
Abstract
Background
Acute liver failure (ALF) is an acute inflammatory liver disease with high mortality. Previous preclinical and clinical trials have confirmed that mesenchymal stem cell (MSC) is a promising therapeutic approach; however, the effect is not satisfied as the underlying molecular mechanisms of MSC in treating ALF remain unclear.
Methods
MSC isolated from 4- to 6-week-old C57BL/6 mice were used to treat ALF. Histological and serological parameters were analyzed to evaluate the efficacy of MSC. We explored the molecular mechanism of MSC in the treatment of ALF by detecting liver inflammatory response and hepatocyte death.
Results
In this study, we found that the therapeutic potential of MSC on ALF is dependent on the secretion of prostaglandin E2 (PGE2), a bioactive lipid. MSC-derived PGE2 inhibited TGF-β-activated kinase 1 (TAK1) signaling and NLRP3 inflammasome activation in liver macrophages to decrease the production of inflammatory cytokines. Meanwhile, macrophages in the liver could be induced to anti-inflammatory (M2) macrophages by MSC-derived PGE2 via STAT6 and mechanistic target of rapamycin (mTOR) signaling, which then promote inflammatory resolution and limit liver injury. Finally, administrating EP4 antagonist significantly ameliorated the therapeutic ability of MSC, which promoted liver inflammation and decreased M2 macrophages.
Conclusions
Our results indicate that PGE2 might be a novel important mediator of MSC in treating ALF, which is through inhibiting the liver inflammatory response and hepatocyte death.
Similar content being viewed by others

Introduction
Acute liver failure (ALF) is a clinical syndrome with high mortality rate, and liver transplantation is the only effective way to cure ALF now [1]. Due to the shortage of organ donor resources, alternative treatments are needed urgently [2]. It has been shown that mesenchymal stem cell (MSC) not only rescued several animal models of liver injury, but that it showed an effective therapeutic potential in clinical trials, which is based on their ability to secrete various trophic factors [3]. Our previous study confirmed that MSC-derived PGE2 could promote hepatocyte proliferation in ALF; however, the role of MSC-derived PGE2 on liver inflammation still remains unclear.
Several studies confirmed the cell death and hepatic inflammation in the progression of ALF [4]. Inflammatory mediators of toxic liver injury include cytokines such as tumor necrosis factor (TNF), interleukin (IL)-1β, and reactive oxygen species (ROS) which accelerate liver injury [5]. Most of these inflammatory mediators were secreted by innate immune cells in the liver, including monocytes, macrophages, and dendritic cells (DC), which are activated by damage-associated molecular patterns (DAMPs) released by damaged hepatocytes. Liver macrophages hold central functions in initiating, perpetuating, and restricting inflammation. Macrophages can be classified into pro-inflammatory (M1) or anti-inflammatory (M2) macrophages [6]. M1 cells secrete large amounts of pro-inflammatory cytokines, such as TNF and IL-1β. M2 cells secrete immunosuppressive cytokines, like IL-10, which plays a vital role in the resolution of inflammation. Recent studies have confirmed the role of MSC on macrophage polarization. For example, the therapeutic ability of MSC on cartilage repair and sepsis was dependent on the induction of M2 macrophages [7, 8]. In various liver disease, the therapeutic ability of MSC also relies on macrophage polarization [9, 10]. However, the mechanism of MSC on macrophage polarization remains unclear.
Sterile inflammation, which is defined as an inflammatory response triggered by damage mediated by noninfectious sources, plays a major role in ALF [4]. The nucleotide-binding and oligomerization domain-like receptor 3 (NLRP3) inflammasome is the main kind of sterile inflammation, which contains three parts: NLRP3, apoptosis-associated speck-like protein (ASC), and pro-caspase 1. The inflammasome is activated by DAMPs or pathogen-associated molecular patterns (PAMPs), which contribute to the maturation and secretion of IL-1β and IL-18 [11]. Recent studies confirmed that inflammasome activation contributes to the progression of various liver disorders, like viral fulminant hepatitis and nonalcoholic fatty liver disease [12]. Therefore, targeting the NLRP3 inflammasome might be an effective strategy for reducing liver inflammation.
In this study, we focused on the inflammatory response in ALF. We confirmed that MSC could attenuate liver inflammation and NLRP3 inflammasome activation; meanwhile, it promoted M2 macrophages to resolve inflammation, thus to cure ALF.
Materials and methods
Isolation of mouse bone marrow MSC
MSCs were isolated from 4- to 6-week-old C57BL/6 mice as we previously described [13]. We used flow cytometry (BD FACSAria II; BD Biosciences, San Jose, CA, USA) to identify the MSCs; the antibodies used were against mice antigens CD29, CD34, CD44, CD45, and CD90 (BD Biosciences). Positive cells were counted and compared to the signals of corresponding immunoglobulin isotypes. Mediums were supplemented with 10% fetal calf serum (Sciencell, San Diego, CA, USA), 100 U/ml penicillin, and 100 g/ml streptomycin. To deplete or overexpress COX2 in MSC to inhibit or increase the synthesis and secretion of PGE2, the short hairpin RNA against COX2 or COX2 gene lentivirus (GeneChem, Shanghai, China) was transfected to MSCs following the manufacturer’s instructions [named MSC-COX2(−) and MSC-COX2(+)].
