
Abstract
Obesity is a global epidemic that greatly increases risk for developing cardiovascular disease and type II diabetes. Sex differences in the obese phenotype are well established in experimental animal models and clinical populations. While having higher adiposity and obesity prevalence, females are generally protected from obesity-related metabolic and cardiovascular complications. This protection is, at least in part, attributed to sex differences in metabolic effects of hormonal mediators such as the renin-angiotensin system (RAS). Previous literature has predominantly focused on the vasoconstrictor arm of the RAS and shown that, in contrast to male rodent models of obesity and diabetes, females are protected from metabolic and cardiovascular derangements produced by angiotensinogen, renin, and angiotensin II. A vasodilator arm of the RAS has more recently emerged which includes angiotensin-(1-7), angiotensin-converting enzyme 2 (ACE2), mas receptors, and alamandine. While accumulating evidence suggests that activation of components of this counter-regulatory axis produces positive effects on glucose homeostasis, lipid metabolism, and energy balance in male animal models, female comparison studies and clinical data related to metabolic outcomes are lacking. This review will summarize current knowledge of sex differences in metabolic effects of the RAS, focusing on interactions with gonadal hormones and potential clinical implications.
Similar content being viewed by others


Introduction
The control of energy balance involves complex interactions between behavioral and physiological factors influencing energy intake, expenditure, and storage to maintain body weight and body composition within a homeostatic range [1]. As previously reviewed, several studies have shown sex differences in body composition, body fat distribution, and feeding behavior even in lean humans and rodent models [2]. Females have been shown to have higher adiposity and lower fat-free mass at any given body mass index (BMI) compared with males [3, 4]. Furthermore, females have more subcutaneous adipose tissue, particularly in abdominal and gluteofemoral regions, compared with a greater visceral adipose distribution in males [2]. These sex differences in adipose distribution have been linked with metabolic health, with females having a more favorable lipid and glucose metabolism profile compared with males [2].
Obesity results when food intake exceeds energy expenditure to promote excess energy storage in white adipose tissue [1]. Obesity is a global epidemic, affecting over 600 million individuals worldwide, which greatly increases risk for developing cardiovascular disease and type II diabetes [5, 6]. The energy imbalance seen in obesity is often accompanied by hypertension, chronic low grade systemic and adipose inflammation, macrophage infiltration in adipose tissue, and metabolic derangements such as hyperinsulinemia, hyperglycemia, hyperleptinemia, hyperlipidemia, insulin resistance, and glucose intolerance [7]. Sex differences in the obese phenotype are well recognized in experimental animal models and clinical populations [3, 4, 8]. Indeed, the prevalence of obesity has increased to a greater extent in women over the past decade [3, 4]. Despite this, premenopausal women are protected from the development of obesity-related metabolic and cardiovascular complications. Obese females, for example, generally have lower blood pressure, more tissue distributed to subcutaneous than pro-inflammatory visceral adipose tissue, smaller and more lipogenic and insulin-sensitive adipocytes, increased mass and metabolic activity of brown adipose tissue, higher levels of insulin-sensitizing hormones such as leptin and adiponectin, and greater peripheral insulin sensitivity when compared with obese males [3, 4, 8]. In addition, high-fat diet (HFD) feeding in male rodents increases pro-inflammatory M1 type macrophages in adipose tissue, increases percentage of pro-inflammatory T cells in the aorta and kidney, and reduces anti-inflammatory regulatory T cells (Tregs). In contrast, female HFD-fed rodents exhibit increases in anti-inflammatory M2 macrophages in adipose and maintain more Tregs in the aorta and kidney [8, 9]. These sex differences in macrophage polarization and T cell profile in response to HFD may contribute to metabolic and cardiovascular protection in females.
While still an active area of investigation, emerging evidence suggests that sex differences in obesity are, at least in part, attributed to hormonal mediators such as the renin-angiotensin system (RAS). This review will highlight recent developments in our understanding of sex differences in the metabolic effects of the RAS, including interactions with gonadal hormones and potential therapeutic implications for clinical populations. While not a focus of this review, sex differences in RAS components and actions have also been implicated in conditions closely related to metabolic function including aging [10], cardiovascular and renal diseases [11, 12], developmental programming [13], and hypertension [14,15,16].
RAS pathways for metabolic regulation
Canonical RAS pathways
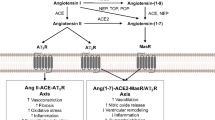
For over a century, the RAS has been recognized for its critical role in blood pressure regulation and the pathogenesis of cardiovascular diseases. Accumulating evidence suggests the RAS is also important in glucose homeostasis and energy balance, and that perturbations in this hormonal system are involved in development of metabolic diseases such as obesity and type II diabetes [17]. In the canonical RAS (Fig. 1), the enzyme renin is secreted into the circulation from renal juxtaglomerular cells in response to stimuli including increased sympathetic activity, local actions of nitric oxide and prostanoids, decreased renal afferent arteriole perfusion pressure, and decreased sodium chloride content in the macula densa of the renal distal tubules [18]. Renin acts upon angiotensinogen to form angiotensin (Ang) I, which is subsequently cleaved by Ang-converting enzyme (ACE) to form Ang II [19]. Ang II acts at cell surface type I G protein-coupled receptors (AT1R) to induce deleterious cardiovascular and metabolic effects including vasoconstriction, sympathetic activation, inflammation, oxidative stress, and insulin resistance [17, 20]. Ang II also binds cell surface type II receptors (AT2R) to counteract AT1R-mediated actions; although these receptors are more limited in tissue expression and affinity [21].

Simplified diagram of the renin-angiotensin system including sites of action for pharmacological agents targeting this hormonal system. A779, mas receptor antagonist [D-Ala7]-angiotensin-(1-7); ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; AT1R, angiotensin II type 1 receptor; AT2R, angiotensin II type 2 receptor; AVE0991, orally active mas receptor agonist; C21, compound 21 (AT2R agonist); DIZE, ACE2 activator diminazene aceturate; EMA401, AT2R agonist; HRP, decoy peptide for handle region of the prorenin prosegment; MasR, angiotensin-(1-7) mas receptor; MLDAD, mononuclear leukocyte-derived aspartate decarboxylase; MrgD, mas-related G protein-coupled receptor; NEP, neprilysin; POP, prolyl oligopeptidase; PRR, prorenin receptor; TOP, thimet oligopeptidase; XNT, ACE2 activator xanthenone
The Ang II-ACE-AT1R arm of the RAS has increased in complexity with recent findings including (1) Ang-(1-12), a C-terminally extended form of Ang I found in plasma and peripheral tissues, which is formed independent of renin and processed to Ang II [22]; (2) prorenin, which in addition to renin can bind the prorenin receptor (PRR) to induce non-proteolytic activation, generating Ang II in tissues and initiating Ang II-independent intracellular signaling [23]; (3) localization of RAS components in tissues (e.g., adipose, brain, kidney, skeletal muscle) [19], although the existence and independence of these local RAS systems from the circulation has been challenged [24]; (4) intracellular RAS capable of generating Ang II within cells (e.g., renal proximal tubule cells, neurons) or internalizing Ang II following cell surface receptor activation to elicit intracrine effects via AT1R-like nuclear receptors [25,26,27]; and (5) ACE-independent pathways for Ang II formation, particularly within tissues, involving actions of proteinases such as chymase, kallikrein, and cathepsin G [22].
Noncanonical RAS pathways
A counter-regulatory arm of the RAS has more recently emerged, which generally opposes actions of the Ang II-ACE-AT1R axis. As shown in Fig. 1, this noncanonical RAS is characterized by Ang-(1-7), which is formed from cleavage of Ang II by ACE2 or cleavage of Ang I by endopeptidases including neprilysin (NEP), prolyl oligopeptidase (POP), and thimet oligopeptidase (TOP) [28, 29]. Ang I can also be converted by ACE2 to Ang-(1-9) and subsequently cleaved by NEP or ACE to form Ang-(1-7). The actions of Ang-(1-7) at cell surface mas G protein-coupled receptors promote positive effects on blood pressure, glucose homeostasis, lipid metabolism, and energy balance [28]. While most physiological actions of Ang-(1-7) have been shown to require mas receptors, a few studies suggest heterodimerization and functional interplay between mas and AT2R [30]. Ang-(1-7) mas receptors may also heterodimerize with AT1R to competitively antagonize Ang II signaling [31]. Additionally, the endogenous heptapeptide alamandine was identified in 2013 in human blood and shown to differ from Ang-(1-7) only in its N-terminal amino acid [Ala1 versus Asp1 for Ang-(1-7)] [32]. As shown in Fig. 1, alamandine is formed through cleavage of Ang II to Ang A via mononuclear leukocyte-derived aspartate decarboxylase (MLDAD) with subsequent cleavage of Ang A via ACE2. Alamandine can also be formed via decarboxylation of Ang-(1-7) and binds mas-related G protein-coupled receptor D (MrgD) to elicit similar cardiovascular actions as Ang-(1-7) [33].
Sex differences in metabolic effects of Ang II pathways
Angiotensinogen
Angiotensinogen, a glycoprotein serving as the main precursor of the RAS, is primarily liver-derived but is also expressed in numerous tissues including adipose [34]. In mice, adipose-derived angiotensinogen has been shown to contribute up to 30% of total circulating levels [35, 36]. Angiotensinogen gene expression in white adipose decreases with fasting and increases with increased nutrient availability or following exposure to long-chain fatty acids, glucocorticoids, cytokines, androgens, and hyperglycemia [34]. In obese animal models, adipose angiotensinogen is increased and correlates with systemic RAS activity and body mass [37]. In male mice, overexpression of angiotensinogen in adipose tissue results in hypertension, increased adiposity, insulin resistance, glucose intolerance, and reduced insulin-stimulated skeletal muscle glucose uptake [36, 38]. This increased adiposity and glucose intolerance is abrogated via ACE inhibition, suggesting Ang II-mediated effects [38]. In contrast, female mice with overexpression of adipose angiotensinogen exhibit normal insulin sensitivity and glucose tolerance [38].
Global deletion of angiotensinogen reduces body mass, adiposity, and circulating insulin and leptin levels in male mice [39]. Adipose-specific angiotensinogen deletion lowers resting blood pressure in male and female mice, with no effect on body weight, fat mass, or adipocyte size [35]. Despite lack of effect on body composition under resting conditions, adipose deletion of angiotensinogen attenuates HFD-induced metabolic dysfunction (e.g., weight gain, glucose intolerance, dyslipidemia) in male mice [40], as well as hypertension in male and female mice [41]. Finally, male transgenic rats with low brain angiotensinogen have reduced body mass and adiposity with improved glucose tolerance [42]. As summarized in Table 1, these overall findings suggest adipose-derived angiotensinogen contributes to hypertension and metabolic derangements and provide evidence for sex differences in the role of this RAS precursor in systemic glucose homeostasis.
Despite strong evidence for upregulation in animal models, inconsistent findings have been reported for adipose angiotensinogen levels in human obesity [34]. Furthermore, clinical studies examining the impact of angiotensinogen on metabolic outcomes are lacking (Table 2). Polymorphisms of the angiotensinogen gene have been associated with insulin resistance in both sexes [100], increased risk for central obesity and dyslipidemia in hypertensive women with metabolic syndrome [101], and with visceral obesity and insulin resistance in obese Japanese women [102]. In addition, plasma angiotensinogen levels are correlated with waist circumference decline during weight loss in obese women [117].
Renin and Prorenin
While renin, prorenin, and the PRR are established to play a role in cardiovascular regulation [118], their contribution to metabolic function is less understood. Renin is an aspartyl protease enzyme that is secreted from renal juxtaglomerular cells to initiate the RAS cascade to ultimately generate Ang II [18]. Additionally, renin is expressed in white adipose tissue, with higher levels in visceral than subcutaneous depots [34]. Sex differences in stimuli influencing renin release have been described with females generally having reduced sympathetic activation (particularly in obesity), increased renal nitric oxide synthesis, and a differential pattern of renal transporters influencing pressure natriuresis and electrolyte homeostasis [15, 119, 120]. Male mice with global deletion of the renin gene are lean due to enhanced energy expenditure, have improved insulin sensitivity, and are resistant to development of HFD-induced obesity [43]. These metabolic effects are reversed by systemic Ang II administration. Similarly, renin inhibition with aliskiren improves insulin sensitivity, skeletal muscle glucose uptake, glucose tolerance, and insulin secretion in male rodent models of hypertension, diabetes, obesity, and metabolic syndrome [44,45,46,47,48]. Conversely, male transgenic rodents overexpressing human renin are obese due to increased food intake and exhibit hyperglycemia, hyperinsulinemia, hyperlipidemia, and insulin resistance [49, 50]. This phenotype is not abrogated by ACE, renin, or prorenin inhibitors, suggesting Ang II-independent mechanisms. In contrast to male mice, female mice overexpressing human renin are protected from HFD-induced obesity [50]. Similar to males, however, renin inhibition with aliskiren improves glucose tolerance and insulin sensitivity in obese female Zucker rats [51]. Clinically, aliskiren is reported to lower blood pressure and improve whole-body insulin sensitivity in male and female hypertensive patients with metabolic syndrome [103].
