
Abstract
Background
Human mesenchymal stem cells are promising tools for regenerative medicine due to their ability to differentiate into many cellular types such as osteocytes, chondrocytes and adipocytes amongst many other cell types. These cells present spontaneous calcium oscillations implicating calcium channels and pumps of the plasma membrane and the endoplasmic reticulum. These oscillations regulate many basic functions in the cell such as proliferation and differentiation. Therefore, the possibility to mimic or regulate these oscillations might be useful to regulate mesenchymal stem cells biological functions.
Methods
One or several electric pulses of 100 μs were used to induce Ca2+ spikes caused by the penetration of Ca2+ from the extracellular medium, through the transiently electropermeabilized plasma membrane, in human adipose mesenchymal stem cells from several donors. Attached cells were preloaded with Fluo-4 AM and exposed to the electric pulse(s) under the fluorescence microscope. Viability was also checked.
Results
According to the pulse(s) electric field amplitude, it is possible to generate a supplementary calcium spike with properties close to those of calcium spontaneous oscillations, or, on the contrary, to inhibit the spontaneous calcium oscillations for a very long time compared to the pulse duration. Through that inhibition of the oscillations, Ca2+ oscillations of desired amplitude and frequency could then be imposed on the cells using subsequent electric pulses. None of the pulses used here, even those with the highest amplitude, caused a loss of cell viability.
Conclusions
An easy way to control Ca2+ oscillations in mesenchymal stem cells, through their cancellation or the addition of supplementary Ca2+ spikes, is reported here. Indeed, the direct link between the microsecond electric pulse(s) delivery and the occurrence/cancellation of cytosolic Ca2+ spikes allowed us to mimic and regulate the Ca2+ oscillations in these cells. Since microsecond electric pulse delivery constitutes a simple technology available in many laboratories, this new tool might be useful to further investigate the role of Ca2+ in human mesenchymal stem cells biological processes such as proliferation and differentiation.
Similar content being viewed by others


Background
Mesenchymal stem cells (MSCs) are multipotent stromal cells [1] originating from the embryonic mesoderm (mesenchyme) and present in many adult tissues such as bone marrow (bMSCs), adipose tissue (aMSCs), muscle, dermis, umbilical cord, etc. [2, 3]. These cells have gained a lot of momentum in the last decade due to their ability to differentiate into a wide variety of cells including osteoblasts, myoblasts, fibroblasts and chondrocytes. They also express key markers of cardiomyocytes, neuronal and endothelial cells [4]. This ability makes them a very promising candidate for cell therapy and regenerative medicine in order to heal damaged tissues and organs. However, MSCs from different tissues are not the same. They have different differentiation capacities and transcriptomic signatures [5].
Human-adipose MSCs (haMSCs), derived from adipose tissue are amongst the most easily accessible MSCs, with high quantities, and without aggressive extraction procedures. They are more available than other MSCs as, for example, the human bMSCs (hbMSCs). In addition, they have a phenotype, surface markers [6], and gene expression profile similar to those of the hbMSCs, and they are easier to maintain and proliferate [3], which make them ideal MSCs to use [7].
These cells present spontaneous Ca2+ oscillations, implicating Ca2+ channels and pumps of the plasma membrane (PM) and the endoplasmic reticulum (ER) [8]. These oscillations seem to start by an ATP autocrine/paracrine signaling [9] followed by inositol triphosphate (IP3)-induced Ca2+ release from the ER and further amplification from plasma membrane store-operated Ca2+ channels (SOCCs). Afterwards, the excess of Ca2+ is removed from the cytosol by the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), the plasma membrane Ca2+ ATPase (PMCA), and the Na+/Ca2+ exchanger (NCX) [10]. Ca2+ is one of the most important second messenger in the cell, and it regulates many important cellular processes such as ATP synthesis, apoptosis, cellular motility, growth, proliferation and gene expression. Hence, Ca2+ oscillations contain embedded information that has to be decoded by the cell, and Ca2+ signalling pathways play a key role in controlling cell behaviour and differentiation processes of MSCs.
It was shown that the Ca2+ oscillations frequency is different between undifferentiated MSCs and MSCs on route to differentiation and it differs between the various outcomes of the differentiation process (the level of differentiation and the differentiated cell type). While the MSCs exhibit regularly repeated Ca2+ oscillations, MSCs undergoing osteodifferentiation display a decrease in the frequency of their spontaneous Ca2+ oscillations while primary myoblasts present still another pattern of oscillations [11]. This shows that each cell type possesses its own pattern of Ca2+ oscillation frequency and shape, but the exact correlation between Ca2+ oscillations and MSCs differentiation is still unclear.
Presently, pulsed electric fields (PEFs) are widely used in research as a non-invasive physical means to permeabilize cellular membranes. Using one or several pulses of ultrashort duration causes changes in the cell membrane structure that permits access to the cell cytosol to molecules that cannot cross the plasma membrane under normal conditions [12]. Normally, Ca2+ is an ion that only crosses the plasma and ER membranes through channel proteins. Applying PEFs to cells in a medium containing Ca2+ (amongst other compounds) allows Ca2+ entry from the cell outside, and, if the electric field amplitude and pulse duration are high enough, Ca2+ is released from the cell inner stores [13]. The electromagnetic fields (EMFs) have been already applied on hbMSCs to test their effect on the differentiation of the latter. Those EMFs were either direct currents of very low electric field amplitude (0.1 V/cm) and a long time exposition (30 minutes) [14] or biphasic electric currents (1.5 μA/cm2 for 250 μs) [15]. These fields either activate voltage-operated Ca2+ (VOCCs) if their amplitude is large enough or do not alter the membrane potential if the electric field is too low. None of the electromagnetic fields used was permeabilizing the cell PM.