For MSC, MSC-COX2(−)- or MSC-COX2 (+)-conditioned medium, MSCs were maintained in serum-free medium for 48 h at 37 °C in a humidified atmosphere containing 5% CO2. Conditioned medium was harvested, clarified by centrifugation, and frozen at − 80 °C until use.
Animal models
Male C57BL/6J wild type mice (6~8 weeks old) were obtained from the Animal Core Facility of Nanjing Medical University. Animals were bred in a pathogen-free facility. Mice were randomly divided into five groups: (1) control group (Ctrl): mice were intraperitoneally (i.p.) injected with PBS only; (2) ALF group (ALF): mice simultaneously received 600 mg/kg D-galactosamine (D-Gal) (Sigma-Aldrich, St. Louis, MO, USA) and 100 μg/kg LPS (Sigma-Aldrich) dissolved in PBS by i.p. injection; (3–5) cell transplantation groups: mice received 600 mg/kg D-Gal and 100 μg/kg LPS via i.p. injection, and then 2 × 106 MSC, MSC-COX2(+), or MSC-COX2(−) were injected via tail vein after 6 h, namely the MSC group, MSC-COX2(+) group, or MSC-COX2(−) group, respectively. For TAK1 inhibition, we administrated the specific TAK1 inhibitor 5z-7-ox (5 mg/kg, Sigma) to mice 1 h before LPS/D-Gal administration. To inhibit the EP4 receptor of PGE2, we administered EP4-specific antagonist (GW627368X, 20 mg/kg, Cayman Chemical, Ann Arbor, MI, USA) 1 h before LPS/D-Gal treatment and every 24 h following MSC infusion. Blood and liver tissues were collected at the indicated time for further analysis.
Bone marrow-derived macrophage (BMDM) isolation and culture
BMDMs were prepared and cultured as described [14]. In brief, BM cells were flushed from femurs and tibias of mice and dispersed mechanically. Cell suspensions were filtered through a 200-mesh filter, and the remaining cells were collected by centrifugation at 300×g for 4 min. After centrifugation, the cells were resuspended and cultured in RIP 1640 supplemented with 10% FBS and macrophage colony-stimulating factor (M-CSF, 20 ng/ml, eBioscience) for 7 days.
For inflammasome activation, BMDMs were treated with LPS (100 ng/ml) for 4 h, followed by 30-min treatment with PGE2 (0.1 μM) or MSC, followed by stimulation with nigericin (4 μM for 1 h, Cayman Chemical). EP4 antagonist was added 30 min prior to treatment with PGE2 or MSC. For macrophage polarization, BMDMs were stimulated with PGE2 or MSC for 24 h.
Real-time quantitative PCR and western blotting
Total RNA or protein for liver tissues and cells were extracted as previously described [13]. The primer sequences and primary antibodies are listed in Supplementary Table S1 and S2.
Small interfering RNA and transfection
siRNA-specific EP4 as well as control siRNAs were obtained from RiboBio (GuangZhou, China). The detailed sequences of siRNAs are listed in Supplementary Table S3. Transfection with siRNAs was completed using riboFECT™ CP (RiboBio) according to the manufacturer’s instruction.
Immunofluorescence and HE analyses
HE staining analysis of the liver sections was conducted as described previously [13]. The images were captured by a Nikon microscope (Japan). Other sets of sections were dewaxed and subjected for immunofluorescence labelling of NLRP3 (1:200, Adipogen Life, San Diego, USA) and F4/80 (1:200, Abcam, Cambridge, UK). Sections were incubated with the primary antibodies at the same working dilutions as noted above overnight at 4 °C, followed by incubation with goat anti-rabbit IgG H&L (Alexa Fluor® 488) (1:500, Abcam) or Goat Anti-Rat IgG H&L (1:500, Alexa Fluor® 594) for 1 h. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, KeyGen BioTECH, Nanjing, China), and images were captured using fluorescence microscopy (Olympus, Japan).
Measurement of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST)
Serum ALT and AST were determined using an automatic biochemical analyzer (Olympus, Tokyo, Japan) in the Affiliated Drum Tower Hospital of Nanjing University Medical School.
TUNEL and PI assay
Cell apoptosis and death in the liver were measured using the one-step TUNEL Apoptosis Assay kit and propidium iodide (PI) staining (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions.
IL-1β and IL-10 analysis
IL-1β levels in serum or cell supernatant and IL-10 levels in serum were detected by an enzyme-linked immunosorbent assay (ELISA) kit (Invitrogen, Thermo Scientific, Vienna, Australia).