Prorenin is an inactive precursor of renin, which contains a 43-amino acid prosegment covering the active cleft and is found in the circulation at concentrations at least tenfold higher than renin [23]. Renin and prorenin both bind the PRR to induce non-proteolytic activation, which generates Ang II in tissues and initiates Ang II-independent intracellular signaling [23]. In addition, PRR can be cleaved intracellularly by furin to secrete a soluble form of PRR in plasma and urine [121]. Increased non-proteolytic activation of prorenin has been observed in skeletal muscle and adipose tissue of male rat models of type II diabetes (fructose-fed and Otsuka Long-Evans Tokushima Fatty rats), in the absence of changes in PRR mRNA levels [52, 122]. In contrast, increased PRR mRNA has been shown in adipose tissue of high-fat/high-carbohydrate diet-induced obese male and female mice [53]. These disparate findings for PRR gene expression in metabolically-sensitive tissues may reflect the differences in sex, species, and use of obese versus diabetic models among these studies. Chronic treatment with HRP (a decoy peptide for the handle region of the prorenin prosegment that inhibits non-proteolytic PRR activation) attenuates weight gain, improves glucose tolerance, and reduces fasting insulin, leptin, triglyceride, and cholesterol levels in obese male rodents [52, 53]. Similarly, male mice with homozygous deletion of the PRR gene in adipocytes exhibit lower body mass and adiposity, higher lean mass, smaller visceral adipocytes, increased metabolic rate, and improved insulin sensitivity under normal diet conditions [54]. Male homozygous adipocyte PRR knockout mice are also resistant to HFD-induced obesity and glucose intolerance, despite elevated blood pressure and lipodystrophy [55]. Female heterozygous adipocyte PRR knockout mice do not exhibit altered body mass or adiposity under normal or HFD conditions, suggesting complete knockout of this gene is needed to influence adipose development [54]. Indeed, similar to males, female mice with homozygous adipose PRR deletion have reduced adiposity and are protected from HFD-induced obesity, despite increased blood pressure and renal cortical Ang II [56].
Clinically, a decrease in active renin and concomitant increase in prorenin is correlated with risk for diabetic nephropathy, retinopathy, and microvascular disease [123, 124]. While these studies included males and females, sex differences have not been explored. A few studies have examined adipose expression in clinical populations, with one study showing no difference in PRR gene expression or adipose depot distribution between lean and obese women [125]. Another study showed increased PRR and AT1R protein expression in subcutaneous adipose tissue of insulin-resistant postmenopausal non-diabetic obese women when compared with matched insulin-sensitive women [53]. Unfortunately, these studies did not include male subjects to determine sex-specific patterns of adipose PRR expression.
Angiotensin II, ACE, and AT1 receptors
The circulating and adipose Ang II-ACE-AT1R axis is activated in obesity and closely correlates with BMI, adiposity, and insulin resistance [17, 104]. Obesity-related hyperinsulinemia can stimulate endogenous Ang II production and subsequent AT1R stimulation [126]. Weight loss conversely decreases adipose angiotensinogen as well as circulating renin activity, Ang II, and aldosterone levels [117]. The overactivation of Ang II in obesity stimulates AT1R to promote hypertension, insulin resistance, and energy imbalance [17, 20]. While acute administration can improve insulin sensitivity in humans, chronic unregulated activation of Ang II pathways produces insulin resistance, glucose intolerance, and oxidative stress [17, 105].
Ang II promotes AT1R-mediated insulin resistance via multiple mechanisms including (1) aldosterone release, (2) direct uncoupling of intracellular insulin signaling pathways, (3) decreasing insulin-stimulated translocation of glucose transporter 4 (Glut4) to the cell membrane to subsequently reduce glucose uptake in peripheral tissues, (4) decreasing microvascular blood volume and flow to reduce glucose delivery, and (5) inhibiting insulin-mediated suppression of endogenous glucose production [17, 127]. In addition, Ang II increases inflammatory cytokine release, lipid transport, and triglyceride accumulation to promote lipotoxicity and impair insulin action in peripheral tissues such as pancreas, liver, and skeletal muscle [17]. RAS components including renin, ACE, and AT1R are also expressed in the pancreas and Ang II reduces pancreatic islet blood flow and induces oxidative stress and inflammation to impair pancreatic β-cell function and decrease glucose-stimulated insulin secretion [17]. For example, acute Ang II infusion decreases spontaneous and glucose-stimulated insulin secretion in healthy male subjects [106].
Ang II, ACE, and AT1R are expressed in white adipose tissue, with higher expression in visceral versus subcutaneous depots [34]. Ang II increases adipose inflammation and stimulates adipocyte differentiation and lipogenesis while inhibiting lipolysis [34]. In terms of energy balance, acute low-dose administration of Ang II reduces food intake and body weight in male rats [128]. More chronically, however, peripheral Ang II infusion promotes a positive energy balance in male rodents. In female atherosclerosis-prone mice, however, chronic Ang II infusion produces no effect on body mass or plasma cholesterol levels [64]. Systemic pharmacological blockade of Ang II activity with ACE inhibitors or angiotensin receptor blockers (ARBs), or global genetic deletion of AT1a receptors (AT1aR), protects male rodents against development of HFD-induced obesity and dyslipidemia by increasing energy expenditure and improving glucose tolerance and insulin sensitivity [57, 58, 129]. Similarly, ACE inhibitors and ARBs improve glucose homeostasis and reduce circulating fatty acid concentrations in obese female Zucker rats [65, 68]. This has also been shown in clinical studies, with the ARB irbesartan-reducing postprandial hypertriglyceridemia in male and female diabetic patients [130]. Enhancement of AT1R-associated protein (ATRAP; Agtrap gene), a local inhibitory protein promoting internalization of AT1R, in adipose tissue also attenuates HFD-induced obesity and insulin resistance in male mice [70].
Global deletion of the ACE gene protects male mice against obesity-related metabolic complications [66]. ACE gene polymorphisms have also been linked with increases in BMI and incidence of obesity in clinical populations [108, 109]. In contrast, male mice harboring an extra copy of the ACE gene have lower adiposity and body mass on HFD, and lower adiposity and increased energy expenditure after prolonged fasting. In contrast to peripheral effects, central Ang II infusion attenuates weight gain in lean and high-calorie cafeteria diet-fed male rats by decreasing food intake, increasing energy expenditure, and improving glycemic control [59, 60]. Furthermore, genetic deletion of AT1aR in either leptin receptor or agouti-related peptide-expressing cells within the hypothalamic arcuate nucleus in male and female mice results in failure to increase resting metabolic rate in response to HFD independent of blood pressure effects, with no gender differences reported [131]. This suggests opposing peripheral versus central Ang II actions on energy balance, as well as anatomical dissociation of cardiovascular versus metabolic control mechanisms. Importantly, most of these studies were performed in male rodents, with female comparisons lacking (Tables 1 and 2).
In male hypertensive rodent models, elevations in ACE activity are observed in the circulation, kidney, and heart [11]. In humans, serum ACE activity is also generally higher in adolescent and adult males versus females [132, 133]. One study also showed sex differences in RAS serum enzyme activity during healthy aging, with reduced ACE and aminopeptidase activity in older men compared with women [10]. Consistent differences in circulating Ang II levels have not been observed, with similar levels between males and females and between untreated and estrogen-treated females [11]. Discrepancies in sex differences in RAS components between humans and animal models may reflect that most clinical studies focus on systemic levels of RAS peptides, whereas animal studies concentrate on tissue levels of these peptides. Further investigation is needed to determine if there are sex differences in systemic and local Ang II concentrations and its effects on metabolic function.
Despite similar basal circulating levels, males appear to exhibit greater sensitivity to Ang II cardiovascular effects, with healthy men having greater pressor and renal vasoconstrictor responses to acute Ang II infusion compared with women [134]. Similarly, chronic Ang II infusion induces hypertension in male but not female rodents [135, 136], perhaps in part due to central estrogen protection shifting the balance from Ang II towards Ang-(1-7) pathways [137, 138]. Similar to findings for HFD exposure [9], recent studies have shown that immune cells may also underlie sex differences in Ang II-mediated hypertension [139]. For example, one study showed that chronic Ang II infusion in rats increases renal pro-inflammatory T cells in males while increasing anti-inflammatory Tregs in females [140]. Furthermore, while females gain more weight and adiposity on HFD, only males exhibit increased circulating Ang II levels and AT1R-mediated hypertension [141]. Male fructose-fed rats develop elevations in blood pressure associated with increased cardiac AT1R and ACE gene expression, with females protected from these derangements [142]. In contrast, despite having lower blood pressure and reduced renal ACE, female rats are not protected from the vascular and renal damage in early-onset diabetes [143]. A recent clinical study showed that in response to exogenous Ang II infusion, insulin resistance in women was associated with lack of response in heart rate variability and arterial stiffness, whereas men exhibited a protective increase in markers of cardiovagal function [107]. In summary, while females appear protected from hypertension resulting from activation of the Ang II-ACE-AT1R axis of the RAS, there is much less information on sex differences in metabolic outcomes (Tables 1 and 2).
AT2 receptors
While generally decreasing after birth, AT2R expression is increased in cardiovascular pathophysiological states as a potential compensatory mechanism to induce vasodilation to counteract AT1R-mediated actions [21]. Recent studies performed selectively in male rodents also implicate a role for AT2R in control of glucose homeostasis and energy balance (Table 1). The trophic actions of Ang II to promote adipocyte differentiation and lipogenesis in vitro are AT2R-mediated [144, 145]. Male mice with global AT2R gene deletion exhibit adipocyte hypotrophy and increased lipid oxidation, suggesting AT2R increases adipose cell mass and negatively regulates lipid utilization [73]. Similarly, global AT2R deletion in male mice protects against HFD-induced obesity, insulin resistance, glucose intolerance, and hypertension [73, 74]. In addition, AT2R deficiency in male mice protects against obesity induced by adipose angiotensinogen overexpression, as well as adipose tissue deletion during prolonged fasting [75, 146]. These anti-obesity effects are associated with reduced food intake and increases in energy expenditure, lipid oxidation, plasma thyroid levels, and urinary estrogen levels. This suggests that AT2R suppresses resting metabolic rate to contribute to obesity in male rodents. Consistent with this, one study showed that AT2R activation reduces differentiation and thermogenic capacity of subcutaneous white adipocytes to suppress resting metabolic rate in male transgenic mice with brain RAS activation [147]. Other studies, however, have shown AT2R activation reduces adiposity, improves glucose uptake and insulin sensitivity, and increases nitric oxide-mediated microvascular perfusion to enhance insulin delivery and action in skeletal muscle of male rodent models with diabetes and metabolic syndrome [61, 76,77,78,79]. AT2R activation has also been shown to improve pancreatic islet insulin biosynthesis and secretion in vitro and in vivo in diabetic male rats, in part by protecting β-cells from oxidative stress and apoptosis [148]. These findings show inconsistent effects of AT2R deletion versus activation on metabolic outcomes in male rodent models.
The AT2R gene is located on the X chromosome [11], with accumulating evidence supporting sex-specific metabolic actions of this receptor. In contrast to male mice, global AT2R deletion exacerbates HFD-induced weight gain, adiposity, hyperinsulinemia, glucose intolerance, and estrogen depletion in female mice [74]. The physiological mechanisms by which AT2R contributes to sex differences in obesity susceptibility in mice remain unclear but may involve differential effects on estrogen levels. Similar to genetic deletion in mice, a polymorphism in the AT2R gene (A/C3123) is associated with modest increases in BMI and hemoglobin A1C levels in healthy Japanese women [114, 115]. Conversely, AT2R activation attenuates HFD-induced weight gain, adiposity, and hyperinsulinemia in female mice independent of urinary estrogen levels [80]. These findings suggest that AT2R may be metabolically protective, particularly in females (Tables 1 and 2).