The novelty of our study is the use of PEFs of a high field amplitude that are capable of permeabilizing the cell PM or both the PM and the ER. Our objectives were to test the effect of these pulses on the Ca2+ oscillations of the cells. Attached haMSCs presenting normal spontaneous Ca2+ oscillations were exposed to one single pulse with a duration of 100 μs and different electric field amplitudes. At low and moderate electric field amplitudes, the Ca2+ spike induced by such PEFs can be very similar to a spontaneous oscillation. At high electric field amplitudes, the electrically induced Ca2+ spikes inhibit the spontaneous oscillations occurrence for some time after which oscillations reappear. This inhibition allowed us to impose on the cells experimentally induced Ca2+ spikes at a desired frequency. Hence, we show here that it is possible to control haMSCs spontaneous Ca2+ oscillations by microsecond pulsed electric fields (μsPEFs) with a complete preservation of the cell viability. The delivery of μsPEFs constitutes an easy way to control Ca2+ oscillations in mesenchymal stem cells, through their cancellation or the addition of supplementary Ca2+ spikes. Indeed, unlike chemical factors, which cause continuous effects, the direct link between the microsecond electric pulse(s) delivery and the ulterior occurrence/cancellation of the cytosolic Ca2+ spikes allowed us to mimic and regulate the Ca2+ oscillations in these cells. Since microsecond electric pulses delivery constitutes a simple technology available in many laboratories, the electrical control of Ca2+ oscillations could be in the future a promising tool to further investigate the role of Ca2+ in MSCs physiology and to better understand the correlation between the Ca2+ oscillations and MSCs biological processes such as proliferation and differentiation.
Methods
Cells and cell culture conditions
HaMSCs were isolated from surgical waste of individuals undergoing elective lipoaspiration. Samples were obtained after written informed consent from all the donors, in accordance with French and European legislations. The lipoaspirates were surgical waste and as such the French legislation (Art.L. 1245-2 du Code de la Santé Publique) establishes that the authorisation from an ethics committee is not required. Every experiment was done on cells before passage 8 from at least two donors (seven different donors in total). All haMSCs reacted similarly to the electric pulses, whoever the donor. The cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with Glutamax and supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 mg/mL streptomycin and were cultured at 37 °C in a humidified incubator with 5% CO2. Cells were passed twice a week (every passage corresponds to one doubling time of the population). The multipotency capabilities of the cells were assessed by submitting them to differentiation conditions as previously reported in Liew et al. [16]. Cells from all the donors have thus been differentiated in osteoblasts, adipocytes and chondrocytes. The cell culture chemicals were purchased from Fischer Scientific (Parc d’innovation Illkirch, France).
Cell staining
One day prior to the experiments, cells were seeded in 24-well plates at a density of 20 × 103 cells/cm2. After 1 day, the cells were incubated for 30 minutes with 5 μM of Fluo-4 AM (Fischer Scientific), a fluorescent Ca2+ marker, in a humidified 5% CO2 atmosphere at 37 °C in complete DMEM. The incubation buffer also contained 375 nM of the nuclear fluorescent dye Hoechst 33342. After incubation, the attached cells were rinsed three times with PBS (phosphate-buffered saline) and 500 μl of complete DMEM were added to the cells.
Microsecond pulse generator and electrodes
One single micropulse of 100 μs was delivered in all the experiments. A Cliniporator™ (Igea, Carpi, Italy) was used for the generation of the microsecond pulsed electric fields (μsPEFs). In order to treat the cells under the microscope, the pulse generator was connected to two parallel stainless steel rods of 1.2 mm diameter used as electrodes. The distance between them was 5 mm, and they were shaped to enter a 24-plate well and to reach the bottom of the dish. The whole system was set under a Zeiss Axiovert S100 epifluorescence inverted microscope.
In order to test the cell viability, another model of electrodes was designed. In this system, a thick cover of 10 cm2 containing two slots was designed to fit into a Petri dish of the same dimensions. Two plate electrodes of 2 mm thickness and 2 cm length were slipped into the slots until they touched the bottom of the Petri dish. This system does not permit observation of the cells under a microscope, it was used because it allows covering a larger surface (in order to have enough cells) than the system described above.
Images acquisition and analyses
Images of the cells were taken every 10 seconds for 10 to 20 minutes with a Zeiss AxioCam Hrc camera controlled by the Axio Vision 4.6 software (Carl Zeiss, Oberkochen, Germany). The electric pulse was delivered after at least 2 minutes of recording, and 2 seconds before the next image. The excitation and emission wavelengths used for Fluo-4 were 496 nm and 515 nm respectively. The nuclear dye Hoechst 33342 (λex = 350 nm, λem = 461 nm) was used to recognise the nuclei and to track the cells using the Cell Profiler (version 2.0) software (Broad Institute, Cambridge, MA, USA). The software allowed the automatic measurement of the fluorescence intensity signal of each cell on every image. Curves were plotted using a Matlab program (version 7.8.0, The MathWorks Inc., Natick, MA, USA). All the observations were done at room temperature. The minimum opening time of the shutter for the fluorescent light was about 500 ms. To decrease the light energy applied on the cells, a 90% density Filter NE110B (Thorlabs, Maisons-Lafitte, France) was used.