Caspase 1 activity
Caspase-1 activity in liver tissues was measured by Caspase 1 Activity Assay Kit (Beyotime Biotechnology) according to the manufacturer’s instructions.
Statistical analysis
Data were expressed as means ± SD. Differences between groups were assessed by a two-tailed t test assuming unequal variance. Five to 8 mice/group were analyzed using Prism software (GraphPad). p values less than 0.05 were considered as statistically significant.
Results
MSC protects against ALF via PGE2
Paralyzed with our previous study [13], MSC protected against ALF through PGE2, as revealed by plasma levels of ALT and AST (Fig. 1a) and liver damage in HE staining (Fig. 1b). TUNEL and PI staining confirmed the hepatocyte necrosis and apoptosis were decreased when infused with MSC, especially in the MSC-COX2(+) group; however, MSC-COX2(−) failed to decrease hepatocyte necrosis and apoptosis (Fig. 1c, d). Taken together, MSC protected hepatocyte death against ALF via PGE2.

MSC protects LPS/D-Gal-induced liver injury via PGE2. a Serum levels of ALT and AST in each group (n = 6). b Representative HE-stained liver sections from each group and quantitation of the necrosis area in each group (n = 4). c Representative images of TUNEL-stained liver sections from each group and the number of TUNEL-positive cells in each group (n = 4). d Representative images of PI staining to indicate necrosis cells and the number of PI-positive cells in each group (n = 4) (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
MSC ameliorates inflammation in ALF
Liver inflammation is associated with hepatocyte damage and death; therefore, we examined the inflammatory response in the liver after MSC infusion. Notably, mRNA expression levels of inflammatory cytokines and chemokines, including CCL2, IL-1β, iNOS, and TNF-α were significantly inhibited in the MSC group, and in the MSC-COX2(+) group, the levels of these cytokines decreased more obviously. By contrast, the MSC-COX2(−) group, which inhibited secretion of PGE2 from MSC, failed to decrease these inflammatory cytokines (Fig. 2a). NF-κB signaling plays a great role in inflammation [15]. We demonstrated that LPS/D-Gal-induced ALF significantly activated NF-κB signaling in the liver, as the increased expression of the phosphorylation of IKK-β and P65 (p-IKK-β and p-P65) (Fig. 2b). The MSC or MSC-COX2(+) group exhibited an ameliorated phenotype, while MSC-COX2(−) did not show this phenotype (Fig. 2b). Next, we aimed to explore the mechanism of MSC on NF-κB signaling inhibition. TGF-β-activated kinase 1 (TAK1) is an intracellular hub molecule that regulates NF-κB signaling pathways which plays a key role in cell survival and death [16]. Our results demonstrated that TAK1 signaling was inhibited after MSC infusion, as the expression of the phosphorylation of TAK1 (p-TAK1), JNK (p-JNK), P38 (p-P38), and c-Jun (p-c-Jun) were significantly reduced in the MSC or MSC-COX2(+) group, while these TAK1 substrates still expressed high in the MSC-COX2(−) group (Fig. 2c).

MSC-derived PGE2 restrains inflammatory responses in the liver during LPS/D-Gal-induced ALF. a The mRNA levels of pro-inflammatory cytokines (IL-1β, CCL2, iNOS, TNF-α) in the liver in each group (n = 4). b The protein expression levels of NF-κB signaling in each group. c The protein expression levels of TAK1 signaling in each group. d Serum levels of ALT and AST in mice pretreated with TAK1 inhibitor, 5Z-7-ox (n = 4). e Representative HE-stained liver sections and quantitation of necrosis area in each group (n = 4). f Protein levels of TAK1 and NF-κB signaling in each group (*p < 0.05, **p < 0.01, ***p < 0.001)
To confirm the role of TAK1 in ALF, we further used 5Z-7-ox, a specific TAK1 inhibitor, to inhibit TAK1 before LPS/D-Gal administration. Notably, TAK1 inhibition protected against liver damage, shown by normalized serum ALT and AST and reduced necrosis on the histological analysis (Fig. 2d, e). Treatment of 5Z-7-ox significantly suppressed TAK1 activation and reduced activation of its downstream signaling pathways, including p-JNK, p-P38, p-c-Jun, and NF-κB signaling (Fig. 2e). Collectively, these observations suggested that MSC-derived PGE2 protected ALF through suppressing liver inflammation in a TAK1-NF-κB pathway.