ACE inhibitors and angiotensin receptor blockers
Pharmacological blockade of Ang II formation and AT1R-mediated actions with ACE inhibitors and ARBs (Fig. 1), respectively, is commonly used for hypertension treatment in obese and type II diabetic patients due to their positive metabolic profile. In addition to cardioprotection, these therapies improve insulin sensitivity via several mechanisms including enhancement of bradykinin-nitric oxide pathways, upregulation of insulin signaling pathways, vasodilation to enhance glucose delivery, and improved Glut4 trafficking to increase whole-body glucose disposal [58, 127]. These therapies also have protective effects on pancreatic β-cells such as increasing islet blood flow and reducing oxidative stress to increase glucose-stimulated insulin release [58]. ACE inhibitors and ARBs reduce incidence of new-onset diabetes in large randomized trials in hypertension, chronic heart failure, and patients at high risk for cardiovascular events [110]. Furthermore, both ACE inhibitors and ARBs protect against HFD-induced weight gain, dyslipidemia, insulin resistance, and glucose intolerance in male rodents [62, 67, 149]. The anti-obesity effects of ARBs in mice may involve induction of thermogenic beige adipocytes to increase energy expenditure [150]. Clinical studies examining chronic effects of RAS blockade have shown improved dyslipidemia but inconsistent results for energy balance, with some studies showing weight loss and others showing no effect on body mass [151,152,153].
There is limited data on the influence of sex on RAS blockade efficacy. In terms of hypertension control, one meta-analysis revealed that sex-specific outcome data were only reported in 43% of clinical trials reviewed, with ACE inhibitors and ARBs showing a small increase in cardiovascular benefit in men versus women [154]. Reduced blood pressure lowering effects of ACE inhibition in females has also been supported in animal studies [155], with ARBs potentially providing more benefit in females. One study showed sex differences in pancreatic blood flow responses to Ang II blockade in diabetic rats, with ACE inhibitors increasing serum insulin only in male rats, and ARBs increasing pancreatic and islet blood flow only in female rats [156]. ARBs also appear metabolically protective in obese female rats to prevent obesity-related metabolic and ovulatory defects [72]. While clinical trials examining incidence of new-onset diabetes with ACE inhibitors and ARBs-enrolled males and females [110], subgroup analysis was only performed in a few of these studies and showed no impact of sex on cardiovascular and metabolic protection [157,158,159]. Similarly, a retrospective study showed no association of sex with the blood glucose-lowering effects of ARBs in hypertensive Japanese patients [160]. These findings highlight the need for further animal and clinical studies examining the impact of sex on effects of RAS blockade in terms of metabolic outcomes.
Sex differences in metabolic effects of Ang-(1-7) pathways
Angiotensin-(1-7)
In contrast to Ang II, activation of Ang-(1-7) pathways promote positive metabolic effects in male rodent models of obesity, diabetes, and cardiometabolic syndrome. There is evidence from both in vitro and in vivo experiments to support a positive influence of Ang-(1-7) on intracellular insulin signaling pathways by increasing phosphorylation of insulin receptor substrate 1 and Akt in the heart, liver, skeletal muscle, and adipose tissues [81]. Ang-(1-7)-mediated Akt activation influences downstream modulators of glucose metabolism including endothelial nitric oxide synthase, AS160 (negative regulator of Glut4 translocation), and glycogen synthase kinase-3β (proline-directed serine-threonine kinase inactivating glycogen synthase). Ang-(1-7) also acts via mas receptors to increase basal and/or insulin-stimulated glucose uptake in cultured adipocytes, hepatocytes, and cardiomyoctes in male rodents [161,162,163]. Importantly, Ang-(1-7) reverses Ang II-mediated inhibition of insulin signaling and glucose transport activity in insulin-sensitive tissues of male rodents [81].
Chronic targeting of Ang-(1-7) improves glucose homeostasis and insulin action in male rodents. In male fructose-fed rats, chronic peripheral or central Ang-(1-7) infusion lowers blood pressure, improves insulin sensitivity and glucose tolerance, reduces insulin levels, and increases insulin signaling in the liver, skeletal muscle, and adipose tissues [82,83,84]. Similarly, male transgenic rats with elevated circulating Ang-(1-7) levels are lean and have improved insulin sensitivity and glucose tolerance in part due to enhanced adipocyte glucose uptake [164]. In HFD-induced obese male mice, plasma Ang-(1-7) is reduced and chronic peripheral restoration of this hormone reverses whole-body insulin resistance by enhancing insulin-stimulated skeletal muscle glucose uptake via enhanced Glut4 translocation independent of body composition or blood pressure [85]. Acute intravenous Ang-(1-7) also enhances insulin action and improves insulin sensitivity in lean male rats by enhancing skeletal muscle glucose uptake [86, 165]. Administration of orally active Ang-(1-7) improves hyperglycemia, hyperinsulinemia, and insulin resistance in male diabetic rats [163]. Ang-(1-7) also improves insulin secretion in vitro and in vivo, regulates development of pancreatic endocrine cells, and protects pancreatic β-cells by attenuating islet endothelial cell dysfunction, reducing β-cell dedifferentiation, and improving microcirculation [87, 91, 166, 167]. In summary, in male obese and diabetic rodents, Ang-(1-7) improves insulin signaling, insulin-stimulated glucose uptake via Glut4, and insulin secretion.
In addition, Ang-(1-7) improves energy balance and lipid metabolism in male rodents. Chronic peripheral Ang-(1-7) infusion induces brown adipocyte differentiation to increase thermogenesis and attenuate weight gain in HFD-induced obese male mice [88]. Chronic central Ang-(1-7) infusion also produces anti-obesity effects in male rats, although potency of these effects is lower than Ang II [59]. Mechanistically, anti-obesity effects of peripherally administered Ang-(1-7) have been linked with regulatory effects on lipid metabolism pathways, particularly in adipose tissue. Noncanonical RAS components such as Ang-(1-7), ACE2, and mas receptor are expressed in adipose tissue. Ang-(1-7) reduces plasma total cholesterol and triglyceride levels [82, 89, 90], decreases lipid accumulation in tissues, protects against adipose inflammation, and preserves insulin signaling in adipocytes in male rodents [88, 168]. The beneficial adipose effects of Ang-(1-7) may involve anti-inflammatory effects as well as modulation of sirtuins or other proteins involved in lipid metabolism (monoglyceride lipase), redox processes (carbonic anhydrases), or energy transduction (annexin A2) [168, 169].
While accumulating evidence shows Ang-(1-7) has beneficial metabolic effects in male rodents, female comparison studies are lacking (Table 1). In hypertensive rats, females have greater circulating and renal Ang-(1-7) levels compared with males, although these sex differences appear strain-specific [25]. One study also showed sex-dependent circulating Ang-(1-7) levels in HFD-induced obese mice [141]. Obese female mice had higher circulating Ang-(1-7) and adipose ACE2 levels and were protected from development of hypertension, despite having more body mass and adiposity compared with males. Ovariectomy or chronic administration of the mas receptor antagonist [D-Ala7]-Ang-(1-7) [A779] elevated nocturnal blood pressure in these obese female mice, with no information on metabolic outcomes, suggesting estrogen and Ang-(1-7) interactions are important in this cardiovascular protection [141].
It is unclear if sex differences similarly exist for Ang-(1-7) levels in clinical populations. While one study reported higher plasma Ang-(1-7) in healthy young adult males versus females [170], another study found that females have higher levels of this hormone in a healthy adult cohort [171]. This could reflect differences in use of protease inhibitors during blood sample collection as well as heterogeneity in terms of geographical location, age, and racial demographics. In the latter study, Ang-(1-7) positively correlated with diastolic blood pressure in females, and with endothelial function in both sexes [171]. An additional study found urinary Ang-(1-7) is higher in females in a normotensive Afro-Caribbean population and positively correlates with systolic blood pressure but not BMI or waist circumference [172]. There were no sex differences in plasma Ang-(1-7), plasma renin activity, or plasma or urinary Ang II levels in this study, consistent with differential processing mechanisms for individual RAS components. During healthy aging, there are no differences in Ang-(1-7)-forming enzyme activities (e.g., ACE2, neprilysin) between men and women [10]. In summary, there are few studies exploring sex differences in Ang-(1-7) in animal models and clinical populations, with most studies focused on differences in levels of this hormone related to cardiovascular regulation. Additional studies examining Ang-(1-7) levels in obesity and type II diabetes, and potential sex differences in terms of metabolic actions, are needed.
While Ang-(1-7) is an attractive therapeutic target for obesity and type II diabetes, this approach is currently limited by the short half-life of the hormone. Oral formulations and stable analogs of Ang-(1-7) are being tested in animal models [173,174,175]; however, presently, there are limited clinical studies. The published clinical studies to date have largely focused effects of intra-arterial or intravenous infusion on cardiovascular outcomes such as vasodilation and blood pressure in healthy subjects and patients with essential hypertension, heart failure, and obesity [29, 176, 177]. Of interest, therapies blocking Ang II activity such as ACE inhibitors and ARBs shift the balance of the RAS to increase Ang-(1-7) levels [29]. While generally attributed to reduced Ang II formation, studies in male rodents have shown that cardiovascular and metabolic effects produced by these therapies may result from this endogenous Ang-(1-7) production [178,179,180]. Furthermore, while ACE inhibitors and ARBs remain mainstays of cardiovascular therapy, a recent meta-analysis provided evidence for an approximate 11% pooled incidence of cough among randomized, controlled clinical trials with ACE inhibitors [181]. These drugs can also rarely cause angioedema related to production of kinins [182]. Direct targeting of Ang-(1-7) may therefore represent an advantageous approach to avoid these side effects.
ACE2
ACE2 is a monocarboxypeptidase that preferentially removes carboxy-terminal amino acids from substrates including Ang II, Ang I, and apelin [183]. While circulating levels are low to undetectable under normal conditions, ACE2 expression and activity is found in multiple tissues including heart, kidney, liver, skeletal muscle, adipose, and pancreas [183]. ACE2 expression is upregulated in the serum, kidney, pancreas, and liver of male and female diabetic rodents suggesting a compensatory protective mechanism [143, 184, 185]. Urinary levels are also elevated in diabetic male mice due to increased proteolytic cleavage of the extracellular catalytic domain of ACE2 [186]. This ACE2 shedding positively correlates with proteinuria, glucose and triglyceride levels. Serum ACE2 activity is also elevated in patients with type I diabetes mellitus and microalbuminuria, and urinary ACE2 is elevated in diabetic renal transplant patients [105].
In preclinical models, the role of ACE2 in glucose homeostasis and energy balance has been limited to studies in males (Table 1). Under normal diet conditions, one study showed lack of a significant metabolic phenotype in male mice with global deletion of the ACE2 gene [92], while another study showed decreased glucose-stimulated first-phase insulin secretion and progressively impaired glucose tolerance with ACE2 deletion [93]. When challenged pharmacologically with Ang II or physiologically with either HFD or high-fat high-sucrose diets, male ACE2 knockout mice exhibit greater impairments in insulin sensitivity, glucose tolerance, and glucose-stimulated insulin secretion when compared with male control mice [91, 92]. Obese male ACE2 knockout mice also have reduced mass and proliferation of β-cells [94], and higher percentage of dedifferentiated β-cells [91], suggesting ACE2 is protective to pancreatic function.
Pharmacological approaches to chronically increase ACE2 have been explored for obesity, diabetes, and hypertension in animal models. The orally active ACE2 activator diminazene aceturate (DIZE) reduces body mass and adiposity, improves plasma lipid profile, and decreases adipogenesis markers in lean and obese male rodents [95, 96]. Furthermore, human recombinant ACE2 administration and ACE2 activators (e.g., DIZE, xanthenone) protect against diabetes-induced complications including cardiac and renal dysfunction and retinopathy in male mice [187, 188]. Human and mouse recombinant ACE2 has also been shown to protect against Ang II-induced hypertension in male mice [189, 190]. Pancreatic-specific adenoviral ACE2 overexpression has no effect on insulin sensitivity but improves glycemia and glucose tolerance in diabetic male mice [97]. The mechanism underlying this improved glycemic control may involve increased β-cell proliferation and glucose-stimulated first-phase insulin secretion and decreased islet apoptosis. ACE2 overexpression also increases basal and insulin-stimulated glucose uptake in isolated adipocytes and hepatocytes from male rodents, in part, by improving insulin signaling and reducing oxidative stress [161, 162]. A limitation of many of these studies remains the unclear conclusions as to whether the beneficial metabolic effects of ACE2 activation in male mice reflects reduced Ang II levels versus increased Ang-(1-7) formation, or a combination of these mechanisms.