Cell viability assessment
One day prior to the experiments, cells were seeded at a density of 20 × 103 cells/cm2 in 10-cm2 Petri dishes containing a 12 × 32 mm cover slide. After 24 hours, medium was removed, cells were washed with PBS, and 1 ml of fresh medium was added. The electrodes were placed on the cover slide using the Petri dish cover containing two slots and a 100-μs pulse was delivered to the attached cells. After the pulse, the Petri dish was put in the incubator for 20 minutes. Then, the non-pulsed cells on the cover slides were scratched under a sterile hood. The area covered by the pulsed cells was recognised due to the loss of cells beneath the electrodes after the removal of the latter. The cover slide with the pulsed cells only was transferred to a Petri dish with fresh medium and placed for 2 hours in the incubator. Then medium was removed and the cells were trypsinised, centrifuged, and resuspended in 300 μl of a fresh medium. This volume was distributed in two wells of an opaque-walled 96-multiwell plate and put in the incubator for 24 hours. After 24 hours, cells were visualised on an epifluorescence microscope to compare their shape and level of confluence, and 150 μl of Cell-Titer Glo Reagent (Promega, Madison, WI, USA) were added to each well according to manufacturer’s protocol. The reagent caused cell lysis and the generation of a luminescent signal proportional to the amount of the ATP present in the medium. A GloMax luminometer (Promega) was used to read the luminescence. The ATP amount and therefore the luminescence intensity reflected the number of cells present in the culture [17].
Statistical analysis
All the experiments were done at least three times. Data are presented as means and standard deviations. The Spearman correlation was used to analyse the data of Figs. 3 and 6.
Results
Spontaneous Ca2+ oscillations in haMSCs in normal conditions and after one 100-μs electric pulse
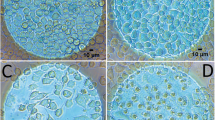
Undifferentiated haMSCs present asynchronous Ca2+ oscillations as seen in Fig. 1 (left column). The oscillation frequencies differed from cell to cell and not all of the cells displayed spontaneous Ca2+ oscillations. When one electric pulse of 100 μs was applied on haMSCs, different situations could be observed. As shown in Fig. 1 (middle column), an electric pulse of 300 V/cm induced a Ca2+ spike in all the cells whatever the stage of their Ca2+ oscillation at the time the pulse was applied. After the pulse, the spontaneous Ca2+ oscillations returned to their original cell-dependent frequency and continued normally. When the electric pulse applied was 900 V/cm (Fig. 1, right), all the cells presented a Ca2+ spike with higher amplitude with respect to 300 V/cm. However, even 750 s after the pulse, the cells still presented a high cytosolic Ca2+ concentration (as shown by the persistent Fluo-4 high fluorescence level) and the spontaneous Ca2+ oscillations did not yet resume.

Time-lapse follow-up of calcium oscillations and spikes in haMSCs. Left: spontaneous calcium oscillations. Middle: spontaneous calcium oscillations before and after a 100-μs pulse of 300 V/cm (the pulse was applied at 630 s and a calcium spike in all the cells follows the pulse delivery at e.g. 650 s). Right: spontaneous calcium oscillations before a 100-μs pulse of 900 V/cm (applied at t = 630 s) followed by a non-oscillating increased calcium signal
Ca2+ spikes in response to different electric field amplitudes
The shapes of the electrically induced Ca2+ spikes in response to a 100-μs duration pulse of different electric field amplitudes are represented in Fig. 2. At a very low electric field amplitude (100 V/cm), no Ca2+ spike was observed. At 150 V/cm, the Ca2+ spikes had a lower amplitude than the spontaneous oscillations and presented a relatively small slope. At moderate electric field amplitudes (200 to 450 V/cm), each electric pulse induced a Ca2+ spike. The Ca2+ spike displayed a higher slope than at 150 V/cm, then reached its maximal amplitude and came back to the normal Ca2+ amplitude as before the pulse. The electro-induced Ca2+ spikes had nearly the same shape and amplitude as the spontaneous oscillations at 200 and 300 V/cm. At 450 V/cm, their amplitudes were higher than the spontaneous ones, but they came back to the original baseline. When high electric field amplitudes were applied (600 to 900 V/cm), the electrically induced Ca2+ spike presented larger slopes (with different degree of sharpness depending on the electric field strength) and displayed a higher amplitude than the spontaneous oscillations; after the Ca2+ spike, the Ca2+ amplitude remained relatively high compared to the initial amplitude, and it took a longer time (several minutes) to come back to normal depending also on the amplitude of the applied electric field.