MSC inhibits inflammasome activation of macrophages in ALF
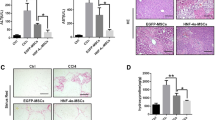
Previous studies have confirmed the role of inflammasome activation in LPS/D-Gal-induced liver injury, which accelerated liver inflammation [17]. Next, we explored whether MSC infusion could inhibit NLRP3 inflammasome activation in the liver. The protein levels of NLRP3, caspase1 p20, and mature-IL-1β were decreased in the MSC or MSC-COX2(+) group (Fig. 3a). In parallel, serum concentrations of IL-1β and the activity of caspase 1 enzyme in liver tissues confirmed the inhibition of NLRP3 inflammasome by MSC-derived PGE2 (Fig. 3b, c). However, MSC-COX2(−) failed to inhibit NLRP3 inflammasome activation in liver tissues. In order to identify the cell population responsible for increased production of NLRP3 inflammasome, the double immunohistochemistry staining was performed. The results demonstrated that NLRP3 was mostly activated in macrophages in the liver and MSC or MSC-COX2(+) could inhibit NLRP3 activation in liver macrophages (Fig. 3d). To confirm this result, we treated mouse hepatocyte cell line AML12 or BMDM with 1 μg/ml LPS for 24 h in vitro. As showed in Fig. 3e and f, the activation of NLRP3 and levels of IL-1β secretion was much higher in BMDM, which indicated that macrophages were more sensitive to DAMPs than hepatocytes. Collectively, our results indicated that MSC-derived PGE2 inhibited liver macrophage inflammasome activation to protect against liver injury.

MSC-derived PGE2 inhibits NLPR3 inflammasome activation in liver macrophages. a Protein levels of NLRP3, Caspase 1 p20, and mature-IL-1β in each group. b Serum levels of IL-1β in each group (n = 6). c Measurement of caspase-1 enzymatic activity in the liver of each group (n = 4). d Representative immunofluorescence staining of co-localization of NLRP3 with F4/80 in the liver of each group. Blue: DAPI; green: NLRP3; red: F4/80. White arrows in the Merge images indicated co-localization. e Protein levels of NLRP3 inflammasome pathway in hepatocyte cell line AML12 and BMDM treated with 1 μg/ml LPS for 24 h. f The levels of IL-1β in supernatants of hepatocyte cell line AML12 and BMDM treated with 1 μg/ml LPS for 24 h (n = 3) (*p < 0.05, **p < 0.01, ***p < 0.001)
To confirm the role of MSC on NLRP3 inhibition, we treated BMDM with LPS and nigericin, a typical inducer of NLPR3. MSC or MSC-COX2(+)-conditioned medium significantly inhibited NLPR3 activation and IL-1β secretion (Fig. 4a, b), while MSC-COX2(−)-conditioned medium did not show this phenomenon. In parallel, exogenous PGE2 exhibited the same effects on NLRP3 (Fig. 4c, d). Previous studies have confirmed the role of NF-κB in NLRP3 inflammasome activation [18]; thus, we explored whether MSC inhibited NLRP3 through TAK1-NF-κB. Our results showed that MSC-conditioned medium or PGE2 could inhibit TAK1 and NF-κB activation in BMDM induced by LPS and nigericin (Fig. 4a, c). Meanwhile, TAK1 inhibitor could also inhibit NLRP3 activation in BMDM (Fig. 4e, f). In vivo study confirmed the effects of TAK1 inhibitor on NLRP3 in the ALF model (Fig. 4g, h). Overall, our results indicated that MSC-derived PGE2 could inhibit NLPR3 inflammasome activation of liver macrophages through TAK1-NF-κB pathway.

MSC-derived PGE2 inhibits NLRP3 inflammasome activation via TAK1. a The protein expression levels of NLRP3 inflammasome and TAK1 signaling in BMDM following indicated condition. b The levels of IL-1β in each supernatant (n = 3). c The protein expression levels of NLRP3 inflammasome and TAK1 signaling in BMDM treated with exogenous PGE2. d The levels of IL-1β in cell supernatant treated with exogenous PGE2 (n = 3). e Effects of TAK1 inhibitor, 5Z-7-ox (TAKi), on NLRP3 inflammasome activation in BMDM. f The levels of IL-1β in cell supernatant treated with TAKi (n = 3). g Expression of NLRP3 inflammasome signaling in the liver pretreated with TAKi. h Measurement of caspase-1 enzymatic activity in the liver of each group (n = 4). i Serum levels of IL-1β in each group (n = 4) (*p < 0.05, **p < 0.01)
MSC induces M2 macrophage polarization in ALF
Mouse models of acute liver injury have revealed the role of macrophages in accelerating damage through cytokines and chemokines releasing. However, liver macrophages are plastic and adapt their phenotype to promote tissue repair according to signals derived from the hepatic microenvironment [6]. Several studies confirmed the role of MSC on macrophage polarization in different models [19, 20]. Thus, we tended to explore whether MSC could induce M2 macrophages in ALF to promote inflammation resolution. We found that gene expression associated with M2-like (Arg1, Mgl1, Mgl2, and Ym1) macrophages were increased in the MSC and MSC-COX2(+) groups (Fig. 5a); meanwhile, the serum levels of IL-10, which is produced by M2 macrophages, were also increased in the MSC and MSC-COX2(+) groups (Fig. 5b). However, MSC-COX2(−) failed to induce M2 macrophages. In vitro studies confirmed the MSC-derived PGE2 on macrophage polarization, as the expression of M2 markers and numbers of CD206-positive cells of BMDM treated with MSC-conditioned medium or PGE2 were increased (Fig. 5c, d). Taken together, our results demonstrated that MSC induced M2 macrophages to resolute inflammation in ALF through PGE2.

