexpected sensitivity of sst2 antagonists to N-terminal radiometal modifications. J Nucl Med. 2012;53:1481–9.
Roosenburg S, Laverman P, Joosten L. PET and SPECT imaging of a radio labeled minigastrin analogue conjugated with DOTA, NOTA, and NODAGA and labeled with Cu-64, Ga-68, and In-111. Mol Pharmaceutics. 2014;11:3930–7.
Maurin M, Garnuszek P, Baran P, Pawlak D, Mikołajczak R. The radiometal makes a difference. Synthesis and preliminary characterisation of DOTA-minigastrin analogue complexes with Ga, Lu and y. Nucl Med Rev. 2015;18:51–5.
Aloj L, Caracò C, Panico M, Zannetti A, Del Vecchio S, Tesauro D, et al. In vitro and in vivo evaluation of 111In-DTPAGlu-G-CCK8 for cholecystokinin-B receptor imaging. J Nucl Med. 2004;45:485–94.
Kaloudi A, Nock BA, Lymperis E, Krenning EP, de Jong M, Maina T. Improving the in vivo profile of minigastrin radiotracers: a comparative study involving the neutral endopeptidase inhibitor phosphoramidon. Cancer Biother Radiopharm. Mary Ann Liebert, Inc. 140 Huguenot Street, 3rd Floor New Rochelle, NY 10801 USA. 2016;31:20–8.
Kokoschka M, Bangert J-A, Stoll R, Sheldrick WS. Sequence-selective organoiridium DNA bis-intercalators with flexible dithiaalkane linker chains. Eur J Inorg Chem. 2010;2010:1507–15.
Nowaczyk M, Berghaus C, Stoll R, R?gner M, Yocum CF, Klinman JP, et al. Preliminary structural characterisation of the 33 kDa protein (PsbO) in solution studied by site-directed mutagenesis and NMR spectroscopy. Phys Chem. 2004;6:4878–81.
Ali Nazif M, Bangert J-A, Ott I, Gust R, Stoll R, Sheldrick WS. Dinuclear organoiridium(III) mono- and bis-intercalators with rigid bridging ligands: synthesis, cytotoxicity and DNA binding. J Inorg Biochem. 2009;103:1405–14.
Glaves R, Baer M, Schreiner E, Stoll R, Marx D. Conformational dynamics of minimal elastin-like polypeptides: the role of proline revealed by molecular dynamics and nuclear magnetic resonance. ChemPhysChem. 2008;9:2759–65.
Stoll R, Voelter W, Holak TA. Conformation of thymosin beta 9 in water/fluoroalcohol solution determined by NMR spectroscopy. Biopolymers. 1997;41:623–34.
Song J, Markley JL. NMR chemical shift mapping of the binding site of a protein proteinase inhibitor: changes in the1H,13C and15N NMR chemical shifts of turkey ovomucoid third domain upon binding to bovine chymotrypsin A? J Mol Recognit. 2001;14:166–71.
Mestrelab Research. MestRe Nova. Santiago De Compostela; 2008. Available from: mestrelab.com/
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–8.
Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–50.
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201.
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61.
Miller LJ, Gao F. Structural basis of cholecystokinin receptor binding and regulation. Pharmacol Ther. 2008;119:83–95.
Heppeler A, Froidevaux S, Mäcke HR, Jermann E, Béhé M, Powell P, et al. Radiometal-labelled macrocyclic chelator-derivatised somatostatin analogue with superb tumor-targeting properties and potential for receptor-mediated internal radiotherapy. Chem - a Eur J. 1999;5:1974–81.
Viola-Villegas N, Doyle RP. The coordination chemistry of 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetra acetic acid (H4DOTA): structural overview and analyses on structure-stability relationships. Coord Chem Rev. 2009;253:1906–25.
Reubi JC, Waser B, Schaer JC, Laederach U, Erion J, Srinivasan A, et al. Unsulfated DTPA- and DOTA-CCK analogs as specific high-affinity ligands for CCK-B receptor-expressing human and rat tissues in vitro and in vivo. Eur J Nucl Med. 1998;25:481–90.
Mather SJ, Mckenzie AJ, Sosabowski JK, Morris TM, Watson SA. Selection of radiolabeled gastrin analogs for peptide receptor—targeted radionuclide therapy. J Nucl Med. 2007;48:615–22.
Grob NM, Behe M, von Guggenberg E, Schibli R, Mindt TL. Methoxinine—an alternative stable amino acid substitute for oxidation-sensitive methionine in radiolabelled peptide conjugates. J Pept Sci. 2017;23:38–44.
Good S, Walter MA, Waser B, Wang X, Müller-Brand J, Béhé MP, et al. Macrocyclic chelator-coupled gastrin-based radiopharmaceuticals for targeting of gastrin receptor-expressing tumours. Eur J Nucl Med Mol Imaging. 2008;35:1868–77.
Kolenc Peitl P, Mansi R, Tamma M, Gmeiner-Stopar T, Sollner-Dolenc M, Waser B, et al. Highly improved metabolic stability and pharmacokinetics of indium-111-DOTA-gastrin conjugates for targeting of the gastrin receptor. J Med Chem. 2011;54:2602–9.
von Guggenberg E, Sallegger W, Helbok A, Ocak M, King R, Mather SJ, et al. Cyclic minigastrin analogues for gastrin receptor scintigraphy with technetium-99m: preclinical evaluation. J Med Chem. 2009;52:4786–93.