The ACE2 gene is located on the X chromosome, with females generally having higher ACE2 activity [11]. Only one study to date has reported sex-specific differences in ACE2 expression and activity in mice, with chronic HFD administration reducing renal ACE2 in males and increasing adipose ACE2 in females [141]. The increased adipose ACE2 in obese female mice was reversed by ovariectomy suggesting estrogen-mediated effects. In addition, global deletion of ACE2 augments HFD-induced obesity hypertension in male mice, and induces obesity hypertension in female mice, by increasing the circulating Ang II:Ang-(1-7) balance [141]. Interestingly, these effects appear independent of body weight, as ACE2 knockout mice had attenuated weight gain during HFD feeding compared with wild-type littermate controls. Similar to males [189, 190], recombinant ACE2 protects female mice against hypertension induced by Ang II infusion or transgenic overexpression of renin [191]. Given findings for sex-specific ACE2 expression in obese and diabetic rodent models, exploring sex differences in the metabolic and cardiovascular therapeutic potential for ACE2 remains a critical area for research.
Mas receptors
In addition to mediating effects of exogenous Ang-(1-7), accumulating evidence suggests endogenous Ang-(1-7) activates mas receptors to influence resting metabolic function. In support of this, pharmacological blockade of mas receptors with A779 reduces insulin sensitivity in diabetic male mice [97] and impairs adipocyte responsiveness to antilipolytic effects of insulin [192]. Additionally, gene silencing of mas receptors in human subcutaneous preadipocytes reduces adipogenic markers [192]. Global deletion of mas receptors in male FVB/N mice results in a metabolic syndrome phenotype characterized by increased adiposity, hyperglycemia, hyperinsulinemia, increased circulating and skeletal muscle triglycerides, insulin resistance, glucose intolerance, and reduced glucose uptake and Glut4 levels in adipose tissue [98]. In contrast, mas receptor deletion in male and female C57Bl/6J mice has no effect on body mass or composition under control diet or HFD conditions [99]. These disparate findings are likely due to differences in background strains but could reflect length of diet administration and age of mice at time of study. Interestingly, a nonpeptide orally active mas receptor agonist, AVE0991, has been developed with beneficial cardiovascular effects shown in hypertensive and diabetic male rodents [174, 193]. One study has shown glucose and lipid lowering effects of AVE0991 in diabetic male rats [175]; however, this compound has not yet been evaluated in humans.
A handful of studies have described sex differences related to mas receptors, primarily related to cardiovascular function. One study demonstrated that obese female mice have elevated plasma Ang-(1-7) levels and are protected from hypertension [141]. This cardiovascular protection was eliminated following chronic systemic blockade of mas receptors with A779 suggesting Ang-(1-7)-mediated effects [141]. Similarly, global mas receptor deletion promotes obesity hypertension in female but not male mice, with no effect on body composition in either sex [99]. In summary, while mas receptors appear important for cardioprotection in female rodents [99, 141], there are limited data on sex differences related to metabolic outcomes in preclinical models (Table 1). As summarized in Table 2, there is limited information in clinical populations, with one study showing that single nucleotide polymorphisms in the mas receptor gene may contribute to obesity risk in a Chinese population-based cohort [116].
Alamandine
Alamandine is a recently discovered component on the noncanonical arm of the RAS, which acts at MrgD receptors to produce vasodilatory and cardioprotective effects similar to Ang-(1-7) in animal models [33]. A recent study shows that mice with global deletion of MrgD receptors develop dilated cardiomyopathy at an early age [194]. This cardiomyopathy was seen to a similar extent in male and female mice, suggesting a sex-independent role for endogenous alamandine in cardiac function. In terms of metabolic function, one study showed alamandine decreases leptin secretion and expression from visceral white adipocytes in vitro and reduces circulating leptin levels in vivo, in male rats [195]. These effects were mediated via activation of mitogen-activated protein kinase pathways and were opposite to effects of Ang-(1-7) to increase leptin secretion and expression. There are currently no preclinical or clinical studies exploring the role of alamandine in glucose homeostasis, lipid metabolism, or energy balance (Tables 1 and 2). Given its recent discovery, an opportunity exists for research exploring effects of alamandine on metabolic function and related sex differences.
Interactions of the RAS with sex hormones
As recently reviewed, premenopausal women have a more favorable lipid and glucose metabolism profile, more adipose tissue distributed to subcutaneous depots, and lower blood pressure compared with men, with protection largely attributed to the presence and positive metabolic and cardiovascular actions of estrogen [2, 196]. Weight gain and blood pressure are enhanced during aging and particularly following menopause, with the incidence of obesity reaching ~ 65% in women over the age of 40 in the USA [197]. Replacement of estrogen may be beneficial for metabolic outcomes as a meta-analysis of randomized controlled trials found that hormone replacement therapy improves fat-free mass and insulin sensitivity and decreases visceral fat and cholesterol levels independent of blood pressure effects, route of administration, or whether a progestin component was included [198].
Accumulating evidence suggests that several components of the RAS are regulated by sex hormones (Table 3), as well as influenced by hormone replacement therapies. The angiotensinogen gene has an estrogen-responsive element, with females generally having higher circulating levels compared with males [199]. In rats, estrogen increasing circulating levels and hepatic gene expression of angiotensinogen [200]. Conversely, in normal and hypertensive rats, angiotensinogen gene expression is decreased in liver and kidney following castration and increased with exogenous testosterone administration [201, 202]. Similar to preclinical models, oral estrogen replacement therapy exerts a positive regulatory influence on angiotensinogen secretion in postmenopausal women irrespective of hypertensive status, with no information on androgen effects on this precursor in clinical populations [203,204,205].
In terms of prorenin, testosterone increases plasma levels in mice and rats [206, 207], with no information on estrogen effects. In clinical populations, women taking estrogen replacement therapy have lower plasma prorenin levels compared with men and women not taking estrogen replacement therapy [133], with no information on androgen effects. In hypertensive rats, estrogen appears to have no influence on plasma renin concentration; however, anti-androgen therapy decreases renin suggesting a positive regulatory relationship with testosterone [201, 208, 209]. In clinical populations, plasma renin concentration is lower in postmenopausal women compared with men, with levels particularly lower in women taking estrogen replacement therapy [133, 203]. While most clinical studies support that estrogen decreases plasma renin concentration [133, 203, 205], estrogen replacement therapy often increases plasma renin activity in postmenopausal women perhaps reflecting higher levels of angiotensinogen substrate [204, 210, 211]. Additionally, there are conflicting views on the impact of oral versus transdermal estrogen replacement therapy on renin. While one study showed that both routes of administration decrease plasma renin activity [203], another showed that oral administration increases plasma renin activity with no effect of transdermal administration [214].
In preclinical models, estrogen has been shown to decrease serum and tissue ACE expression, tissue AT1R expression and density, and aldosterone production, while testosterone conversely increases ACE activity and tissue AT1R expression [208, 212, 213, 216, 218,219,220,221, 226]. The influence of estrogen on circulating Ang II levels is less clear, with one study showing a decrease following estrogen replacement therapy in hypertensive rats [208], while another study showed an increase following estrogen treatment in normal rats [217]. One study has also shown no effect of gestational exposure to testosterone on circulating Ang II levels in rats [216]. In clinical populations, ACE activity is similar among age-matched women and men, regardless of menopausal status [203]. Plasma Ang II levels are also similar in normotensive premenopausal women compared with men [227]. Oral estrogen replacement therapy decreases circulating ACE activity and increases Ang II levels in postmenopausal women, with no effect of transdermal administration on these RAS components [204, 205, 214, 215].
There is currently limited data on interactions of gonadal hormones with counter-regulatory RAS components, with research mostly limited to animal models. Estrogen upregulates AT2R binding and expression in adrenal and renal tissues in male and female rats, suggesting a positive regulatory interaction [219, 222]. Reciprocally, AT2R stimulation increases ovarian estrogen production and stimulates ovulation and oocyte maturation in rabbits [228]. Testosterone conversely downregulates AT2R gene and protein expression levels in rat aorta [223]. Circulating Ang-(1-7) levels appear higher in obese female mice and in healthy women when compared with males [141, 171]. Estradiol administration increases circulating Ang-(1-7) levels and renal ACE2 gene expression in hypertensive rats and amplifies the vasodilator properties of Ang-(1-7) in ovariectomized rats [208, 224, 229]. Additionally, plasma Ang-(1-7) levels and adipose ACE2 activity are reduced by ovariectomy in obese female mice [141]. Testosterone has no effect on ACE2 mRNA in cultured adipocytes, or on testicular ACE2 activity in lean rats [225]. Overall, these data suggest that in animal models, estrogen shifts the balance of the RAS from the deleterious Ang II-ACE-AT1R axis to the beneficial Ang-(1-7)-ACE2-mas receptor axis, an effect which could promote positive cardiometabolic effects.
Conclusions
The identification of sex-specific mechanisms underlying metabolic effects of the RAS, as well as beneficial effects of therapies targeting the RAS, remains an active area of research. Sex differences in expression, activity, and tissue responsiveness of several RAS components are apparent, with estrogen downregulating Ang II and upregulating Ang-(1-7) pathways [11]. In animal models of obesity, females appear to maintain circulating Ang-(1-7) levels [141] and are protected from hypertension and metabolic complications induced by angiotensinogen, renin, angiotensin II, and AT1R activation [38, 50, 142]. While inconsistent effects are observed in males, stimulation of counter-regulatory AT2R appears metabolically protective in female rodents [80, 114, 115, 147]. Activation of Ang-(1-7) pathways is also an attractive target to improve glucose homeostasis, lipid metabolism, and energy balance in male rodent models of obesity [28]. While development of pharmacotherapies activating Ang-(1-7) pathways may broaden therapeutic potential for RAS targeting, few studies have been performed in female animal models, with no clinical data in either sex supporting efficacy on metabolic outcomes. This underlines the important issue of translating findings related to sex differences in RAS therapies from experimental animal models to clinical practice. Current therapeutic recommendations for metabolic and cardiovascular complications in obesity are not specific to men versus women, even though sex differences in RAS pathways are evident. Furthermore, while large controlled clinical trials for RAS inhibition enroll both men and women, few studies have assessed sex-specific effects on cardiovascular and metabolic outcomes. These collective findings illustrate the critical need for additional mechanistic and clinical research to determine the impact of sex on metabolic effects of the RAS.
Availability of data and materials
Not Applicable
Abbreviations
- A779:
-
[D-Ala7]-angiotensin-(1-7)
- ACE:
-
Angiotensin-converting enzyme
- ACE2:
-
Angiotensin-converting enzyme 2
- Ang:
-
Angiotensin
- ARB:
-
Angiotensin receptor blocker
- AT1R:
-
Angiotensin II type 1 receptor
- AT2R:
-
Angiotensin II type 2 receptor
- AVE0991:
-
Orally active mas receptor agonist
- BMI:
-
Body mass index
- DIZE:
-
Diminazene aceturate
- Glut4:
-
Glucose transporter 4
- HFD:
-
High-fat diet
- MLDAD:
-
Mononuclear leukocyte-derived aspartate decarboxylase
- MrgD:
-
Mas-related G protein-coupled receptor
- PRR:
-
Prorenin receptor
- RAS:
-
Renin-angiotensin system
References
Hill JO, Wyatt HR, Peters JC. Energy balance and obesity. Circulation. 2012;126(1):126–32.
Varlamov O, Bethea CL, Roberts CT Jr. Sex-specific differences in lipid and glucose metabolism. Front Endocrinol (Lausanne). 2014;5:241.
Mauvais-Jarvis F. Sex differences in metabolic homeostasis, diabetes, and obesity. Biol Sex Differ. 2015;6:14.
Lovejoy JC, Sainsbury A. Stock conference working G. sex differences in obesity and the regulation of energy homeostasis. Obes Rev. 2009;10(2):154–67.
Sullivan PW, Morrato EH, Ghushchyan V, Wyatt HR, Hill JO. Obesity, inactivity, and the prevalence of diabetes and diabetes-related cardiovascular comorbidities in the U.S., 2000-2002. Diabetes Care. 2005;28(7):1599–603.
Yach D, Stuckler D, Brownell KD. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat Med. 2006;12(1):62–6.
Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem. 2006;75:367–401.
Griffin C, Lanzetta N, Eter L, Singer K. Sexually dimorphic myeloid inflammatory and metabolic responses to diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2016;311(2):R211–6.
Taylor LE, Gillis EE, Musall JB, Baban B, Sullivan JC. High-fat diet-induced hypertension is associated with a proinflammatory T cell profile in male and female Dahl salt-sensitive rats. Am J Physiol Heart Circ Physiol. 2018;315(6):H1713–H23.
Fernandez-Atucha A, Izagirre A, Fraile-Bermudez AB, Kortajarena M, Larrinaga G, Martinez-Lage P, et al. Sex differences in the aging pattern of renin-angiotensin system serum peptidases. Biol Sex Differ. 2017;8:5.