MSC-derived PGE2 promotes an M2 macrophage phenotype in the liver. a The mRNA levels of M2 markers (Arg1, Mgl1, Mgl2, Ym1, and IL-10) in the liver of each group (n = 4). b Serum levels of IL-10 in each group (n = 4). c Representative immunofluorescence staining of M2 marker, CD206, and F4/80 in BMDM following indicated condition. d The mRNA levels of M2 markers in BMDM following indicated condition (n = 3) (*p < 0.05, **p < 0.01, ***p < 0.001)
Next, we investigated the mechanism of MSC-derived PGE2 on macrophage polarization. Western blotting results showed that not only the classical M2 pathway, STAT6, was activated when treated with MSC or PGE2, but also resulted in mTOR activation, as increased phosphorylation of the mTOR substrates mTOR, S6K, AKT, and GSK-3β (Fig. 6a), which correlated with previous studies [21]. In contrast, MSC-COX2(−)-conditioned medium failed to activate mTOR and STAT6 pathway (Fig. 6a). Treatment of BMDM with mTOR inhibitor, rapamycin (Rap), or AKT inhibitor, MK-2206, confirmed the role of mTOR signaling in MSC-derived PGE2 on M2 macrophage polarization, as the mRNA levels of M2 markers were significantly increased after administration of Rap or MK-2206 (Fig. 6b–d). Taken together, our results indicated that MSC-derived PGE2 induced M2 macrophages through mTOR and classical STAT6 pathway.

MSC-derived PGE2 promotes M2 macrophage through STAT6 and mTOR signaling. a Protein levels of STAT6 and mTOR signaling in BMDM following indicated condition. b Protein levels of mTOR signaling in BMDM treated with mTOR inhibitor (rapamycin, Rap) and AKT inhibitor (MK-2206) following indicated condition. c The mRNA levels of M2 markers (Arg1, Mgl1, Mgl2, and Ym1) in BMDM treated with Rap following indicated condition (n = 3). d The mRNA levels of M2 markers (Arg1, Mgl1, Mgl2, and Ym1) in BMDM treated with MK-2206 following indicated condition (n = 3) (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
MSC protects against liver inflammation via PGE2 receptor (EP) 4
Our previous studies showed that MSC-derived PGE2 promoted hepatocyte proliferation through EP4 [13]; we explored whether EP4 also played a role in liver inflammation resolution. We treated mice with EP4 inhibitor (EP4i), GW627368X. The mRNA levels of inflammatory cytokines increased when treated with EP4i (Fig. 7a). Western blot results confirmed that inhibition of TAK1 and NF-κB signaling by MSC was also mediated by EP4, as the expression of TAK1 and NF-κB signaling substrates was higher when EP4i was administered (Fig. 7b, c). Meanwhile, the expression of NLRP3 and the activity of caspase 1 enzyme were also higher when EP4i was administered, even in the MSC-COX2(+) group (Fig. 7d, e). In vitro studies using EP4i or siRNA to inhibit or deplete EP4 in BMDM, the western blot and IL-1β secretion confirmed the role of EP4 on NLRP3 inflammasome inhibition (Fig. 7f, g). Taken together, these results showed that MSC-derived PGE2 ameliorated liver inflammation through EP4.

MSC-derived PGE2 protects against liver inflammation via EP4. a The mRNA levels of inflammatory cytokines (IL-1β, CCL2, iNOS, TNF-α) in the liver from each group pretreated with EP4 inhibitor (EP4i) (n = 4). b The protein expression levels of NF-κB signaling in each group pretreated with EP4i. c The protein expression levels of TAK1 signaling in each group pretreated with EP4i. d The protein expression levels of NLRP3 inflammasome signaling in each group pretreated with EP4i. e Measurement of caspase-1 enzymatic activity in the liver of each group pretreated with EP4i (n = 4). f Protein levels of NLRP3 inflammasome and TAK1 signaling in BMDM pretreated with EP4i or knocking down of EP4 via siRNA. g The levels of IL-1β in supernatants of BMDM pretreated with EP4i or knocking down of EP4 via siRNA (*p < 0.05, **p < 0.01)
EP4 mediates MSC-derived PGE2 on macrophage polarization
Next, we explored whether M2 macrophage polarization was also mediated by EP4. The mRNA levels of M2 macrophage markers and serum levels of IL-10 were also inhibited when treated with EP4i (Fig. 8a, b). And mRNA levels of PGE2 receptors (EP1-EP4) in BMDM confirmed the role of EP4 on M2 macrophage polarization (Fig. 8c). EP4i or siRNA treatment demonstrated the role of EP4 in macrophage polarization, as the mRNA levels of M2 markers were decreased when treated with EP4i or siRNA (Fig. 8e, g). Meanwhile, the protein levels of STAT6 and mTOR signaling substrates confirmed the role of EP4 on STAT6 and mTOR signaling (Fig. 8d, f). Overall, these results demonstrated that EP4 played a major role in MSC-derived PGE2 on macrophage polarization.