Calcium spikes in response to different electric field amplitudes. For each panel, two separate adherent cells, displaying the typical behaviour of the cells after the delivery of one 100-μs electric pulse, were chosen to illustrate that behaviour. In all the cases, the pulse was applied at time = 390 s (black arrows)
According to Fig. 3, only 10% of the cells presented a Ca2+ spike in response to the electric pulse at 150 V/cm. This percentage increased proportionally to the electric field amplitude, mainly between 150 and 300 V/cm. One hundred percent of the cells responded to the pulse at 450 V/cm. According to Spearman correlation analysis, there was a perfect correlation between the electric field amplitude and the percentage of cells displaying a Ca2+ spike (r = 0.96).

Percentage of cells presenting a calcium spike in response to one 100-μs electric pulse as a function of the field amplitude. r = 0.96 according to Spearman correlation
Different outcomes on Ca2+ oscillations according to the electric field amplitude
According to the electric field amplitude, the pulse-induced Ca2+ spike had different effects on Ca2+ oscillations. At a relatively low electric field amplitude (300 V/cm), the pulse-induced Ca2+ spike displayed a very similar shape to the spontaneous Ca2+ oscillations (Fig. 4a), and it could be generated between two oscillations or during an oscillation. After the 100-μs pulse-induced Ca2+ spike, the spontaneous oscillations continued normally at their proper initial rhythm with no significant modification of the amplitude or duration of the oscillations. At relatively high electric field amplitude such as 600 V/cm, the pulse-induced Ca2+ spike displayed higher amplitude than the spontaneous oscillations and three different outcomes could be observed after the electro-induced spike. The oscillations could continue normally, with a minor change of the oscillation shape (Fig. 4b, upper trace). Or, second, a short inhibition of the spontaneous oscillations could be observed for some minutes, after which the oscillations reappeared (Fig. 4b, lower trace). The first oscillations that appeared after such an inhibition period did not display the same shape as the original ones. The third possible outcome was a longer inhibition of the Ca2+ oscillations, for a duration of longer than 10 minutes (Fig. 4c). The application of higher electric field amplitude (750 and 900 V/cm) resulted in the inhibition of the spontaneous Ca2+ oscillations for longer times. For example, only some cells had recovered their oscillations 45 to 60 minutes after the pulse delivery. However, after 24 hours all cells recovered their oscillations whatever the electric field applied (data not shown).

Examples of changes in the pattern of Ca2+ oscillations caused by the delivery of one 100-μs electric pulse of different field amplitudes, a 300 V/cm, b and c 600 V/cm, d 900 V/cm
The percentage of cells with no spontaneous oscillation for at least 10 minutes after the electric pulse delivery increased as a function of the electric field amplitude (Fig. 5). While no inhibition of the spontaneous oscillations was observed at 300 V/cm, 25% of the cells had their Ca2+ oscillations inhibited after one electric pulse at 450 V/cm and nearly all the cells at 750 V/cm. Twenty-four hours after the pulse delivery, the cells remained viable under all the conditions tested, even for the highest electric field amplitude (Fig. 6). Photos taken before the Cell-Titer Glo assay showed no difference in the shape and level of confluence of the cells between the pulsed and the control wells (data not shown). The Spearman correlation showed no effect of the electric field amplitude on the cell viability (r = 0.43).

Percentage of cells displaying no Ca2+ oscillation within a period of at least 10 minutes after the delivery of one 100-μs electric pulse of different field amplitudes

haMSCs cell viability after the delivery of one 100-μs electric pulse of different field amplitudes, r = 0.43 according to Spearman correlation
Electrically mediated Ca2+ spikes manipulation after the 900 V/cm inhibition
To test whether the cells were in a “refractory” state after the inhibition of their Ca2+ oscillations, or if they were still able to respond to the electric pulses, pulses of different field amplitudes were applied after a pulse of 900 V/cm (which provokes the absence of further spontaneous oscillations in 100% of the cells). For example (Fig. 7), a sequence of three pulses with descending electric field amplitudes (750 followed by 600 and 450 V/cm) was applied successively: the cells responded to the electric pulses as if each pulse would have been applied alone, or independently. The amplitude of the electrically induced Ca2+ spike was again proportional to the electric field amplitude like in Fig. 2. Thus calibrated electric pulses allow restoration of Ca2+ spikes at the time and with the amplitude chosen by the investigators.

Two examples of calcium spike manipulation in haMSCs. After the delivery of one 100-μs electric pulse at 900 V/cm (that abolishes the spontaneous Ca2+ oscillations), pulses of various field amplitudes were delivered to mimic Ca2+ oscillations at experimentally controlled times
Discussion
Mesenchymal stem cells (MSCs) are multipotent stromal cells that have the ability to self-renew and to differentiate into a wide variety of cells of the mesodermal, ectodermal, and endodermal lineages [18]. This is why they represent an important tool for cell therapy and regenerative medicine. However, the differentiation procedures may be long and not able to respond to the demand. The improvement and optimisation of these procedures is of a crucial importance to ensure the differentiated cells in a reasonable time. Many studies have tested the effects of physical stimuli on the differentiation outcomes. Altman et al. showed that the application of a cyclic mechanical stimulation (translational and rotational strain) to undifferentiated MSCs (embedded in a collagen gel) over a period of 21 days resulted in ligament cell lineage differentiation even without differentiation factors [19]. Ward and colleagues studied the effect of the application of a 3–5% tensile strain to a collagen I substrate and concluded that it stimulated osteogenesis of attached hMSCs [20]. Low-intensity ultrasounds, another form of mechanical stress, have been shown to enhance the chondrogenic differentiation [21]. Sun et al. demonstrated that 0.1 V/cm electrical stimulation applied for 30 minutes per day for 10 days, on attached MSCs cultured with osteoinductive factors, accelerated their osteodifferentiation [14].