Komukai K, Mochizuki S, Yoshimura M. Gender and the renin-angiotensin-aldosterone system. Fundam Clin Pharmacol. 2010;24(6):687–98.
Sullivan JC, Gillis EE. Sex and gender differences in hypertensive kidney injury. Am J Physiol Renal Physiol. 2017;313(4):F1009–F17.
Dasinger JH, Alexander BT. Gender differences in developmental programming of cardiovascular diseases. Clin Sci (Lond). 2016;130(5):337–48.
Maric-Bilkan C, Manigrasso MB. Sex differences in hypertension: contribution of the renin-angiotensin system. Gend Med. 2012;9(4):287–91.
Faulkner JL, Belin de Chantemele EJ. Sex differences in mechanisms of hypertension associated with obesity. Hypertension. 2018;71(1):15–21.
Ahmed S, Hu R, Leete J, Layton AT. Understanding sex differences in long-term blood pressure regulation: insights from experimental studies and computational modeling. Am J Physiol Heart Circ Physiol. 2019;316(5):H1113–H23.
Luther JM, Brown NJ. The renin-angiotensin-aldosterone system and glucose homeostasis. Trends Pharmacol Sci. 2011;32(12):734–9.
Kurtz A. Renin release: sites, mechanisms, and control. Annu Rev Physiol. 2011;73:377–99.
Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system--an endocrine and paracrine system. Endocrinology. 2003;144(6):2179–83.
Unger T. The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol. 2002;89(2A):3A–9A discussion 10A.
Lemarie CA, Schiffrin EL. The angiotensin II type 2 receptor in cardiovascular disease. J Renin-Angiotensin-Aldosterone Syst. 2010;11(1):19–31.
Ferrario CM, Ahmad S, Nagata S, Simington SW, Varagic J, Kon N, et al. An evolving story of angiotensin-II-forming pathways in rodents and humans. Clin Sci (Lond). 2014;126(7):461–9.
Campbell DJ. Critical review of prorenin and (pro) renin receptor research. Hypertension. 2008;51(5):1259–64.
Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res. 2015;116(6):960–75.
Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the angiotensin converting enzyme 2-angiotensin (1-7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol (Lausanne). 2014;4:201.
Lavoie JL, Liu X, Bianco RA, Beltz TG, Johnson AK, Sigmund CD. Evidence supporting a functional role for intracellular renin in the brain. Hypertension. 2006;47(3):461–6.
Li XC, Zhu D, Zheng X, Zhang J, Zhuo JL. Intratubular and intracellular renin-angiotensin system in the kidney: a unifying perspective in blood pressure control. Clin Sci (Lond). 2018;132(13):1383–401.
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1-7). Physiol Rev. 2018;98(1):505–53.
Schindler C, Bramlage P, Kirch W, Ferrario CM. Role of the vasodilator peptide angiotensin-(1-7) in cardiovascular drug therapy. Vasc Health Risk Manag. 2007;3(1):125–37.
Leonhardt J, Villela DC, Teichmann A, Munter LM, Mayer MC, Mardahl M, et al. Evidence for heterodimerization and functional interaction of the angiotensin type 2 receptor and the receptor MAS. Hypertension. 2017;69(6):1128–35.
Gaidarov I, Adams J, Frazer J, Anthony T, Chen X, Gatlin J, et al. Angiotensin (1-7) does not interact directly with MAS1, but can potently antagonize signaling from the AT1 receptor. Cell Signal. 2018;50:9–24.
Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, et al. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res. 2013;112(8):1104–11.
Villela DC, Passos-Silva DG, Santos RA. Alamandine: a new member of the angiotensin family. Curr Opin Nephrol Hypertens. 2014;23(2):130–4.
Pahlavani M, Kalupahana NS, Ramalingam L, Moustaid-Moussa N. Regulation and functions of the renin-angiotensin system in white and brown adipose tissue. Compr Physiol. 2017;7(4):1137–50.
Yiannikouris F, Karounos M, Charnigo R, English VL, Rateri DL, Daugherty A, et al. Adipocyte-specific deficiency of angiotensinogen decreases plasma angiotensinogen concentration and systolic blood pressure in mice. Am J Physiol Regul Integr Comp Physiol. 2012;302(2):R244–51.
Massiera F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, et al. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001;15(14):2727–9.
Hainault I, Nebout G, Turban S, Ardouin B, Ferre P, Quignard-Boulange A. Adipose tissue-specific increase in angiotensinogen expression and secretion in the obese (fa/fa) Zucker rat. Am J Physiol Endocrinol Metab. 2002;282(1):E59–66.
Kalupahana NS, Massiera F, Quignard-Boulange A, Ailhaud G, Voy BH, Wasserman DH, et al. Overproduction of angiotensinogen from adipose tissue induces adipose inflammation, glucose intolerance, and insulin resistance. Obesity (Silver Spring). 2012;20(1):48–56.
Kim S, Soltani-Bejnood M, Quignard-Boulange A, Massiera F, Teboul M, Ailhaud G, et al. The adipose renin-angiotensin system modulates systemic markers of insulin sensitivity and activates the intrarenal renin-angiotensin system. J Biomed Biotechnol. 2006;2006(5):27012.
Massiera F, Seydoux J, Geloen A, Quignard-Boulange A, Turban S, Saint-Marc P, et al. Angiotensinogen-deficient mice exhibit impairment of diet-induced weight gain with alteration in adipose tissue development and increased locomotor activity. Endocrinology. 2001;142(12):5220–5.
Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL, et al. Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension. 2012;60(6):1524–30.
Kasper SO, Carter CS, Ferrario CM, Ganten D, Ferder LF, Sonntag WE, et al. Growth, metabolism, and blood pressure disturbances during aging in transgenic rats with altered brain renin-angiotensin systems. Physiol Genomics. 2005;23(3):311–7.
Takahashi N, Li F, Hua K, Deng J, Wang CH, Bowers RR, et al. Increased energy expenditure, dietary fat wasting, and resistance to diet-induced obesity in mice lacking renin. Cell Metab. 2007;6(6):506–12.
Chou CL, Lin H, Chen JS, Fang TC. Renin inhibition improves metabolic syndrome, and reduces angiotensin II levels and oxidative stress in visceral fat tissues in fructose-fed rats. PLoS One. 2017;12(7):e0180712.
Habibi J, Whaley-Connell A, Hayden MR, DeMarco VG, Schneider R, Sowers SD, et al. Renin inhibition attenuates insulin resistance, oxidative stress, and pancreatic remodeling in the transgenic Ren2 rat. Endocrinology. 2008;149(11):5643–53.
Kang YS, Lee MH, Song HK, Hyun YY, Cha JJ, Ko GJ, et al. Aliskiren improves insulin resistance and ameliorates diabetic vascular complications in db/db mice. Nephrol Dial Transplant. 2011;26(4):1194–204.
Lastra G, Habibi J, Whaley-Connell AT, Manrique C, Hayden MR, Rehmer J, et al. Direct renin inhibition improves systemic insulin resistance and skeletal muscle glucose transport in a transgenic rodent model of tissue renin overexpression. Endocrinology. 2009;150(6):2561–8.
Lee KC, Hsieh YC, Yang YY, Chan CC, Huang YH, Lin HC. Aliskiren reduces hepatic steatosis and epididymal fat mass and increases skeletal muscle insulin sensitivity in high-fat diet-fed mice. Sci Rep. 2016;6:18899.
Gratze P, Boschmann M, Dechend R, Qadri F, Malchow J, Graeske S, et al. Energy metabolism in human renin-gene transgenic rats: does renin contribute to obesity? Hypertension. 2009;53(3):516–23.
Uehara S, Tsuchida M, Kanno T, Sasaki M, Nishikibe M, Fukamizu A. Late-onset obesity in mice transgenic for the human renin gene. Int J Mol Med. 2003;11(6):723–7.
Marchionne EM, Diamond-Stanic MK, Prasonnarong M, Henriksen EJ. Chronic renin inhibition with aliskiren improves glucose tolerance, insulin sensitivity, and skeletal muscle glucose transport activity in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2012;302(1):R137–42.
Nagai Y, Ichihara A, Nakano D, Kimura S, Pelisch N, Fujisawa Y, et al. Possible contribution of the non-proteolytic activation of prorenin to the development of insulin resistance in fructose-fed rats. Exp Physiol. 2009;94(9):1016–23.
Tan P, Shamansurova Z, Bisotto S, Michel C, Gauthier MS, Rabasa-Lhoret R, et al. Impact of the prorenin/renin receptor on the development of obesity and associated cardiometabolic risk factors. Obesity (Silver Spring). 2014;22(10):2201–9.
Shamansurova Z, Tan P, Ahmed B, Pepin E, Seda O, Lavoie JL. Adipose tissue (P) RR regulates insulin sensitivity, fat mass and body weight. Mol Metab. 2016;5(10):959–69.
Wu CH, Mohammadmoradi S, Thompson J, Su W, Gong M, Nguyen G, et al. Adipocyte (pro)renin-receptor deficiency induces lipodystrophy, liver steatosis and increases blood pressure in male mice. Hypertension. 2016;68(1):213–9.
Gatineau E, Cohn DM, Poglitsch M, Loria AS, Gong M, Yiannikouris F. Losartan prevents the elevation of blood pressure in adipose-PRR deficient female mice while elevated circulating sPRR activates the renin-angiotensin system. Am J Physiol Heart Circ Physiol. 2019;316(3):H506–H15.
Kouyama R, Suganami T, Nishida J, Tanaka M, Toyoda T, Kiso M, et al. Attenuation of diet-induced weight gain and adiposity through increased energy expenditure in mice lacking angiotensin II type 1a receptor. Endocrinology. 2005;146(8):3481–9.
Henriksen EJ. Improvement of insulin sensitivity by antagonism of the renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2007;293(3):R974–80.
Winkler M, Bader M, Schuster F, Stolting I, Binder S, Raasch W. Development of obesity can be prevented in rats by chronic icv infusions of AngII but less by Ang(1-7). Pflugers Arch. 2018;470(6):867–81.
Porter JP, Anderson JM, Robison RJ, Phillips AC. Effect of central angiotensin II on body weight gain in young rats. Brain Res. 2003;959(1):20–8.
Chai W, Wang W, Dong Z, Cao W, Liu Z. Angiotensin II receptors modulate muscle microvascular and metabolic responses to insulin in vivo. Diabetes. 2011;60(11):2939–46.
Keidar S, Attias J, Heinrich R, Coleman R, Aviram M. Angiotensin II atherogenicity in apolipoprotein E deficient mice is associated with increased cellular cholesterol biosynthesis. Atherosclerosis. 1999;146(2):249–57.
Ogihara T, Asano T, Ando K, Chiba Y, Sakoda H, Anai M, et al. Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension. 2002;40(6):872–9.
Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105(11):1605–12.
Henriksen EJ, Jacob S, Kinnick TR, Teachey MK, Krekler M. Selective angiotensin II receptor antagonism reduces insulin resistance in obese Zucker rats. Hypertension. 2001;38(4):884–90.
Jayasooriya AP, Mathai ML, Walker LL, Begg DP, Denton DA, Cameron-Smith D, et al. Mice lacking angiotensin-converting enzyme have increased energy expenditure, with reduced fat mass and improved glucose clearance. Proc Natl Acad Sci U S A. 2008;105(18):6531–6.
de Kloet AD, Krause EG, Kim DH, Sakai RR, Seeley RJ, Woods SC. The effect of angiotensin-converting enzyme inhibition using captopril on energy balance and glucose homeostasis. Endocrinology. 2009;150(9):4114–23.
Dal Ponte DB, Fogt DL, Jacob S, Henriksen EJ. Interactions of captopril and verapamil on glucose tolerance and insulin action in an animal model of insulin resistance. Metabolism. 1998;47(8):982–7.
Henriksen EJ, Jacob S. Effects of captopril on glucose transport activity in skeletal muscle of obese Zucker rats. Metabolism. 1995;44(2):267–72.
Azushima K, Ohki K, Wakui H, Uneda K, Haku S, Kobayashi R, et al. Adipocyte-specific enhancement of angiotensin II type 1 receptor-associated protein ameliorates diet-induced visceral obesity and insulin resistance. J Am Heart Assoc. 2017;6(3):e004488.
Munoz MC, Giani JF, Dominici FP, Turyn D, Toblli JE. Long-term treatment with an angiotensin II receptor blocker decreases adipocyte size and improves insulin signaling in obese Zucker rats. J Hypertens. 2009;27(12):2409–20.