M2 macrophage polarization depends on EP4. a The mRNA levels of M2 markers (Arg1, Mgl1, Mgl2, and Ym1) in the liver of each group pretreated with EP4i (n = 4). b Serum levels of IL-10 in each group (n = 4). c The mRNA levels of (EP1, EP2, EP3, and EP4) in BMDM under indicated condition (n = 3). d Protein levels of STAT6 and mTOR signaling in BMDM treated with EP4i. e The mRNA levels of M2 markers (Arg1, Mgl1, Mgl2, and Ym1) in BMDM treated with EP4i (n = 3). f Protein levels of STAT6 and mTOR signaling in BMDM knocked down EP4 via siRNA. g The mRNA levels of M2 markers (Arg1, Mgl1, Mgl2, and Ym1) in BMDM knocked down EP4 via siRNA (n = 3) (*p < 0.05, **p < 0.01)
Discussion
Cell death and inflammation play great roles in the progression of ALF [4]. Accordingly, we speculated that reduced cell death and hepatic inflammation would be beneficial for the prevention and treatment of ALF. As described previously, MSC could regulate these important pro-inflammatory cytokines via a variety of effector mechanisms [22]. In our study, we clarified that MSC-derived PGE2 reduced hepatocyte death and liver inflammation to attenuate ALF. Meanwhile, MSC-derived PGE2 also modulated macrophage polarization to promote inflammation resolution in ALF.
PGE2 is a bioactive lipid which is catalyzed by the COX2 from arachidonic acid. PGE2 exerts a wild range of biological effects associated with inflammation and cancer [23]. Several studies confirmed that PGE2 promoted the resolution of inflammation and tissue repair. In a concanavalin A (ConA)-induced liver injury model, depleted COX2 in mice accelerated liver injury [24]. Hepatocyte COX2 expression protected against liver ischemia-reperfusion injury (IRI); meanwhile, in patients who underwent liver transplantation, there was a significant positive correlation of plasma PGE2 levels and graft function [25]. These results indicated that PGE2 might have great therapeutic potentials in treating inflammatory liver diseases. In this study, we demonstrated the therapeutic role of MSC secreted PGE2 on LPS/D-Gal-induced ALF based on its anti-apoptosis and anti-inflammation ability.
We confirmed that MSC-derived PGE2 reduced pro-inflammatory cytokine production in the liver through inhibiting TAK1-NF-κB signaling, which is important in regulating the expression of pro-inflammatory cytokines. TAK1 is a central regulator of cell death and inflammation through activating downstream effectors such as NF-κB and mitogen-activated protein kinases (MAPKs) [16]. Recent studies have demonstrated the role of TAK1 in the progression of various liver diseases, like liver IRI, nonalcoholic steatohepatitis (NASH), and hepatocellular carcinoma [26,27,28]. In our study, we found that MSC infusion could decrease the expression of TAK1 downstream substrates and NF-κB, thus alleviating liver inflammation. In parallel, administration of the TAK1-specific inhibitor, 5Z-7-ox, could also protect against ALF in vivo.
Inflammasome activation has been identified as a major contributor to hepatocyte damage in ALF [29]. Our results showed that administration of MSC, especially MSC-COX2(+), significantly suppressed LPS/D-Gal-induced inflammasome activation, while MSC-COX2(−) failed to show this phenomenon, which demonstrated the role of PGE2 on NLRP3 inflammasome inhibition. Next, we confirmed that macrophages in the liver are the main source of NLRP3, which then accelerated liver inflammatory response. A previous study has showed that TAK1 restricted NLRP3 activation and NF-κB was important for NLRP3 activation [18, 30]. We found that mice pretreated with 5Z-7-ox significantly inhibited NLRP3 inflammasome activation, which correlated with the in vitro model results, showing the role of TAK1 on NLRP3 inflammation activation. These results indicated that MSC-derived PGE2 restricted macrophage NLRP3 activation through TAK1.