Ca2+ oscillations play an important role in the transduction of many physical stimuli in the cells. For example, tissue strain and shear stress on mouse tibia are transduced by repetitive Ca2+ spikes in the osteocytes. The spikes frequency and amplitude depend on the mechanical magnitude [22]. Electromagnetic fields (EMFs) can also promote MSCs differentiation to osteoblasts through Ca2+-related mechanisms. The exposition of MSCs to pulsed EMFs for 10 minutes every day results in the enhancement of osteogenesis early stages [23].
Ca2+ oscillations are a universal mode of Ca2+ signalling in both excitable [24, 25], and non-excitable cells [26]. In non-excitable cells such as hMSCs, Ca2+ oscillations are typically initiated by a receptor-triggered production of IP3 (after the binding of an agonist to the receptor) [27] and the subsequent Ca2+ release from the ER [28], which stimulates the store-operated calcium entry (SOCE) via the SOCCs [29]. It was shown that the Ca2+ oscillation frequency is different between undifferentiated MSCs and MSCs on route to differentiation and it differs between the various differentiated cell types [11]. According to Titushkin et al., the MSCs Ca2+ oscillations are suppressed after the addition of neuroinductive factors, but reappear in less than 7 days after neurodifferentiation [11]. Sun and colleagues observed that multipotent MSCs present 8.06 ± 2.64 Ca2+ spikes per 30 minutes of observation, and that this number decreases to 3.66 ± 2.42 after 28 days of incubation with osteoinductive factors [14].
In our study, we decided to analyse the effect of a type of short electric pulses termed microsecond pulsed electric fields (μsPEFs) on the spontaneous Ca2+ oscillations of haMSCs because many of the physical stimuli are transduced by cytosolic Ca2+ concentration rapid changes (oscillations) in the cells. The ultrashort pulses (one thousandth to one ten thousandth shorter than the pulses of 100 μs used in our study) called nanopulses or nanosecond pulsed electric fields (nsPEFs) have been used already to induce Ca2+ bursts in the cell, which resulted from the electropermeabilization of the ER, the plasma membrane or both [13, 30]. However, contrary to the 100-μs electric pulses, the technology to deliver nanopulses is not yet spread in the laboratories and is not simple to use.
The haMSCs loaded with the fluorescent marker Fluo-4 asynchronously displayed periodic changes in their cytosolic Ca2+ concentration detected by the periodic increase of their fluorescence. The videos of the haMSCs showed that not all the cells presented Ca2+ oscillations at a specific time. Some of the cells never displayed oscillations during the recording time (20 minutes). This observation can be related to the fact that MSCs display Ca2+ oscillations only during the phases G1 and S of the cell cycle, and not all the cells in the visualisation area of an experiment were in those phases of the cell cycle. Ca2+ oscillations increase the levels of cell cycle regulators such as cyclins A and E and probably control cell cycle progression and cell proliferation, via the regulation of cyclin levels (amongst other mechanisms) [31]. In addition, the haMSCs presenting Ca2+ oscillations displayed them at different rhythms and frequencies: the oscillations were asynchronous, and at a given time, each cell was in a different phase of a Ca2+ oscillation and the oscillations frequencies displayed wide variations between cells. The average duration of an oscillation was around 2 minutes.
Electric pulses of 100 μs duration are already applied in many biotechnological and medical applications, notably in anticancer electrochemotherapy [32], tumour ablation [33], and cell transfection [34]: by permeabilizing the plasma membrane temporally they allow the internalisation of non-permeant molecules of interest like drugs or nucleic acids. Classically, eight successive pulses of 100 μs are used in biomedical sciences. The technology is spread in many laboratories and is very simple to use. In our study, we applied a single electric pulse because, when several pulses are delivered, pulses repetition frequency may impact the efficacy of cell membrane permeabilization as we have recently shown in our group [35]. When an electric pulse of 100 μs was applied to the cells, an electrically induced Ca2+ spike was observed synchronously in a given percentage of the cells (as a function of the electric field amplitude). Moreover, the electric field amplitude should be enough to cause the transmembrane voltage to reach a permeabilization threshold. This threshold depends on the size of the molecule to be internalised. This is why, at 100 V/cm, which is a very low electric field amplitude, no Ca2+ spike was observed because there was no permeabilization and hence no Ca2+ entry. 