Sagae SC, Lubaczeuski C, Zacharias P, Bonfleur ML, Franci CR, Sanvitto GL. Prevention of metabolic disorders and reproductive performance deficits by the blockade of angiotensin II AT1 receptor in female rats fed with cafeteria diet. Physiol Behav. 2013;119:1–8.
Yvan-Charvet L, Even P, Bloch-Faure M, Guerre-Millo M, Moustaid-Moussa N, Ferre P, et al. Deletion of the angiotensin type 2 receptor (AT2R) reduces adipose cell size and protects from diet-induced obesity and insulin resistance. Diabetes. 2005;54(4):991–9.
Samuel P, Khan MA, Nag S, Inagami T, Hussain T. Angiotensin AT(2) receptor contributes towards gender bias in weight gain. PLoS One. 2013;8(1):e48425.
Yvan-Charvet L, Massiera F, Lamande N, Ailhaud G, Teboul M, Moustaid-Moussa N, et al. Deficiency of angiotensin type 2 receptor rescues obesity but not hypertension induced by overexpression of angiotensinogen in adipose tissue. Endocrinology. 2009;150(3):1421–8.
Ohshima K, Mogi M, Jing F, Iwanami J, Tsukuda K, Min LJ, et al. Direct angiotensin II type 2 receptor stimulation ameliorates insulin resistance in type 2 diabetes mice with PPARgamma activation. PLoS One. 2012;7(11):e48387.
Yan F, Yuan Z, Wang N, Carey RM, Aylor KW, Chen L, et al. Direct activation of angiotensin II type 2 receptors enhances muscle microvascular perfusion, oxygenation, and insulin delivery in male rats. Endocrinology. 2018;159(2):685–95.
Quiroga DT, Munoz MC, Gil C, Pffeifer M, Toblli JE, Steckelings UM, et al. Chronic administration of the angiotensin type 2 receptor agonist C21 improves insulin sensitivity in C57BL/6 mice. Physiol Rep. 2018;6(16):e13824.
Shum M, Pinard S, Guimond MO, Labbe SM, Roberge C, Baillargeon JP, et al. Angiotensin II type 2 receptor promotes adipocyte differentiation and restores adipocyte size in high-fat/high-fructose diet-induced insulin resistance in rats. Am J Physiol Endocrinol Metab. 2013;304(2):E197–210.
Nag S, Khan MA, Samuel P, Ali Q, Hussain T. Chronic angiotensin AT2R activation prevents high-fat diet-induced adiposity and obesity in female mice independent of estrogen. Metabolism. 2015;64(7):814–25.
Dominici FP, Burghi V, Munoz MC, Giani JF. Modulation of the action of insulin by angiotensin-(1-7). Clin Sci (Lond). 2014;126(9):613–30.
Giani JF, Mayer MA, Munoz MC, Silberman EA, Hocht C, Taira CA, et al. Chronic infusion of angiotensin-(1-7) improves insulin resistance and hypertension induced by a high-fructose diet in rats. Am J Physiol Endocrinol Metab. 2009;296(2):E262–71.
Guimaraes PS, Oliveira MF, Braga JF, Nadu AP, Schreihofer A, Santos RA, et al. Increasing angiotensin-(1-7) levels in the brain attenuates metabolic syndrome-related risks in fructose-fed rats. Hypertension. 2014;63(5):1078–85.
Marcus Y, Shefer G, Sasson K, Kohen F, Limor R, Pappo O, et al. Angiotensin 1-7 as means to prevent the metabolic syndrome: lessons from the fructose-fed rat model. Diabetes. 2013;62(4):1121–30.
Williams IM, Otero YF, Bracy DP, Wasserman DH, Biaggioni I, Arnold AC. Chronic angiotensin-(1-7) improves insulin sensitivity in high-fat fed mice independent of blood pressure. Hypertension. 2016;67(5):983–91.
Echeverria-Rodriguez O, Del Valle-Mondragon L, Hong E. Angiotensin 1-7 improves insulin sensitivity by increasing skeletal muscle glucose uptake in vivo. Peptides. 2014;51:26–30.
He J, Yang Z, Yang H, Wang L, Wu H, Fan Y, et al. Regulation of insulin sensitivity, insulin production, and pancreatic beta cell survival by angiotensin-(1-7) in a rat model of streptozotocin-induced diabetes mellitus. Peptides. 2015;64:49–54.
Morimoto H, Mori J, Nakajima H, Kawabe Y, Tsuma Y, Fukuhara S, et al. Angiotensin 1-7 stimulates brown adipose tissue and reduces diet-induced obesity. Am J Physiol Endocrinol Metab. 2018;314(2):E131–E8.
Feltenberger JD, Andrade JM, Paraiso A, Barros LO, Filho AB, Sinisterra RD, et al. Oral formulation of angiotensin-(1-7) improves lipid metabolism and prevents high-fat diet-induced hepatic steatosis and inflammation in mice. Hypertension. 2013;62(2):324–30.
Santos SH, Andrade JM, Fernandes LR, Sinisterra RD, Sousa FB, Feltenberger JD, et al. Oral angiotensin-(1-7) prevented obesity and hepatic inflammation by inhibition of resistin/TLR4/MAPK/NF-kappaB in rats fed with high-fat diet. Peptides. 2013;46:47–52.
Xuan X, Gao F, Ma X, Huang C, Wang Y, Deng H, et al. Activation of ACE2/angiotensin (1-7) attenuates pancreatic beta cell dedifferentiation in a high-fat-diet mouse model. Metabolism. 2018;81:83–96.
Takeda M, Yamamoto K, Takemura Y, Takeshita H, Hongyo K, Kawai T, et al. Loss of ACE2 exaggerates high-calorie diet-induced insulin resistance by reduction of GLUT4 in mice. Diabetes. 2013;62(1):223–33.
Niu MJ, Yang JK, Lin SS, Ji XJ, Guo LM. Loss of angiotensin-converting enzyme 2 leads to impaired glucose homeostasis in mice. Endocrine. 2008;34(1–3):56–61.
Shoemaker R, Yiannikouris F, Thatcher S, Cassis L. ACE2 deficiency reduces beta-cell mass and impairs beta-cell proliferation in obese C57BL/6 mice. Am J Physiol Endocrinol Metab. 2015;309(7):E621–31.
Bruce EB, Sakarya Y, Kirichenko N, Toklu HZ, Sumners C, Morgan D, et al. ACE2 activator diminazene aceturate reduces adiposity but preserves lean mass in young and old rats. Exp Gerontol. 2018;111:133–40.
de Macedo SM, Guimarares TA, Andrade JM, Guimaraes AL, Batista de Paula AM, Ferreira AJ, et al. Angiotensin converting enzyme 2 activator (DIZE) modulates metabolic profiles in mice, decreasing lipogenesis. Protein Pept Lett. 2015;22(4):332–40.
Bindom SM, Hans CP, Xia H, Boulares AH, Lazartigues E. Angiotensin I-converting enzyme type 2 (ACE2) gene therapy improves glycemic control in diabetic mice. Diabetes. 2010;59(10):2540–8.
Santos SH, Fernandes LR, Mario EG, Ferreira AV, Porto LC, Alvarez-Leite JI, et al. Mas deficiency in FVB/N mice produces marked changes in lipid and glycemic metabolism. Diabetes. 2008;57(2):340–7.
Wang Y, Shoemaker R, Powell D, Su W, Thatcher S, Cassis L. Differential effects of Mas receptor deficiency on cardiac function and blood pressure in obese male and female mice. Am J Physiol Heart Circ Physiol. 2017;312(3):H459–H68.
Guo X, Cheng S, Taylor KD, Cui J, Hughes R, Quinones MJ, et al. Hypertension genes are genetic markers for insulin sensitivity and resistance. Hypertension. 2005;45(4):799–803.
Procopciuc LM, Sitar-Taut A, Pop D, Sitar-Taut DA, Olteanu I, Zdrenghea D. Renin angiotensin system polymorphisms in patients with metabolic syndrome (MetS). Eur J Intern Med. 2010;21(5):414–8.
Takakura Y, Yoshida T, Yoshioka K, Umekawa T, Kogure A, Toda H, et al. Angiotensinogen gene polymorphism (Met235Thr) influences visceral obesity and insulin resistance in obese Japanese women. Metabolism. 2006;55(6):819–24.
Fogari R, Zoppi A, Mugellini A, Lazzari P, Derosa G. Different effects of aliskiren and losartan on fibrinolysis and insulin sensitivity in hypertensive patients with metabolic syndrome. Horm Metab Res. 2010;42(12):892–6.
Saiki A, Ohira M, Endo K, Koide N, Oyama T, Murano T, et al. Circulating angiotensin II is associated with body fat accumulation and insulin resistance in obese subjects with type 2 diabetes mellitus. Metabolism. 2009;58(5):708–13.
Underwood PC, Adler GK. The renin angiotensin aldosterone system and insulin resistance in humans. Curr Hypertens Rep. 2013;15(1):59–70.
Fliser D, Schaefer F, Schmid D, Veldhuis JD, Ritz E. Angiotensin II affects basal, pulsatile, and glucose-stimulated insulin secretion in humans. Hypertension. 1997;30(5):1156–61.
Rannelli LA, MacRae JM, Mann MC, Ramesh S, Hemmelgarn BR, Rabi D, et al. Sex differences in associations between insulin resistance, heart rate variability, and arterial stiffness in healthy women and men: a physiology study. Can J Physiol Pharmacol. 2017;95(4):349–55.
Strazzullo P, Iacone R, Iacoviello L, Russo O, Barba G, Russo P, et al. Genetic variation in the renin-angiotensin system and abdominal adiposity in men: the Olivetti Prospective Heart Study. Ann Intern Med. 2003;138(1):17–23.
Kramer H, Wu X, Kan D, Luke A, Zhu X, Adeyemo A, et al. Angiotensin-converting enzyme gene polymorphisms and obesity: an examination of three black populations. Obes Res. 2005;13(5):823–8.
Abuissa H, Jones PG, Marso SP, O'Keefe JH Jr. Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers for prevention of type 2 diabetes: a meta-analysis of randomized clinical trials. J Am Coll Cardiol. 2005;46(5):821–6.
Grassi G, Seravalle G, Dell'Oro R, Trevano FQ, Bombelli M, Scopelliti F, et al. Comparative effects of candesartan and hydrochlorothiazide on blood pressure, insulin sensitivity, and sympathetic drive in obese hypertensive individuals: results of the CROSS study. J Hypertens. 2003;21(9):1761–9.
Jin HM, Pan Y. Angiotensin type-1 receptor blockade with losartan increases insulin sensitivity and improves glucose homeostasis in subjects with type 2 diabetes and nephropathy. Nephrol Dial Transplant. 2007;22(7):1943–9.
van der Zijl NJ, Moors CC, Goossens GH, Hermans MM, Blaak EE, Diamant M. Valsartan improves {beta}-cell function and insulin sensitivity in subjects with impaired glucose metabolism: a randomized controlled trial. Diabetes Care. 2011;34(4):845–51.
Kotani K, Fujiwara S, Tsuzaki K, Sano Y, Matsuoka Y, Hamada T, et al. An association between angiotensin II type 2 receptor gene A/C3123 polymorphism and glycemic control marker in a general Japanese population. Mol Biol Rep. 2009;36(5):917–20.
Kotani K, Sakane N, Saiga K, Tsuzaki K, Sano Y, Mu H, et al. The angiotensin II type 2 receptor gene polymorphism and body mass index in healthy Japanese women. Ann Clin Biochem. 2007;44(Pt 1):83–5.
Zhou JB, Liu C, Niu WY, Xin Z, Yu M, Feng JP, et al. Contributions of renin-angiotensin system-related gene interactions to obesity in a Chinese population. PLoS One. 2012;7(8):e42881.
Engeli S, Bohnke J, Gorzelniak K, Janke J, Schling P, Bader M, et al. Weight loss and the renin-angiotensin-aldosterone system. Hypertension. 2005;45(3):356–62.
Hennrikus M, Gonzalez AA, Prieto MC. The prorenin receptor in the cardiovascular system and beyond. Am J Physiol Heart Circ Physiol. 2018;314(2):H139–H45.
Neugarten J, Ding Q, Friedman A, Lei J, Silbiger S. Sex hormones and renal nitric oxide synthases. J Am Soc Nephrol. 1997;8(8):1240–6.
Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, et al. Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol. 2017;28(12):3504–17.
Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, Nguyen G. Soluble form of the (pro) renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension. 2009;53(6):1077–82.