Liver macrophages hold a central position in maintaining homeostasis in the liver as well as in the pathogenesis of acute or chronic liver injury. We showed that MSC-derived PGE2 ameliorated macrophage activation-induced inflammatory response. Recent studies have revealed macrophages to be heterogeneous; M2 macrophages protected against exacerbated inflammation and thus limited tissue injury [6]. Several studies have showed the role of MSC on macrophage polarization [19, 20]. Our studies demonstrated the role of MSC-derived PGE2 on M2 macrophage induction in ALF thus limiting liver injury, as the increased expression of M2 markers and plasma levels of IL-10. Next, we found that MSC-derived PGE2 not only activated STAT6 signaling in BMDM, the classical pathway on M2 macrophage polarization [31], but also enhanced the expression of mTOR signaling substrates, which has shown to control macrophage metabolism and thus to shape their properties [21, 32]. Administration of the mTOR inhibitor or AKT-specific inhibitor significantly inhibited the ability of MSC-derived PGE2 on M2 macrophage polarization.
PGE2 elicits a wide range of biological effects through its receptors, EP1-EP4 [23]. Our previous study found that MSC-derived PGE2 promoted hepatocyte proliferation via EP4. In this study, we administed EP4 antagonist to mice, which significantly inhibited the effects of MSC on attenuating liver inflammation and M2 macrophage polarization. And the administration of an agonist for EP4 has been found to confer protection against IRI in the liver, ischemic heart diseases, and intestinal injury [33,34,35]. Collectively, MSC-derived PGE2 protected against ALF through EP4.
In conclusion, our data confirmed that MSC attenuated ALF dependent on the secretion of PGE2. MSC-derived PGE2 inhibited TAK1 signaling and NLRP3 inflammasome activation in liver macrophages; meanwhile, MSC-derived PGE2 could also induce M2 macrophages to secret anti-inflammatory cytokines, like IL-10 to promote inflammation resolution and limit liver injury through activating STAT6 and mTOR signaling in macrophages, finally to inhibit the liver inflammatory response and hepatocyte apoptosis induced by LPS/D-Gal. All these phenomena were relied on the PGE2 receptor, EP4. Inhibiting EP4 significantly alleviated the therapeutic potential of MSC on ALF. This study provided a new therapeutic mechanism of MSC on ALF, which might provide a new direction to enhance the functionality of MSC.
Availability of data and materials
The authors confirm that all data underlying the findings are fully available.
Change history
23 November 2023
A Correction to this paper has been published: https://doi.org/10.1186/s13287-023-03570-7
Abbreviations
- MSC:
-
Mesenchymal stem cell
- ALF:
-
Acute liver failure
- PGE2 :
-
Prostaglandin E2
- TAK1:
-
TGF-β-activated kinase 1
- mTOR:
-
Mechanistic target of rapamycin
- TNF:
-
Tumor necrosis factor
- IL:
-
Interleukin
- ROS:
-
Reactive oxygen species
- DAMPs:
-
Damage-associated molecular patterns
- DC:
-
Dendritic cells
- NLRP3:
-
Nucleotide-binding and oligomerization domain-like receptor 3
- ASC:
-
Apoptosis-associated speck-like protein
- PAMPs:
-
Pathogen-associated molecular patterns
- ALT:
-
Alanine aminotransaminase
- AST:
-
Aspartate aminotransferase
- BMDM:
-
Bone marrow-derived macrophage
- M-CSF:
-
Macrophage colony-stimulating factor
- ConA:
-
Concanavalin A
- IRI:
-
Ischemia-reperfusion injury
- MAPKs:
-
Mitogen-activated protein kinases
- NASH:
-
Nonalcoholic steatohepatitis
References
Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol. 2015;62:S112–20.
European Association for the Study of the Liver. Electronic address, easloffice easloffice eu EASL Clinical Practice Guidelines: liver transplantation. J Hepatol. 2016;64:433–85.
Alfaifi M, Eom YW, Newsome PN, Baik SK. Mesenchymal stromal cell therapy for liver diseases. J Hepatol. 2018;68:1272–85.
Chung RT, et al. Pathogenesis of liver injury in acute liver failure. Gastroenterology. 2012;143:e1–7.
Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–94.
Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306–21.
Zhang S, et al. MSC exosomes mediate cartilage repair by enhancing proliferation, attenuating apoptosis and modulating immune reactivity. Biomaterials. 2018;156:16–27.
Song Y, et al. Exosomal miR-146a contributes to the enhanced therapeutic efficacy of interleukin-1beta-primed mesenchymal stem cells against sepsis. Stem Cells. 2017;35:1208–21.
Luo XY, et al. Transplantation of bone marrow mesenchymal stromal cells attenuates liver fibrosis in mice by regulating macrophage subtypes. Stem Cell Res Ther. 2019;10:16.
Lee KC, Lin HC, Huang YH, Hung SC. Allo-transplantation of mesenchymal stem cells attenuates hepatic injury through IL1Ra dependent macrophage switch in a mouse model of liver disease. J Hepatol. 2015;63:1405–12.
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87.
Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol. 2015;12:387–400.
Liu Y, et al. Prostaglandin E2 secreted by mesenchymal stem cells protects against acute liver failure via enhancing hepatocyte proliferation. FASEB J. 2019;33:2514–25.