120 V/cm was the lowest electric field amplitude at which some cells presented Ca2+ spikes. This threshold is very low compared to other ones previously reported, because the Ca2+ has a small size compared to other classical markers such as yo-pro-1 iodide, propidium iodide or bleomycin that are those frequently used (Hanna et al., submitted). At 450 V/cm, 100% of the cells responded to the electric pulse. The origin of the Ca2+ causing the Ca2+ spike was not always the same: it could be the result of Ca2+ entry from the external medium only, or the combination of Ca2+ entry from cells outside and Ca2+ release from the ER. Our group demonstrated recently that the μsPEFs could permeabilize not only the cell membrane (as usually observed and stated) but also the internal organelles membranes (Hanna et al., submitted). The origin of the Ca2+ spike depends on the electric field amplitude. If the latter is below 500 V/cm, the pulse permeabilizes only the plasma membrane (Hanna et al., submitted), and the Ca2+ spike is primarily the result of Ca2+ entry through the plasma membrane followed by an amplification due to Ca2+ entry from the voltage-operated Ca2+ channels (VOCCs), activated by the membrane depolarisation [36]. However, above 500 V/cm, the electric pulses will also affect the ER membranes, permeabilize them and cause Ca2+ release from the ER (Hanna et al., submitted). Hence, the Ca2+ spike at high electric field amplitudes, resulted primarily of a massive Ca2+ entry through the plasma membrane (due to the generation of larger pores at the higher field amplitudes) also followed by the external Ca2+ entry through the VOCCs, the Ca2+ release from the ER and the subsequent stimulation of the store-operated Ca2+ channels (SOCCs) [37]. This explains why the Ca2+ spike induced by electric fields below 500 V/cm had a very similar shape to the spontaneous Ca2+ oscillations with a gradual increase and same amplitude, whereas the Ca2+ spike induced by electric fields above 500 V/cm had higher amplitude than the spontaneous oscillations, and presented a sharper rise (due to the abrupt penetration of Ca2+ through the permeabilized membranes). At 600 V/cm, three different outcomes were observed, mainly due to four factors: first the distribution of the electric field which results in field amplitude small differences in the area between the two electrodes; second, the cells are heterogeneous in size and according to Schwan equation [38], the electric field impact will be different between the smallest and the largest cells; third, the orientation of the cell with respect to the electric field lines, which has an impact on the induced transmembrane potential that will lead to the cell membrane permeabilization [39]; fourth, the position of the cells are in the cell cycle that could lead to different responses to the pulses. However, at 750 V/cm, it seems that the electric field has exceeded a certain threshold above which no more heterogeneity was observed and nearly all the cells had their Ca2+ oscillations inhibited.
The Ca2+ spikes obtained looked like the Ca2+ spikes induced by even shorter electric pulses (the so-called nanopulses, of a duration of e.g. 10 ns, that can directly affect the cell internal membranes such as those of the endoplasmic reticulum [30]). In our study, an electrically induced Ca2+ spike with properties very near to the oscillations did not inhibit the latter, could be applied at any time of an oscillation without interfering with the latter, and after the pulse, the oscillations continued normally at their own rhythm, and with their normal shape. Nevertheless, a high-amplitude Ca2+ spike inhibited the Ca2+ oscillations. The percentage of cells that were unable to display further Ca2+ oscillations increased proportionally with the electric field amplitude of the inhibitory electric pulse. The Ca2+ oscillations inhibition could last some minutes or be longer, to reach tens of minutes, also depending on the electric field amplitude of the inhibitory electric pulse.
The inhibition of the Ca2+ oscillations for some time allowed us to study the state of the cell during this period, to determine whether the cell was in a “refractory state” or if it was still able to respond to an electric pulse. The application of several pulses after a 900 V/cm pulse inhibition showed that the cells could still react to the pulse and present Ca2+ spikes with different amplitudes depending on the applied electric field amplitude.
Last but not least, the application of one 100-μs electric pulse did not affect the cell viability 24 hours after treatment (Fig. 6), even when applying a much stronger electric field than the one used to inhibit the calcium oscillations for several minutes. Moreover, in a previous study [16] we showed that haMSCs maintain their stem cell phenotype after being exposed to even stronger electric field conditions (eight electric pulses of 100 μs at 1500 V/cm and at a 1Hz frequency). More precisely:
-
1.
Seven days after exposure, MSCs showed similar stem cell surface antigens expression compared with non-treated MSCs.
-
2.
Four days after exposure to the eight electric pulses, MSCs were cultured in osteogenic and adipogenic differentiation media and successfully differentiated into osteoblasts and adipocytes.
-
3.
Eleven days after exposure to the electric pulses, MSCs cultures yielded similar cell growth rates compared with non-treated MSCs cultures.
The electrically induced Ca2+ spikes can have wide effects on the Ca2+ oscillations frequency, amplitude and duration. Whereas the nanosecond pulses allowed only to add a Ca2+ spike to the oscillations [30], the microsecond pulses display more complex effects. The electric field amplitude allowed us either to induce a Ca2+ spike without affecting the oscillations, or to inhibit the Ca2+ oscillations and impose on the cell Ca2+ oscillations at a desired frequency and amplitude. It is worth noting that the microsecond technology is cheaper than the nanosecond one, it is more available in the laboratories, and simple generators can deliver it. These considerations add further value to the observations reported here. Taking into account that each type of differentiated cell from MSCs has its own Ca2+ oscillation characteristics, and that these oscillations may play an important role in the decision of MSCs fate, the μsPEFs constitute a novel useful tool to control the oscillations and the differentiation of MSCs.