Laverman P, Béhé M, Oyen WJG, Willems PHGM, Corstens FHM, Behr TM, et al. Two technetium-99m-labeled cholecystokinin-8 (CCK8) peptides for scintigraphic imaging of CCK receptors. Bioconjug Chem. 2004;15:561–8.
von Guggenberg E, Behe M, Behr TM, Saurer M, Seppi T, Decristoforo C. 99m Tc-labeling and in vitro and in vivo evaluation of HYNIC- and ( Nα-His ) acetic acid-modified [D-Glu 1]-minigastrin. Bioconjugate Chem. 2004;15:864–71.
Pfister J, Summer D, Rangger C, Petrik M, von Guggenberg E, Minazzi P, et al. Influence of a novel, versatile bifunctional chelator on theranostic properties of a minigastrin analogue. EJNMMI Res. 2015;5:74.
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42.
Dong M, Miller LJ. Direct demonstration of unique mode of natural peptide binding to the type 2 cholecystokinin receptor using photoaffinity labeling. Peptides. 2013;46:143–9.
Dong M, Liu G, Pinon DI, Miller LJ. Differential docking of high-affinity peptide ligands to type A and B cholecystokinin receptors demonstrated by photoaffinity labeling. Biochemistry. 2005;44:6693–700.
Harikumar KG, Miller LJ. Fluorescence resonance energy transfer analysis of the antagonist- and partial agonist-occupied states of the cholecystokinin receptor. J Biol Chem. 2005;280:18631–5.
Escrieut C, De K, Clerc P, Niu F, Azema J, Masri B, et al. Distinct CCK-2 receptor conformations associated with β-arrestin-2 recruitment or phospholipase-C activation revealed by a biased antagonist. 2012;
Marco E, Foucaud M, Langer I, Escrieut C, Tikhonova IG, Fourmy D. Mechanism of activation of a G protein-coupled receptor, the human cholecystokinin-2 receptor. J Biol Chem. 2007;282:28779–90.
Cawston EE, Lam PCH, Harikumar KG, Dong M, Ball AM, Augustine ML, et al. Molecular basis for binding and subtype selectivity of 1,4-benzodiazepine antagonist ligands of the cholecystokinin receptor. J Biol Chem. 2012;287:18618–35.
Gupta AK, Varshney K, Singh N, Mishra V, Saxena M, Palit G, et al. Identification of novel amino acid derived CCK-2R antagonists as potential antiulcer agent: homology modeling, design, synthesis, and pharmacology. J Chem Inf Model 2013;53:176-87.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Steardo L, Knight M, Tamminga CA, Chase TN. Products of cholecystokinin (CCK)-octapeptide proteolysis interact with central CCK receptors. Neurosci Lett. 1985;54:319–25.
TILLEY JW, DANHO W, S -J SHIUEY, KULESHA I, SARABU R, SWISTOK J, et al. Structure activity of C-terminal modified analogs of Ac-CCK-7. Int J Pept Protein Res. 1992;39:322–36.
Corringer PJ, Weng JH, Ducos B, Durieux C, Boudeau P, Bohme A, et al. CCK-B agonist or antagonist activities of structurally hindered and peptidase-resistant Boc-CCK4 derivatives. J Med Chem. 1993;36:166–72.
Silvente-Poirot S, Escrieut C, Galès C, Fehrentz JA, Escherich A, Wank SA, et al. Evidence for a direct interaction between the penultimate aspartic acid of cholecystokinin and histidine 207, located in the second extracellular loop of the cholecystokinin B receptor. J Biol Chem. 1999;274:23191–7.
Marseigne I, Dor A, Begue D, Reibaud M, Zundel JL, Blanchard JC, et al. Synthesis and biological activity of CCK26-33-related analogs modified in position 31. J Med Chem. 1988;31:966–70.
Danho W, Tilley JW, Shiuey S-J, Kulesha I, Swistok J, Makofske R, et al. Structure activity studies of tryptophan30 modified analogs of Ac-CCK-7. Int J Pept Protein Res. 1992;39:337–47.
Rodriguez M, Bernad N, Galas M, Lignon M, Laur J, Aumelas A, et al. Synthesis and biological activities of cholecystokinin analogues substituted in position 30 by 3-(1-naphthyl)-l-alanine [Nal(1)] or 3-(2-naphthyl)-l-alanine [Nal(2)]. Eur J Med Chem. 1991;26:245–53.
Acknowledgements
Dr. Luigi Aloj is acknowledged for providing the cell lines used in the study. The simulations were performed in Świerk Computing Centre of the National Centre for Nuclear Research in Świerk, Poland. Molecular graphics was prepared with the UCSF Chimera package [47]. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Funding
This project was financed from the funds of the National Science Centre (Poland) allocated on the basis of the decision number DEC-2011/03/B/ST5/02734. The collaboration within WG3 of COST CM1105 “Functional Metal Complexes that Bind toBiomolecules” is acknowledged.
Availability of data and materials
Please contact the corresponding author for data requests.
Author information
Authors and Affiliations
Contributions
PFJL carried out the molecular modeling and wrote the manuscript. PG designed the study, managed the project, and revised the manuscript. MM performed the in vitro studies and radiolabeling. RS and NMN performed the NMR measurements. AW carried out a part of the molecular modeling. JCzD consulted the molecular modeling and revised the manuscript. MKD interpreted the NMR data. MO conducted the in vitro studies, especially the binding assays. RM designed the study, managed the project, and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lipiński, P.F.J., Garnuszek, P., Maurin, M. et al. Structural studies on radiopharmaceutical DOTA-minigastrin analogue (CP04) complexes and their interaction with CCK2 receptor. EJNMMI Res 8, 33 (2018). https://doi.org/10.1186/s13550-018-0387-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13550-018-0387-3