Ma PF, et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J Hepatol. 2017;67:770–9.
Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309–24.
Mihaly SR, Ninomiya-Tsuji J, Morioka S. TAK1 control of cell death. Cell Death Differ. 2014;21:1667–76.
Gehrke N, et al. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J Hepatol. 2018;68:986–95.
Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat Immunol. 2017;18:861–9.
Yin Y, et al. Human umbilical cord-derived mesenchymal stem cells direct macrophage polarization to alleviate pancreatic islets dysfunction in type 2 diabetic mice. Cell Death Dis. 2018;9:760.
Chiossone L, Conte R, Spaggiari GM, et al. Mesenchymal stromal cells induce peculiar alternatively activated macrophages capable of dampening both innate and adaptive immune responses. Stem Cells. 2016;34(7):1909–21.
Byles V, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun. 2013;4:2834.
Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13:392–402.
Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol. 2013;35:123–37.
Yin H, Cheng L, Langenbach R, Ju C. Prostaglandin I (2) and E (2) mediate the protective effects of cyclooxygenase-2 in a mouse model of immune-mediated liver injury. Hepatology. 2007;45:159–69.
Motiño O, Francés DE, Casanova N, et al. Protective role of hepatocyte cyclooxygenase-2 expression against liver ischemia-reperfusion injury in mice [published correction appears in Hepatology. 2020;71(1):404]. Hepatology. 2019;70(2):650–65.
Yang L, et al. Creg in hepatocytes ameliorates liver ischemia/reperfusion injury in a TAK1-dependent manner in mice. Hepatology. 2019;69:294–313.
Yang L, et al. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology. 2014;59:483–95.
Inokuchi-Shimizu S, et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest. 2014;124:3566–78.
Woolbright BL, Jaeschke H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol. 2017;66:836–48.
Malireddi RKS, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med. 2018;215:1023–34.
Porta C, Riboldi E, Ippolito A, Sica A. Molecular and epigenetic basis of macrophage polarized activation. Semin Immunol. 2015;27:237–48.
Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol. 2015;27:286–96.
Miyoshi H, et al. Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. EMBO J. 2017;36:5–24.
Kuzumoto Y, et al. Significance and therapeutic potential of prostaglandin E2 receptor in hepatic ischemia/reperfusion injury in mice. Hepatology. 2005;42:608–17.
Pang L, et al. Prostaglandin E receptor subtype 4 signaling in the heart: role in ischemia/reperfusion injury and cardiac hypertrophy. J Diabetes Res. 2016;2016:1324347.
Acknowledgements
Not applicable.
Funding
This work was funded by grants from the National Natural Science Foundation of China (81872359, 81670566), Jiangsu Province’s Key Provincial Talents Program (ZDRCA2016066), the Certificate of China Postdoctoral Science Foundation Grant (2018 M642222), Jiangsu Province Natural Science Foundation (BK20190114), the Nanjing Medical Science and Technique Development Foundation (QRX17129), the Nanjing health science and technology development project for Distinguished Young Scholars (JQX19002), the Nanjing health science and technology development project for Medical and health research (YKK19070), the Nanjing Science and technology project (201911039), and Project of Nanjing University Hospital Management Institute, Aid project of Nanjing Drum Tower Hospital Health, Education & Research Foundation (NDYG2020047).
Author information
Authors and Affiliations
Contributions
Jinglin Wang, Yang Liu, and Haoran Ding performed the experiments. Yang Liu analyzed the data and wrote the manuscript. Jinglin Wang and Haoran Ding contributed in the animal experiments and manuscript writing. Haozhen Ren and Xiaolei Shi designed the project and coordinated the execution of the experimental plan. Xiaolei Shi obtained funding and directed the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Nanjing Drum Tower Hospital (Approval No. 20160601).
Consent for publication
This manuscript is approved by all authors for publication. I would like to declare on behalf of my co-authors that the work described was original research that has not been published previously, and not under consideration for publication elsewhere, in whole or in part. The materials in this manuscript are not reproduced from other articles. All the authors listed have approved the manuscript that is enclosed.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1 Table S1.
Antibodies for immunoblots and immunohistochemistry. Table S2. Sequence of primers used in experiments. Table S3. Primers of siRNAs. Figure S1: Identification of mesenchymal stem cells (MSCs). Flow cytometry analysis of MSCs. PE, phycoerythrin. The immunophenotypic characterization by flow cytometry showed positive stromal marker expression (CD29, CD44, and CD90), but little or no hematopoietic marker expression (CD34 and CD45), which means a high purity after the third passage (Fig. S1).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, J., Liu, Y., Ding, H. et al. Mesenchymal stem cell-secreted prostaglandin E2 ameliorates acute liver failure via attenuation of cell death and regulation of macrophage polarization. Stem Cell Res Ther 12, 15 (2021). https://doi.org/10.1186/s13287-020-02070-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-020-02070-2