Rafiq K, Hitomi H, Nakano D, Ichihara A, Nishiyama A. Possible involvement of the (pro) renin receptor-dependent system in the development of insulin resistance. Front Biosci (Schol Ed). 2011;3:1478–85.
Deinum J, Tarnow L, van Gool JM, de Bruin RA, Derkx FH, Schalekamp MA, et al. Plasma renin and prorenin and renin gene variation in patients with insulin-dependent diabetes mellitus and nephropathy. Nephrol Dial Transplant. 1999;14(8):1904–11.
Stankovic AR, Fisher ND, Hollenberg NK. Prorenin and angiotensin-dependent renal vasoconstriction in type 1 and type 2 diabetes. J Am Soc Nephrol. 2006;17(12):3293–9.
Achard V, Boullu-Ciocca S, Desbriere R, Nguyen G, Grino M. Renin receptor expression in human adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R274–82.
Tuck ML, Bounoua F, Eslami P, Nyby MD, Eggena P, Corry DB. Insulin stimulates endogenous angiotensin II production via a mitogen-activated protein kinase pathway in vascular smooth muscle cells. J Hypertens. 2004;22(9):1779–85.
Henriksen EJ, Prasannarong M. The role of the renin-angiotensin system in the development of insulin resistance in skeletal muscle. Mol Cell Endocrinol. 2013;378(1–2):15–22.
Cassis LA, Marshall DE, Fettinger MJ, Rosenbluth B, Lodder RA. Mechanisms contributing to angiotensin II regulation of body weight. Am J Phys. 1998;274(5 Pt 1):E867–76.
Hayek T, Attias J, Coleman R, Brodsky S, Smith J, Breslow JL, et al. The angiotensin-converting enzyme inhibitor, fosinopril, and the angiotensin II receptor antagonist, losartan, inhibit LDL oxidation and attenuate atherosclerosis independent of lowering blood pressure in apolipoprotein E deficient mice. Cardiovasc Res. 1999;44(3):579–87.
Ceriello A, Assaloni R, Da Ros R, Maier A, Piconi L, Quagliaro L, et al. Effect of atorvastatin and irbesartan, alone and in combination, on postprandial endothelial dysfunction, oxidative stress, and inflammation in type 2 diabetic patients. Circulation. 2005;111(19):2518–24.
Claflin KE, Sandgren JA, Lambertz AM, Weidemann BJ, Littlejohn NK, Burnett CM, et al. Angiotensin AT1A receptors on leptin receptor-expressing cells control resting metabolism. J Clin Invest. 2017;127(4):1414–24.
Landazuri P, Granobles C, Loango N. Gender differences in serum angiotensin-converting enzyme activity and blood pressure in children: an observational study. Arq Bras Cardiol. 2008;91(6):352–7.
Danser AH, Derkx FH, Schalekamp MA, Hense HW, Riegger GA, Schunkert H. Determinants of interindividual variation of renin and prorenin concentrations: evidence for a sexual dimorphism of (pro)renin levels in humans. J Hypertens. 1998;16(6):853–62.
Toering TJ, van der Graaf AM, Visser FW, Buikema H, Navis G, Faas MM, et al. Gender differences in response to acute and chronic angiotensin II infusion: a translational approach. Physiol Rep. 2015;3(7):e12434.
Tatchum-Talom R, Eyster KM, Martin DS. Sexual dimorphism in angiotensin II-induced hypertension and vascular alterations. Can J Physiol Pharmacol. 2005;83(5):413–22.
Xue B, Pamidimukkala J, Hay M. Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am J Physiol Heart Circ Physiol. 2005;288(5):H2177–84.
Xue B, Pamidimukkala J, Lubahn DB, Hay M. Estrogen receptor-alpha mediates estrogen protection from angiotensin II-induced hypertension in conscious female mice. Am J Physiol Heart Circ Physiol. 2007;292(4):H1770–6.
Jonklaas J, Buggy J. Angiotensin-estrogen interaction in female brain reduces drinking and pressor responses. Am J Phys. 1984;247(1 Pt 2):R167–72.
Ramirez LA, Sullivan JC. Sex differences in hypertension: where we have been and where we are going. Am J Hypertens. 2018;31(12):1247–54.
Zimmerman MA, Baban B, Tipton AJ, O'Connor PM, Sullivan JC. Chronic ANG II infusion induces sex-specific increases in renal T cells in Sprague-Dawley rats. Am J Physiol Renal Physiol. 2015;308(7):F706–12.
Gupte M, Thatcher SE, Boustany-Kari CM, Shoemaker R, Yiannikouris F, Zhang X, et al. Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol. 2012;32(6):1392–9.
Bundalo MM, Zivkovic MD, Romic S, Tepavcevic SN, Koricanac GB, Djuric TM, et al. Fructose-rich diet induces gender-specific changes in expression of the renin-angiotensin system in rat heart and upregulates the ACE/AT1R axis in the male rat aorta. J Renin-Angiotensin-Aldosterone Syst. 2016;17(2):1470320316642915.
Yamaleyeva LM, Gilliam-Davis S, Almeida I, Brosnihan KB, Lindsey SH, Chappell MC. Differential regulation of circulating and renal ACE2 and ACE in hypertensive mRen2.Lewis rats with early-onset diabetes. Am J Physiol Renal Physiol. 2012;302(11):F1374–84.
Jones BH, Standridge MK, Moustaid N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology. 1997;138(4):1512–9.
Saint-Marc P, Kozak LP, Ailhaud G, Darimont C, Negrel R. Angiotensin II as a trophic factor of white adipose tissue: stimulation of adipose cell formation. Endocrinology. 2001;142(1):487–92.
Yvan-Charvet L, Even P, Lamande N, Ferre P, Quignard-Boulange A. Prevention of adipose tissue depletion during food deprivation in angiotensin type 2 receptor-deficient mice. Endocrinology. 2006;147(11):5078–86.
Littlejohn NK, Keen HL, Weidemann BJ, Claflin KE, Tobin KV, Markan KR, et al. Suppression of resting metabolism by the angiotensin AT2 receptor. Cell Rep. 2016;16(6):1548–60.
Shao C, Yu L, Gao L. Activation of angiotensin type 2 receptors partially ameliorates streptozotocin-induced diabetes in male rats by islet protection. Endocrinology. 2014;155(3):793–804.
Oliveira-Junior SA, Martinez PF, Guizoni DM, Campos DH, Fernandes T, Oliveira EM, et al. AT1 receptor blockade attenuates insulin resistance and myocardial remodeling in rats with diet-induced obesity. PLoS One. 2014;9(1):e86447.
Graus-Nunes F, Rachid TL, de Oliveira Santos F, Barbosa-da-Silva S, Souza-Mello V. AT1 receptor antagonist induces thermogenic beige adipocytes in the inguinal white adipose tissue of obese mice. Endocrine. 2017;55(3):786–98.
Dominguez JR, de la Calle H, Hurtado A, Robles RG, Sancho-Rof J. Effect of converting enzyme inhibitors in hypertensive patients with non-insulin-dependent diabetes mellitus. Postgrad Med J. 1986;62(Suppl 1):66–8.
Nazzaro P, Manzari M, Merlo M, Triggiani R, Scarano A, Ciancio L, et al. Distinct and combined vascular effects of ACE blockade and HMG-CoA reductase inhibition in hypertensive subjects. Hypertension. 1999;33(2):719–25.
Enalapril in essential hypertension: a comparative study with propranolol. Enalapril in Hypertension Study Group (UK). Br J Clin Pharmacol. 1984;18(1):51–6.
Rabi DM, Khan N, Vallee M, Hladunewich MA, Tobe SW, Pilote L. Reporting on sex-based analysis in clinical trials of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker efficacy. Can J Cardiol. 2008;24(6):491–6.
Sullivan JC. Sex and the renin-angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol. 2008;294(4):R1220–6.
Huang Z, Jansson L, Sjoholm A. Gender-specific regulation of pancreatic islet blood flow, insulin levels and glycaemia in spontaneously diabetic Goto-Kakizaki rats. Clin Sci (Lond). 2008;115(1):35–42.
Heart Outcomes Prevention Evaluation Study IYusuf S, Sleight P, Pogue J, Bosch J, Davies R, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000;342(3):145–53.
Officers A, Coordinators for the ACRGTA, Lipid-Lowering Treatment to Prevent Heart Attack T. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA. 2002;288(23):2981–97.
Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, Michelson EL, et al. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362(9386):759–66.
Kitamura N, Takahashi Y, Yamadate S, Asai S. Angiotensin II receptor blockers decreased blood glucose levels: a longitudinal survey using data from electronic medical records. Cardiovasc Diabetol. 2007;6:26.
Cao X, Yang FY, Xin Z, Xie RR, Yang JK. The ACE2/Ang-(1-7)/Mas axis can inhibit hepatic insulin resistance. Mol Cell Endocrinol. 2014;393(1–2):30–8.
Liu C, Lv XH, Li HX, Cao X, Zhang F, Wang L, et al. Angiotensin-(1-7) suppresses oxidative stress and improves glucose uptake via Mas receptor in adipocytes. Acta Diabetol. 2012;49(4):291–9.
Santos SH, Giani JF, Burghi V, Miquet JG, Qadri F, Braga JF, et al. Oral administration of angiotensin-(1-7) ameliorates type 2 diabetes in rats. J Mol Med (Berl). 2014;92(3):255–65.
Santos SH, Braga JF, Mario EG, Porto LC, Rodrigues-Machado Mda G, Murari A, et al. Improved lipid and glucose metabolism in transgenic rats with increased circulating angiotensin-(1-7). Arterioscler Thromb Vasc Biol. 2010;30(5):953–61.
Fu Z, Zhao L, Aylor KW, Carey RM, Barrett EJ, Liu Z. Angiotensin-(1-7) recruits muscle microvasculature and enhances insulin's metabolic action via mas receptor. Hypertension. 2014;63(6):1219–27.
Lu CL, Wang Y, Yuan L, Li Y, Li XY. The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects the function of pancreatic beta cells by improving the function of islet microvascular endothelial cells. Int J Mol Med. 2014;34(5):1293–300.
Sahr A, Wolke C, Maczewsky J, Krippeit-Drews P, Tetzner A, Drews G, et al. The angiotensin-(1-7)/Mas axis improves pancreatic beta-cell function in vitro and in vivo. Endocrinology. 2016;157(12):4677–90.
Santos SH, Fernandes LR, Pereira CS, Guimaraes AL, de Paula AM, Campagnole-Santos MJ, et al. Increased circulating angiotensin-(1-7) protects white adipose tissue against development of a proinflammatory state stimulated by a high-fat diet. Regul Pept. 2012;178(1–3):64–70.
Andrade JM, Lemos Fde O, da Fonseca Pires S, Millan RD, de Sousa FB, Guimaraes AL, et al. Proteomic white adipose tissue analysis of obese mice fed with a high-fat diet and treated with oral angiotensin-(1-7). Peptides. 2014;60:56–62.
Reyes-Engel A, Morcillo L, Aranda FJ, Ruiz M, Gaitan MJ, Mayor-Olea A, et al. Influence of gender and genetic variability on plasma angiotensin peptides. J Renin-Angiotensin-Aldosterone Syst. 2006;7(2):92–7.
Sullivan JC, Rodriguez-Miguelez P, Zimmerman MA, Harris RA. Differences in angiotensin (1-7) between men and women. Am J Physiol Heart Circ Physiol. 2015;308(9):H1171–6.
Cohall DH, Scantlebury-Manning T, James S, Hall K, Ferrario CM. Renin-angiotensin-aldosterone system gender differences in an Afro-Caribbean population. J Renin-Angiotensin-Aldosterone Syst. 2015;16(3):539–46.
Ferreira AJ, Santos RA, Bradford CN, Mecca AP, Sumners C, Katovich MJ, et al. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension. 2010;55(2):207–13.
Santos RA, Ferreira AJ. Pharmacological effects of AVE 0991, a nonpeptide angiotensin-(1-7) receptor agonist. Cardiovasc Drug Rev. 2006;24(3–4):239–46.
Ebermann L, Spillmann F, Sidiropoulos M, Escher F, Heringer-Walther S, Schultheiss HP, et al. The angiotensin-(1-7) receptor agonist AVE0991 is cardioprotective in diabetic rats. Eur J Pharmacol. 2008;590(1–3):276–80.
Schinzari F, Tesauro M, Veneziani A, Mores N, Di Daniele N, Cardillo C. Favorable vascular actions of angiotensin-(1-7) in human obesity. Hypertension. 2018;71(1):185–91.
van Twist DJ, Houben AJ, de Haan MW, Mostard GJ, Kroon AA, de Leeuw PW. Angiotensin-(1-7)-induced renal vasodilation in hypertensive humans is attenuated by low sodium intake and angiotensin II co-infusion. Hypertension. 2013;62(4):789–93.