Conclusions
hMSCs show great promise for regenerative medicine. As a function of the pulse(s) electric field amplitude, we report here that it is possible to generate in hMSCs a supplementary calcium spike with properties close to those of their spontaneous calcium oscillations, or, on the contrary, to inhibit their spontaneous calcium oscillations for a very long time compared to the pulse duration. Through that inhibition of the oscillations, Ca2+ oscillations of desired amplitude and frequency could then be imposed on the cells using subsequent electric pulses. The delivery of short PEFs of 100 μs, a robust technology easy to apply, could be therefore a promising way to investigate the role of the Ca2+ in MSCs physiology and biological processes such as proliferation and differentiation.
Abbreviations
- aMSCs:
-
Adipose tissue mesenchymal stem cells
- bMSCs:
-
Bone marrow mesenchymal stem cells
- DMEM:
-
Dulbecco’s Modified Eagle Medium
- EMFs:
-
Electromagnetic fields
- ER:
-
Endoplasmic reticulum
- haMSCs:
-
Human aMSC
- hbMSCs:
-
Human bMSC
- IP3:
-
Inositol triphosphate
- MSCs:
-
Mesenchymal stem cells
- NCX:
-
Na+/Ca2+ exchanger
- PBS:
-
Phosphate-buffered saline
- PEFs:
-
Pulsed electric fields
- PM:
-
Plasma membrane
- PMCA:
-
Plasma membrane Ca2+ ATPase
- SERCA:
-
Sarco/endoplasmic reticulum Ca2+ ATPase
- SOCCs:
-
Store-operated Ca2+ channels
- VOCCs:
-
Voltage-operated Ca2+ channels
- μsPEFs:
-
Microsecond pulsed electric fields
References
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–7.
Malgieri A, Kantzari E, Patrizi MP, Gambardella S. Bone marrow and umbilical cord blood human mesenchymal stem cells: state of the art. Int J Clin Exp Med. 2010;3:248–69.
Lee RH, Kim B, Choi I, Kim H, Choi HS, Suh K, et al. Characterization and expression analysis of mesenchymal stem cells from human bone marrow and adipose tissue. Cell Physiol Biochem. 2004;14:311–24.
Marion NW, Mao JJ. Mesenchymal stem cells and tissue engineering. Methods Enzymol. 2006;420:339–61.
Sacchetti B, Funari A, Remoli C, Giannicola G, Kogler G, Liedtke S, et al. No identical “mesenchymal stem cells” at different times and sites: human committed progenitors of distinct origin and differentiation potential are incorporated as adventitial cells in microvessels. Stem Cell Rep. 2016;6:897–913.
Gronthos S, Franklin DM, Leddy HA, Robey PG, Storms RW, Gimble JM. Surface protein characterization of human adipose tissue-derived stromal cells. J Cell Physiol. 2001;189:54–63.
Lindroos B, Suuronen R, Miettinen S. The potential of adipose stem cells in regenerative medicine. Stem Cell Rev. 2011;7:269–91.
Kawano S, Shoji S, Ichinose S, Yamagata K, Tagami M, Hiraoka M. Characterization of Ca(2+) signaling pathways in human mesenchymal stem cells. Cell Calcium. 2002;32:165–74.
Kawano S, Otsu K, Kuruma A, Shoji S, Yanagida E, Muto Y, et al. ATP autocrine/paracrine signaling induces calcium oscillations and NFAT activation in human mesenchymal stem cells. Cell Calcium. 2006;39:313–24.
Kawano S, Otsu K, Shoji S, Yamagata K, Hiraoka M. Ca(2+) oscillations regulated by Na(+)-Ca(2+) exchanger and plasma membrane Ca(2+) pump induce fluctuations of membrane currents and potentials in human mesenchymal stem cells. Cell Calcium. 2003;34:145–56.
Titushkin I, Sun S, Shin J, Cho M. Physicochemical control of adult stem cell differentiation: shedding light on potential molecular mechanisms. BioMed Res Int. 2010;2010:e743476.
Mir LM, Banoun H, Paoletti C. Introduction of definite amounts of nonpermeant molecules into living cells after electropermeabilization: direct access to the cytosol. Exp Cell Res. 1988;175:15–25.
Vernier PT, Sun Y, Marcu L, Salemi S, Craft CM, Gundersen MA. Calcium bursts induced by nanosecond electric pulses. Biochem Biophys Res Commun. 2003;310:286–95.
Sun S, Liu Y, Lipsky S, Cho M. Physical manipulation of calcium oscillations facilitates osteodifferentiation of human mesenchymal stem cells. FASEB J. 2007;21:1472–80.
Kim IS, Song JK, Song YM, Cho TH, Lee TH, Lim SS, et al. Novel effect of biphasic electric current on in vitro osteogenesis and cytokine production in human mesenchymal stromal cells. Tissue Eng Part A. 2009;15:2411–22.
Liew A, André FM, Lesueur LL, De Ménorval M-A, O’Brien T, Mir LM. Robust, efficient, and practical electrogene transfer method for human mesenchymal stem cells using square electric pulses. Hum Gene Ther Methods. 2013;24:289–97.
Crouch SP, Kozlowski R, Slater KJ, Fletcher J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J Immunol Methods. 1993;160:81–8.
Woodbury D, Reynolds K, Black IB. Adult bone marrow stromal stem cells express germline, ectodermal, endodermal, and mesodermal genes prior to neurogenesis. J Neurosci Res. 2002;69:908–17.