Benter IF, Yousif MH, Al-Saleh FM, Raghupathy R, Chappell MC, Diz DI. Angiotensin-(1-7) blockade attenuates captopril- or hydralazine-induced cardiovascular protection in spontaneously hypertensive rats treated with NG-nitro-L-arginine methyl ester. J Cardiovasc Pharmacol. 2011;57(5):559–67.
Loloi J, Miller AJ, Bingaman SS, Silberman Y, Arnold AC. Angiotensin-(1-7) contributes to insulin-sensitizing effects of angiotensin-converting enzyme inhibition in obese mice. Am J Physiol Endocrinol Metab. 2018;315(6):E1204–E11.
Schuchard J, Winkler M, Stolting I, Schuster F, Vogt FM, Barkhausen J, et al. Lack of weight gain after angiotensin AT1 receptor blockade in diet-induced obesity is partly mediated by an angiotensin-(1-7)/Mas-dependent pathway. Br J Pharmacol. 2015;172(15):3764–78.
Bangalore S, Kumar S, Messerli FH. Angiotensin-converting enzyme inhibitor associated cough: deceptive information from the Physicians’ Desk Reference. Am J Med. 2010;123(11):1016–30.
Sica DA, Black HR. Current concepts of pharmacotherapy in hypertension: ACE inhibitor-related angioedema: can angiotensin-receptor blockers be safely used? J Clin Hypertens (Greenwich). 2002;4(5):375–80.
Burrell LM, Johnston CI, Tikellis C, Cooper ME. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol Metab. 2004;15(4):166–9.
Roca-Ho H, Riera M, Palau V, Pascual J, Soler MJ. Characterization of ACE and ACE2 expression within different organs of the NOD mouse. Int J Mol Sci. 2017;18(3):563.
Ye M, Wysocki J, Naaz P, Salabat MR, LaPointe MS, Batlle D. Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice: a renoprotective combination? Hypertension. 2004;43(5):1120–5.
Salem ES, Grobe N, Elased KM. Insulin treatment attenuates renal ADAM17 and ACE2 shedding in diabetic Akita mice. Am J Physiol Renal Physiol. 2014;306(6):F629–39.
Murca TM, Moraes PL, Capuruco CA, Santos SH, Melo MB, Santos RA, et al. Oral administration of an angiotensin-converting enzyme 2 activator ameliorates diabetes-induced cardiac dysfunction. Regul Pept. 2012;177(1–3):107–15.
Verma A, Shan Z, Lei B, Yuan L, Liu X, Nakagawa T, et al. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Mol Ther. 2012;20(1):28–36.
Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55(1):90–8.
Ye M, Wysocki J, Gonzalez-Pacheco FR, Salem M, Evora K, Garcia-Halpin L, et al. Murine recombinant angiotensin-converting enzyme 2: effect on angiotensin II-dependent hypertension and distinctive angiotensin-converting enzyme 2 inhibitor characteristics on rodent and human angiotensin-converting enzyme 2. Hypertension. 2012;60(3):730–40.
Liu P, Wysocki J, Souma T, Ye M, Ramirez V, Zhou B, et al. Novel ACE2-Fc chimeric fusion provides long-lasting hypertension control and organ protection in mouse models of systemic renin angiotensin system activation. Kidney Int. 2018;94(1):114–25.
Than A, Leow MK, Chen P. Control of adipogenesis by the autocrine interplays between angiotensin 1-7/Mas receptor and angiotensin II/AT1 receptor signaling pathways. J Biol Chem. 2013;288(22):15520–31.
Benter IF, Yousif MH, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol. 2007;292(1):H666–72.
Oliveira AC, Melo MB, Motta-Santos D, Peluso AA, Souza-Neto F, da Silva RF, et al. Genetic deletion of the alamandine receptor MRGD leads to dilated cardiomyopathy in mice. Am J Physiol Heart Circ Physiol. 2019;316(1):H123–H33.
Uchiyama T, Okajima F, Mogi C, Tobo A, Tomono S, Sato K. Alamandine reduces leptin expression through the c-Src/p38 MAP kinase pathway in adipose tissue. PLoS One. 2017;12(6):e0178769.
Colafella KMM, Denton KM. Sex-specific differences in hypertension and associated cardiovascular disease. Nat Rev Nephrol. 2018;14(3):185–201.
Kozakowski J, Gietka-Czernel M, Leszczynska D, Majos A. Obesity in menopause - our negligence or an unfortunate inevitability? Prz Menopauzalny. 2017;16(2):61–5.
Salpeter SR, Walsh JM, Ormiston TM, Greyber E, Buckley NS, Salpeter EE. Meta-analysis: effect of hormone-replacement therapy on components of the metabolic syndrome in postmenopausal women. Diabetes Obes Metab. 2006;8(5):538–54.
Clauser E, Gaillard I, Wei L, Corvol P. Regulation of angiotensinogen gene. Am J Hypertens. 1989;2(5 Pt 1):403–10.
Klett C, Ganten D, Hellmann W, Kaling M, Ryffel GU, Weimar-Ehl T, et al. Regulation of hepatic angiotensinogen synthesis and secretion by steroid hormones. Endocrinology. 1992;130(6):3660–8.
Chen YF, Naftilan AJ, Oparil S. Androgen-dependent angiotensinogen and renin messenger RNA expression in hypertensive rats. Hypertension. 1992;19(5):456–63.
Ellison KE, Ingelfinger JR, Pivor M, Dzau VJ. Androgen regulation of rat renal angiotensinogen messenger RNA expression. J Clin Invest. 1989;83(6):1941–5.
Schunkert H, Danser AH, Hense HW, Derkx FH, Kurzinger S, Riegger GA. Effects of estrogen replacement therapy on the renin-angiotensin system in postmenopausal women. Circulation. 1997;95(1):39–45.
Harvey PJ, Morris BL, Miller JA, Floras JS. Estradiol induces discordant angiotensin and blood pressure responses to orthostasis in healthy postmenopausal women. Hypertension. 2005;45(3):399–405.
Seely EW, Brosnihan KB, Jeunemaitre X, Okamura K, Williams GH, Hollenberg NK, et al. Effects of conjugated oestrogen and droloxifene on the renin-angiotensin system, blood pressure and renal blood flow in postmenopausal women. Clin Endocrinol. 2004;60(3):315–21.
Johannessen A, Nielsen AH, Poulsen K. Sexual dimorphism of inactive renin in rat plasma. Clin Exp Hypertens A. 1990;12(8):1405–17.
Nielsen AH, Johannessen A, Poulsen K. Inactive plasma renin exhibits sex difference in mice. Clin Sci (Lond). 1989;76(4):439–46.
Brosnihan KB, Li P, Ganten D, Ferrario CM. Estrogen protects transgenic hypertensive rats by shifting the vasoconstrictor-vasodilator balance of RAS. Am J Physiol-Reg I. 1997;273(6):R1908–R15.
Baltatu O, Cayla C, Iliescu R, Andreev D, Bader M. Abolition of end-organ damage by antiandrogen treatment in female hypertensive transgenic rats. Hypertension. 2003;41(3 Pt 2):830–3.
Umeda M, Ichikawa S, Kanda T, Sumino H, Kobayashi I. Hormone replacement therapy increases plasma level of angiotensin II in postmenopausal hypertensive women. Am J Hypertens. 2001;14(3):206–11.
Kang AK, Duncan JA, Cattran DC, Floras JS, Lai V, Scholey JW, et al. Effect of oral contraceptives on the renin angiotensin system and renal function. Am J Physiol Regul Integr Comp Physiol. 2001;280(3):R807–13.
Seltzer A, Pinto JE, Viglione PN, Correa FM, Libertun C, Tsutsumi K, et al. Estrogens regulate angiotensin-converting enzyme and angiotensin receptors in female rat anterior pituitary. Neuroendocrinology. 1992;55(4):460–7.
Gallagher PE, Li P, Lenhart JR, Chappell MC, Brosnihan KB. Estrogen regulation of angiotensin-converting enzyme mRNA. Hypertension. 1999;33(1):323–8.
Ichikawa J, Sumino H, Ichikawa S, Ozaki M. Different effects of transdermal and oral hormone replacement therapy on the renin-angiotensin system, plasma bradykinin level, and blood pressure of normotensive postmenopausal women. Am J Hypertens. 2006;19(7):744–9.
Proudler AJ, Ahmed AI, Crook D, Fogelman I, Rymer JM, Stevenson JC. Hormone replacement therapy and serum angiotensin-converting-enzyme activity in postmenopausal women. Lancet. 1995;346(8967):89–90.
Chinnathambi V, More AS, Hankins GD, Yallampalli C, Sathishkumar K. Gestational exposure to elevated testosterone levels induces hypertension via heightened vascular angiotensin II type 1 receptor signaling in rats. Biol Reprod. 2014;91(1):6.
Xu X, Xiao JC, Luo LF, Wang S, Zhang JP, Huang JJ, et al. Effects of ovariectomy and 17beta-estradiol treatment on the renin-angiotensin system, blood pressure, and endothelial ultrastructure. Int J Cardiol. 2008;130(2):196–204.
Nickenig G, Baumer AT, Grohe C, Kahlert S, Strehlow K, Rosenkranz S, et al. Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation. 1998;97(22):2197–201.
Baiardi G, Macova M, Armando I, Ando H, Tyurmin D, Saavedra JM. Estrogen upregulates renal angiotensin II AT1 and AT2 receptors in the rat. Regul Pept. 2005;124(1–3):7–17.
Leung PS, Wong TP, Chung YW, Chan HC. Androgen dependent expression of AT1 receptor and its regulation of anion secretion in rat epididymis. Cell Biol Int. 2002;26(1):117–22.
Zhang X, Thatcher SE, Rateri DL, Bruemmer D, Charnigo R, Daugherty A, et al. Transient exposure of neonatal female mice to testosterone abrogates the sexual dimorphism of abdominal aortic aneurysms. Circ Res. 2012;110(11):e73–85.
Macova M, Armando I, Zhou J, Baiardi G, Tyurmin D, Larrayoz-Roldan IM, et al. Estrogen reduces aldosterone, upregulates adrenal angiotensin II AT2 receptors and normalizes adrenomedullary Fra-2 in ovariectomized rats. Neuroendocrinology. 2008;88(4):276–86.
Mishra JS, Hankins GD, Kumar S. Testosterone downregulates angiotensin II type-2 receptor via androgen receptor-mediated ERK1/2 MAP kinase pathway in rat aorta. J Renin-Angiotensin-Aldosterone Syst. 2016;17(4):1470320316674875.
Ji H, Menini S, Zheng W, Pesce C, Wu X, Sandberg K. Role of angiotensin-converting enzyme 2 and angiotensin(1-7) in 17beta-oestradiol regulation of renal pathology in renal wrap hypertension in rats. Exp Physiol. 2008;93(5):648–57.
Douglas GC, O'Bryan MK, Hedger MP, Lee DK, Yarski MA, Smith AI, et al. The novel angiotensin-converting enzyme (ACE) homolog, ACE2, is selectively expressed by adult Leydig cells of the testis. Endocrinology. 2004;145(10):4703–11.
Freshour JR, Chase SE, Vikstrom KL. Gender differences in cardiac ACE expression are normalized in androgen-deprived male mice. Am J Physiol Heart Circ Physiol. 2002;283(5):H1997–2003.
Miller JA, Anacta LA, Cattran DC. Impact of gender on the renal response to angiotensin II. Kidney Int. 1999;55(1):278–85.
Yoshimura Y, Karube M, Aoki H, Oda T, Koyama N, Nagai A, et al. Angiotensin II induces ovulation and oocyte maturation in rabbit ovaries via the AT2 receptor subtype. Endocrinology. 1996;137(4):1204–11.
Neves LAA, Averill DB, Ferrario CM, Aschner JL, Brosnihan KB. Vascular responses to angiotensin-(1-7) during the estrous cycle. Endocrine. 2004;24(2):161–5.
Acknowledgements
Not Applicable
Funding
ACA is supported by an Early Career Development Award from the Central Society for Clinical and Translational Research and by NIH grants R00HL122507 and UL1TR002014.
Author information
Authors and Affiliations
Contributions
All authors were major contributors in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable
Consent for publication
Not Applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
White, M.C., Fleeman, R. & Arnold, A.C. Sex differences in the metabolic effects of the renin-angiotensin system. Biol Sex Differ 10, 31 (2019). https://doi.org/10.1186/s13293-019-0247-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13293-019-0247-5