Altman GH, Horan RL, Martin I, Farhadi J, Stark PRH, Volloch V, et al. Cell differentiation by mechanical stress. FASEB J. 2002;16:270–2.
Ward DF, Salasznyk RM, Klees RF, Backiel J, Agius P, Bennett K, et al. Mechanical strain enhances extracellular matrix-induced gene focusing and promotes osteogenic differentiation of human mesenchymal stem cells through an extracellular-related kinase-dependent pathway. Stem Cells Dev. 2007;16:467–80.
Ebisawa K, Hata K-I, Okada K, Kimata K, Ueda M, Torii S, et al. Ultrasound enhances transforming growth factor beta-mediated chondrocyte differentiation of human mesenchymal stem cells. Tissue Eng. 2004;10:921–9.
Jing D, Baik AD, Lu XL, Zhou B, Lai X, Wang L, et al. In situ intracellular calcium oscillations in osteocytes in intact mouse long bones under dynamic mechanical loading. FASEB J. 2014;28:1582–92.
Petecchia L, Sbrana F, Utzeri R, Vercellino M, Usai C, Visai L, et al. Electro-magnetic field promotes osteogenic differentiation of BM-hMSCs through a selective action on Ca2 + -related mechanisms. Sci Rep. 2015;5:13856.
Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–12.
Tsien RW. Calcium channels in excitable cell membranes. Annu Rev Physiol. 1983;45:341–58.
Fewtrell C. Ca2+ oscillations in non-excitable cells. Annu Rev Physiol. 1993;55:427–54.
Nash MS, Young KW, John Challiss RA, Nahorski SR. Intracellular signalling: Receptor-specific messenger oscillations. Nature. 2001;413:381–2.
Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, et al. STIM is a Ca2+ sensor essential for Ca2 + -store-depletion-triggered Ca2+ influx. Curr Biol CB. 2005;15:1235–41.
Parekh AB, Putney JW. Store-operated calcium channels. Physiol Rev. 2005;85:757–810.
de Menorval M-A, Andre FM, Silve A, Dalmay C, Français O, Le Pioufle B, et al. Electric pulses: a flexible tool to manipulate cytosolic calcium concentrations and generate spontaneous-like calcium oscillations in mesenchymal stem cells. Sci Rep. 2016;6:32331.
Resende RR, Adhikari A, da Costa JL, Lorençon E, Ladeira MS, Guatimosim S, et al. Influence of spontaneous calcium events on cell-cycle progression in embryonal carcinoma and adult stem cells. Biochim Biophys Acta. 2010;1803:246–60.
Belehradek M, Domenge C, Luboinski B, Orlowski S, Belehradek J, Mir LM. Electrochemotherapy, a new antitumor treatment. First clinical phase I-II trial. Cancer. 1993;72:3694–700.
Al-Sakere B, André F, Bernat C, Connault E, Opolon P, Davalos RV, et al. Tumor ablation with irreversible electroporation. PLoS One. 2007;2:e1135.
Potter H, Heller R. Transfection by electroporation. Curr Protoc Cell Biol. 2011;52:20.5.1–20.5.10.
Silve A, Guimerà Brunet A, Al-Sakere B, Ivorra A, Mir LM. Comparison of the effects of the repetition rate between microsecond and nanosecond pulses: electropermeabilization-induced electro-desensitization? Biochim Biophys Acta. 2014;1840:2139–51.
Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3:a003947.
Smyth JT, Hwang S-Y, Tomita T, DeHaven WI, Mercer JC, Putney JW. Activation and regulation of store-operated calcium entry. J Cell Mol Med. 2010;14:2337–49.
Schwan HP. Electrical properties of tissue and cell suspensions. Adv Biol Med Phys. 1957;5:147–209.
Kotnik T, Pucihar G, Miklavcic D. Induced transmembrane voltage and its correlation with electroporation-mediated molecular transport. J Membr Biol. 2010;236:3–13.
Acknowledgements
The authors would like to thank Dr. Bassim Al Sakere for the lipoaspirates.
Funding
This work was supported by CNRS, Gustave Roussy, Université Paris-Sud, the ITMO Cancer in the frame of the Plan Cancer 2015-2019 (project PC201517) and the Fondation EDF. The research was also conducted in the scope of the EBAM European Associated Laboratory (LEA) and of the COST action BM 1309 EMF-MED.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request
Authors’ contributions
HH, FMA and LMM designed the study, contributed to data interpretation, analysed the data and drafted the manuscript. HH performed the experiments. All authors edited and approved the manuscript.
Authors’ information
Not applicable
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All the authors have read and approved the manuscript for publication.
Ethics approval and consent to participate
HaMSCs were isolated from surgical waste of individuals undergoing elective lipoaspiration. Samples were obtained after written informed consent from all the donors, in accordance with French and European legislations. The lipoaspirates were surgical waste and as such the French legislation (Art.L. 1245-2 du Code de la Santé Publique) establishes that the authorization from an ethics committee is not required.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hanna, H., Andre, F.M. & Mir, L.M. Electrical control of calcium oscillations in mesenchymal stem cells using microsecond pulsed electric fields. Stem Cell Res Ther 8, 91 (2017). https://doi.org/10.1186/s13287-017-0536-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-017-0536-z